DUX4 Expression in FSHD Muscles: Focus on Its mRNA Regulation



{kind=link}
Abstract
:1. Introduction
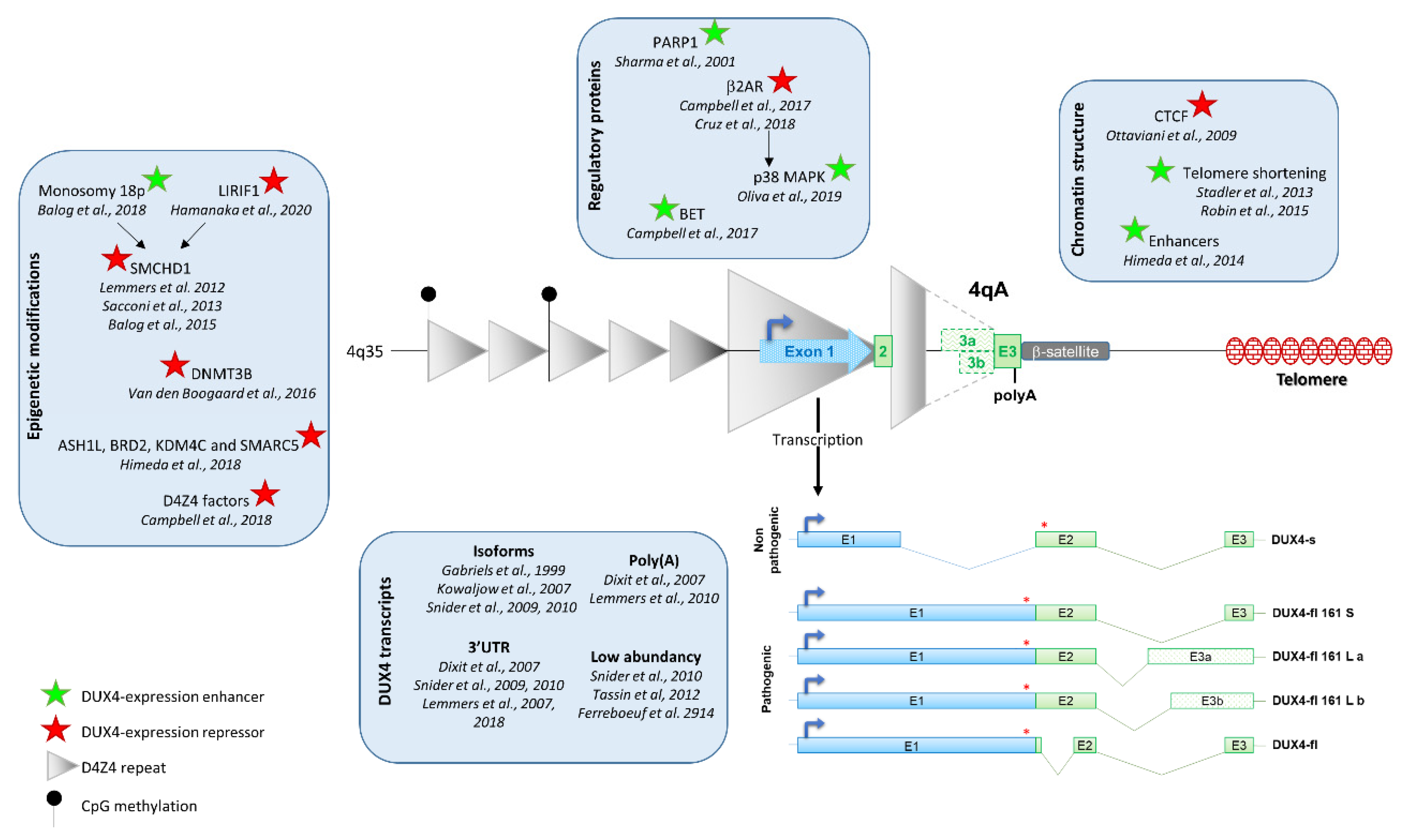
2. FSHD
3. Regulation of DUX4 Expression
3.1. D4Z4 Epigenetic Modification
3.2. Chromatin Conformation
3.3. Regulatory Proteins of DUX4 Expression
4. DUX4 mRNA
4.1. DUX4 Transcription
4.2. DUX4 Isoforms
5. DUX4 Low Abundancy and Stochastic Expression
6. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Greco, A.; Goossens, R.; van Engelen, B.; van der Maarel, S.M. Consequences of epigenetic derepression in facioscapulohumeral muscular dystrophy. Clin. Genet. 2020, 97, 799–814. [Google Scholar] [CrossRef]
- De Iaco, A.; Planet, E.; Coluccio, A.; Verp, S.; Duc, J.; Trono, D. DUX-family transcription factors regulate zygotic genome activation in placental mammals. Nat. Genet. 2017, 49, 941–945. [Google Scholar] [CrossRef]
- Whiddon, J.L.; Langford, A.T.; Wong, C.J.; Zhong, J.W.; Tapscott, S.J. Conservation and innovation in the DUX4-family gene network. Nat. Genet. 2017, 49, 935–940. [Google Scholar] [CrossRef] [Green Version]
- Hendrickson, P.G.; Dorais, J.A.; Grow, E.J.; Whiddon, J.L.; Lim, J.W.; Wike, C.L.; Weaver, B.D.; Pflueger, C.; Emery, B.R.; Wilcox, A.L.; et al. Conserved roles of mouse DUX and human DUX4 in activating cleavage-stage genes and MERVL/HERVL retrotransposons. Nat. Genet. 2017, 49, 925–934. [Google Scholar] [CrossRef]
- Snider, L.; Geng, L.N.; Lemmers, R.J.; Kyba, M.; Ware, C.B.; Nelson, A.M.; Tawil, R.; Filippova, G.N.; van der Maarel, S.M.; Tapscott, S.J.; et al. Facioscapulohumeral dystrophy: Incomplete suppression of a retrotransposed gene. PLoS Genet. 2010, 6, e1001181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chew, G.L.; Campbell, A.E.; De Neef, E.; Sutliff, N.A.; Shadle, S.C.; Tapscott, S.J.; Bradley, R.K. DUX4 Suppresses MHC Class I to Promote Cancer Immune Evasion and Resistance to Checkpoint Blockade. Dev. Cell 2019, 50, 658–671.e7. [Google Scholar] [CrossRef] [PubMed]
- Gabriels, J.; Beckers, M.C.; Ding, H.; De Vriese, A.; Plaisance, S.; van der Maarel, S.M.; Padberg, G.W.; Frants, R.R.; Hewitt, J.E.; Collen, D.; et al. Nucleotide sequence of the partially deleted D4Z4 locus in a patient with FSHD identifies a putative gene within each 3.3 kb element. Gene 1999, 236, 25–32. [Google Scholar] [CrossRef]
- Lim, K.R.Q.; Nguyen, Q.; Yokota, T. DUX4 Signalling in the Pathogenesis of Facioscapulohumeral Muscular Dystrophy. Int. J. Mol. Sci. 2020, 21, 729. [Google Scholar] [CrossRef] [Green Version]
- Salsi, V.; Magdinier, F.; Tupler, R. Does DNA Methylation Matter in FSHD? Genes 2020, 11, 258. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tawil, R.; Van Der Maarel, S.r.M. Facioscapulohumeral muscular dystrophy. Muscle Nerve 2006, 34, 1–15. [Google Scholar] [CrossRef]
- Wijmenga, C.; Hewitt, J.E.; Sandkuijl, L.A.; Clark, L.N.; Wright, T.J.; Dauwerse, H.G.; Gruter, A.M.; Hofker, M.H.; Moerer, P.; Williamson, R.; et al. Chromosome 4q DNA rearrangements associated with facioscapulohumeral muscular dystrophy. Nat. Genet. 1992, 2, 26–30. [Google Scholar] [CrossRef] [PubMed]
- van Deutekom, J.C.; Wijmenga, C.; van Tienhoven, E.A.; Gruter, A.M.; Hewitt, J.E.; Padberg, G.W.; van Ommen, G.J.; Hofker, M.H.; Frants, R.R. FSHD associated DNA rearrangements are due to deletions of integral copies of a 3.2 kb tandemly repeated unit. Hum. Mol. Genet. 1993, 2, 2037–2042. [Google Scholar] [CrossRef] [PubMed]
- van der Maarel, S.M.; Frants, R.R. The D4Z4 repeat-mediated pathogenesis of facioscapulohumeral muscular dystrophy. Am. J. Hum. Genet. 2005, 76, 375–386. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmers, R.J.; van der Vliet, P.J.; Klooster, R.; Sacconi, S.; Camano, P.; Dauwerse, J.G.; Snider, L.; Straasheijm, K.R.; van Ommen, G.J.; Padberg, G.W.; et al. A unifying genetic model for facioscapulohumeral muscular dystrophy. Science 2010, 329, 1650–1653. [Google Scholar] [CrossRef] [Green Version]
- van Overveld, P.G.; Lemmers, R.J.; Deidda, G.; Sandkuijl, L.; Padberg, G.W.; Frants, R.R.; van der Maarel, S.M. Interchromosomal repeat array interactions between chromosomes 4 and 10: A model for subtelomeric plasticity. Hum. Mol. Genet. 2000, 9, 2879–2884. [Google Scholar] [CrossRef] [Green Version]
- van Deutekom, J.C.; Bakker, E.; Lemmers, R.J.; van der Wielen, M.J.; Bik, E.; Hofker, M.H.; Padberg, G.W.; Frants, R.R. Evidence for subtelomeric exchange of 3.3 kb tandemly repeated units between chromosomes 4q35 and 10q26: Implications for genetic counselling and etiology of FSHD1. Hum. Mol. Genet. 1996, 5, 1997–2003. [Google Scholar] [CrossRef] [Green Version]
- van der Maarel, S.M.; Miller, D.G.; Tawil, R.; Filippova, G.N.; Tapscott, S.J. Facioscapulohumeral muscular dystrophy: Consequences of chromatin relaxation. Curr. Opin. Neurol. 2012, 25, 614–620. [Google Scholar] [CrossRef] [Green Version]
- Lemmers, R.J.; Tawil, R.; Petek, L.M.; Balog, J.; Block, G.J.; Santen, G.W.; Amell, A.M.; van der Vliet, P.J.; Almomani, R.; Straasheijm, K.R.; et al. Digenic inheritance of an SMCHD1 mutation and an FSHD-permissive D4Z4 allele causes facioscapulohumeral muscular dystrophy type 2. Nat. Genet. 2012, 44, 1370–1374. [Google Scholar] [CrossRef] [Green Version]
- Van den Boogaard, M.L.; Lemmers, R.; Balog, J.; Wohlgemuth, M.; Auranen, M.; Mitsuhashi, S.; van der Vliet, P.J.; Straasheijm, K.R.; van den Akker, R.F.P.; Kriek, M.; et al. Mutations in DNMT3B Modify Epigenetic Repression of the D4Z4 Repeat and the Penetrance of Facioscapulohumeral Dystrophy. Am. J. Hum. Genet. 2016, 98, 1020–1029. [Google Scholar] [CrossRef] [Green Version]
- Lemmers, R.J.; van der Vliet, P.J.; van der Gaag, K.J.; Zuniga, S.; Frants, R.R.; de Knijff, P.; van der Maarel, S.M. Worldwide population analysis of the 4q and 10q subtelomeres identifies only four discrete interchromosomal sequence transfers in human evolution. Am. J. Hum. Genet. 2010, 86, 364–377. [Google Scholar] [CrossRef] [Green Version]
- Tupler, R.; Berardinelli, A.; Barbierato, L.; Frants, R.; Hewitt, J.E.; Lanzi, G.; Maraschio, P.; Tiepolo, L. Monosomy of distal 4q does not cause facioscapulohumeral muscular dystrophy. J. Med. Genet. 1996, 33, 366–370. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas, N.S.T.; Wiseman, K.; Spurlock, G.; MacDonald, M.; Ustek, D.; Upadhyaya, M. A large patient study confirming that facioscapulohumeral muscular dystrophy (FSHD) disease expression is almost exclusively associated with an FSHD locus located on a 4qA-defined 4qter subtelomere. J. Med. Genet. 2007, 44, 215–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmers, R.J.; de Kievit, P.; Sandkuijl, L.; Padberg, G.W.; van Ommen, G.J.; Frants, R.R.; van der Maarel, S.M. Facioscapulohumeral muscular dystrophy is uniquely associated with one of the two variants of the 4q subtelomere. Nat. Genet. 2002, 32, 235–236. [Google Scholar] [CrossRef] [PubMed]
- Snider, L.; Asawachaicharn, A.; Tyler, A.E.; Geng, L.N.; Petek, L.M.; Maves, L.; Miller, D.G.; Lemmers, R.J.L.F.; Winokur, S.T.; Tawil, R.; et al. RNA transcripts, miRNA-sized fragments and proteins produced from D4Z4 units: New candidates for the pathophysiology of facioscapulohumeral dystrophy. Hum. Mol. Genet. 2009, 18, 2414–2430. [Google Scholar] [CrossRef]
- Geng, L.N.; Yao, Z.; Snider, L.; Fong, A.P.; Cech, J.N.; Young, J.M.; van der Maarel, S.M.; Ruzzo, W.L.; Gentleman, R.C.; Tawil, R.; et al. DUX4 Activates Germline Genes, Retroelements, and Immune Mediators: Implications for Facioscapulohumeral Dystrophy. Dev. Cell 2012, 22, 38–51. [Google Scholar] [CrossRef] [Green Version]
- Sacconi, S.; Lemmers, R.J.; Balog, J.; van der Vliet, P.J.; Lahaut, P.; van Nieuwenhuizen, M.P.; Straasheijm, K.R.; Debipersad, R.D.; Vos-Versteeg, M.; Salviati, L.; et al. The FSHD2 gene SMCHD1 is a modifier of disease severity in families affected by FSHD1. Am. J. Hum. Genet. 2013, 93, 744–751. [Google Scholar] [CrossRef] [Green Version]
- Balog, J.; Thijssen, P.E.; Shadle, S.; Straasheijm, K.R.; van der Vliet, P.J.; Krom, Y.D.; van den Boogaard, M.L.; de Jong, A.; Lemmers, R.J.L.F.; Tawil, R.; et al. Increased DUX4 expression during muscle differentiation correlates with decreased SMCHD1 protein levels at D4Z4. Epigenetics 2015, 10, 1133–1142. [Google Scholar] [CrossRef] [Green Version]
- Hamanaka, K.; Sikrova, D.; Mitsuhashi, S.; Masuda, H.; Sekiguchi, Y.; Sugiyama, A.; Shibuya, K.; Lemmers, R.; Goossens, R.; Ogawa, M.; et al. Homozygous nonsense variant in LRIF1 associated with facioscapulohumeral muscular dystrophy. Neurology 2020, 94, e2441–e2447. [Google Scholar] [CrossRef]
- Zeng, W.; Chen, Y.Y.; Newkirk, D.A.; Wu, B.; Balog, J.; Kong, X.; Ball, A.R., Jr.; Zanotti, S.; Tawil, R.; Hashimoto, N.; et al. Genetic and Epigenetic Characteristics of FSHD-Associated 4q and 10q D4Z4 that are Distinct from Non-4q/10q D4Z4 Homologs. Hum. Mutat 2014, 35, 998–1010. [Google Scholar] [CrossRef]
- Balog, J.; Goossens, R.; Lemmers, R.; Straasheijm, K.R.; van der Vliet, P.J.; Heuvel, A.V.D.; Cambieri, C.; Capet, N.; Feasson, L.; Manel, V.; et al. Monosomy 18p is a risk factor for facioscapulohumeral dystrophy. J. Med. Genet. 2018, 55, 469–478. [Google Scholar] [CrossRef]
- Himeda, C.L.; Jones, T.I.; Virbasius, C.M.; Zhu, L.J.; Green, M.R.; Jones, P.L. Identification of Epigenetic Regulators of DUX4-fl for Targeted Therapy of Facioscapulohumeral Muscular Dystrophy. Mol. Ther. 2018, 26, 1797–1807. [Google Scholar] [CrossRef] [Green Version]
- Cabianca, D.S.; Casa, V.; Bodega, B.; Xynos, A.; Ginelli, E.; Tanaka, Y.; Gabellini, D. A Long ncRNA Links Copy Number Variation to a Polycomb/Trithorax Epigenetic Switch in FSHD Muscular Dystrophy. Cell 2012, 149, 819–831. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huichalaf, C.; Micheloni, S.; Ferri, G.; Caccia, R.; Gabellini, D. DNA methylation analysis of the macrosatellite repeat associated with FSHD muscular dystrophy at single nucleotide level. PLoS ONE 2014, 9, e115278. [Google Scholar] [CrossRef] [PubMed]
- Campbell, A.E.; Shadle, S.C.; Jagannathan, S.; Lim, J.W.; Resnick, R.; Tawil, R.; van der Maarel, S.M.; Tapscott, S.J. NuRD and CAF-1-mediated silencing of the D4Z4 array is modulated by DUX4-induced MBD3L proteins. Elife 2018, 7. [Google Scholar] [CrossRef]
- Krom, Y.D.; Thijssen, P.E.; Young, J.M.; den Hamer, B.; Balog, J.; Yao, Z.; Maves, L.; Snider, L.; Knopp, P.; Zammit, P.S.; et al. Intrinsic Epigenetic Regulation of the D4Z4 Macrosatellite Repeat in a Transgenic Mouse Model for FSHD. PLoS Genet. 2013, 9, e1003415. [Google Scholar] [CrossRef] [Green Version]
- Dion, C.; Roche, S.; Laberthonniere, C.; Broucqsault, N.; Mariot, V.; Xue, S.; Gurzau, A.D.; Nowak, A.; Gordon, C.T.; Gaillard, M.C.; et al. SMCHD1 is involved in de novo methylation of the DUX4-encoding D4Z4 macrosatellite. Nucleic Acids Res. 2019, 47, 2822–2839. [Google Scholar] [CrossRef] [Green Version]
- Ottaviani, A.; Rival-Gervier, S.; Boussouar, A.; Foerster, A.M.; Rondier, D.; Sacconi, S.; Desnuelle, C.; Gilson, E.; Magdinier, F. The D4Z4 macrosatellite repeat acts as a CTCF and A-type lamins-dependent insulator in facio-scapulo-humeral dystrophy. PLoS Genet. 2009, 5, e1000394. [Google Scholar] [CrossRef]
- Haynes, P.; Bomsztyk, K.; Miller, D.G. Sporadic DUX4 expression in FSHD myocytes is associated with incomplete repression by the PRC2 complex and gain of H3K9 acetylation on the contracted D4Z4 allele. Epigenetics Chromatin 2018, 11, 47. [Google Scholar] [CrossRef]
- Stadler, G.; Rahimov, F.; King, O.D.; Chen, J.C.; Robin, J.D.; Wagner, K.R.; Shay, J.W.; Emerson, C.P., Jr.; Wright, W.E. Telomere position effect regulates DUX4 in human facioscapulohumeral muscular dystrophy. Nat. Struct. Mol. Biol. 2013, 20, 671–678. [Google Scholar] [CrossRef] [Green Version]
- Stadler, G.; King, O.D.; Robin, J.D.; Shay, J.W.; Wright, W.E. Facioscapulohumeral muscular dystrophy: Are telomeres the end of the story? Rare Dis. 2013, 1, e26142. [Google Scholar] [CrossRef] [Green Version]
- Robin, J.D.; Ludlow, A.T.; Batten, K.; Gaillard, M.C.; Stadler, G.; Magdinier, F.; Wright, W.; Shay, J.W. SORBS2 transcription is activated by telomere position effect-over long distance upon telomere shortening in muscle cells from patients with facioscapulohumeral dystrophy. Genome Res. 2015, 25, 1781–1790. [Google Scholar] [CrossRef] [Green Version]
- Zeng, W.; de Greef, J.C.; Chen, Y.Y.; Chien, R.; Kong, X.; Gregson, H.C.; Winokur, S.T.; Pyle, A.; Robertson, K.D.; Schmiesing, J.A.; et al. Specific loss of histone H3 lysine 9 trimethylation and HP1gamma/cohesin binding at D4Z4 repeats is associated with facioscapulohumeral dystrophy (FSHD). PLoS Genet. 2009, 5, e1000559. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, T.I.; Yan, C.; Sapp, P.C.; McKenna-Yasek, D.; Kang, P.B.; Quinn, C.; Salameh, J.S.; King, O.D.; Jones, P.L. Identifying diagnostic DNA methylation profiles for facioscapulohumeral muscular dystrophy in blood and saliva using bisulfite sequencing. Clin. Epigenetics 2014, 6, 23. [Google Scholar] [CrossRef] [Green Version]
- Jones, T.I.; King, O.D.; Himeda, C.L.; Homma, S.; Chen, J.C.; Beermann, M.L.; Yan, C.; Emerson, C.P., Jr.; Miller, J.B.; Wagner, K.R.; et al. Individual epigenetic status of the pathogenic D4Z4 macrosatellite correlates with disease in facioscapulohumeral muscular dystrophy. Clin. Epigenetics 2015, 7, 37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Das, S.; Chadwick, B.P. Influence of Repressive Histone and DNA Methylation upon D4Z4 Transcription in Non-Myogenic Cells. PLoS ONE 2016, 11, e0160022. [Google Scholar] [CrossRef] [PubMed]
- Himeda, C.L.; Debarnot, C.; Homma, S.; Beermann, M.L.; Miller, J.B.; Jones, P.L.; Jones, T.I. Myogenic enhancers regulate expression of the facioscapulohumeral muscular dystrophy-associated DUX4 gene. Mol. Cell Biol. 2014, 34, 1942–1955. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemmers, R.J.; Osborn, M.; Haaf, T.; Rogers, M.; Frants, R.R.; Padberg, G.W.; Cooper, D.N.; van der Maarel, S.M.; Upadhyaya, M. D4F104S1 deletion in facioscapulohumeral muscular dystrophy: Phenotype, size, and detection. Neurology 2003, 61, 178–183. [Google Scholar] [CrossRef]
- Nguyen, K.; Broucqsault, N.; Chaix, C.; Roche, S.; Robin, J.D.; Vovan, C.; Gerard, L.; Megarbane, A.; Urtizberea, J.A.; Bellance, R.; et al. Deciphering the complexity of the 4q and 10q subtelomeres by molecular combing in healthy individuals and patients with facioscapulohumeral dystrophy. J. Med. Genet. 2019, 56, 590–601. [Google Scholar] [CrossRef] [Green Version]
- Sharma, V.; Pandey, S.N.; Khawaja, H.; Brown, K.J.; Hathout, Y.; Chen, Y.W. PARP1 Differentially Interacts with Promoter region of DUX4 Gene in FSHD Myoblasts. J. Genet. Syndr. Gene Ther. 2016, 7. [Google Scholar] [CrossRef]
- Block, G.J.; Narayanan, D.; Amell, A.M.; Petek, L.M.; Davidson, K.C.; Bird, T.D.; Tawil, R.; Moon, R.T.; Miller, D.G. Wnt/beta-catenin signaling suppresses DUX4 expression and prevents apoptosis of FSHD muscle cells. Hum. Mol. Genet. 2013, 22, 390–396. [Google Scholar] [CrossRef] [Green Version]
- Campbell, A.E.; Oliva, J.; Yates, M.P.; Zhong, J.W.; Shadle, S.C.; Snider, L.; Singh, N.; Tai, S.; Hiramuki, Y.; Tawil, R.; et al. BET bromodomain inhibitors and agonists of the beta-2 adrenergic receptor identified in screens for compounds that inhibit DUX4 expression in FSHD muscle cells. Skelet. Muscle 2017, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Cruz, J.M.; Hupper, N.; Wilson, L.S.; Concannon, J.B.; Wang, Y.; Oberhauser, B.; Patora-Komisarska, K.; Zhang, Y.; Glass, D.J.; Trendelenburg, A.U.; et al. Protein kinase A activation inhibits DUX4 gene expression in myotubes from patients with facioscapulohumeral muscular dystrophy. J. Biol. Chem. 2018, 293, 11837–11849. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yamauchi, J.; Nagao, M.; Kaziro, Y.; Itoh, H. Activation of p38 mitogen-activated protein kinase by signaling through G protein-coupled receptors. Involvement of Gbetagamma and Galphaq/11 subunits. J. Biol. Chem. 1997, 272, 27771–27777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oliva, J.; Galasinski, S.; Richey, A.; Campbell, A.E.; Meyers, M.J.; Modi, N.; Zhong, J.W.; Tawil, R.; Tapscott, S.J.; Sverdrup, F.M. Clinically Advanced p38 Inhibitors Suppress DUX4 Expression in Cellular and Animal Models of Facioscapulohumeral Muscular Dystrophy. J. Pharmacol Exp. Ther. 2019, 370, 219–230. [Google Scholar] [CrossRef]
- Lee, J.H.; Goto, K.; Matsuda, C.; Arahata, K. Characterization of a tandemly repeated 3.3-kb Kpnl unit in the facioscapulohumeral muscular dystrophy (FSHD) gene region on chromosome 4q35. Muscle Nerve Suppl. 1995, 2, S6–S13. [Google Scholar] [CrossRef]
- Kowaljow, V.; Marcowycz, A.; Ansseau, E.; Conde, C.B.; Sauvage, S.; Matteotti, C.; Arias, C.; Corona, E.D.; Nunez, N.G.; Leo, O.; et al. The DUX4 gene at the FSHD1A locus encodes a pro-apoptotic protein. Neuromuscul. Disord. 2007, 17, 611–623. [Google Scholar] [CrossRef] [Green Version]
- Dixit, M.; Ansseau, E.; Tassin, A.; Winokur, S.; Shi, R.; Qian, H.; Sauvage, S.; Matteotti, C.; van Acker, A.M.; Leo, O.; et al. DUX4, a candidate gene of facioscapulohumeral muscular dystrophy, encodes a transcriptional activator of PITX1. Proc. Natl. Acad. Sci. USA 2007, 104, 18157–18162. [Google Scholar] [CrossRef] [Green Version]
- Lemmers, R.J.; Wohlgemuth, M.; Frants, R.R.; Padberg, G.W.; Morava, E.; van der Maarel, S.M. Contractions of D4Z4 on 4qB subtelomeres do not cause facioscapulohumeral muscular dystrophy. Am. J. Hum. Genet. 2004, 75, 1124–1130. [Google Scholar] [CrossRef] [Green Version]
- Lemmers, R.J.; Wohlgemuth, M.; van der Gaag, K.J.; van der Vliet, P.J.; van Teijlingen, C.M.; de Knijff, P.; Padberg, G.W.; Frants, R.R.; van der Maarel, S.M. Specific sequence variations within the 4q35 region are associated with facioscapulohumeral muscular dystrophy. Am. J. Hum. Genet. 2007, 81, 884–894. [Google Scholar] [CrossRef] [Green Version]
- Lemmers, R.J.; van der Vliet, P.J.; Balog, J.; Goeman, J.J.; Arindrarto, W.; Krom, Y.D.; Straasheijm, K.R.; Debipersad, R.D.; Ozel, G.; Sowden, J.; et al. Deep characterization of a common D4Z4 variant identifies biallelic DUX4 expression as a modifier for disease penetrance in FSHD2. Eur. J. Hum. Genet. 2018, 26, 94–106. [Google Scholar] [CrossRef] [Green Version]
- Mitsuhashi, H.; Ishimaru, S.; Homma, S.; Yu, B.; Honma, Y.; Beermann, M.L.; Miller, J.B. Functional domains of the FSHD-associated DUX4 protein. Biol. Open 2018, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, T.I.; Chen, J.C.; Rahimov, F.; Homma, S.; Arashiro, P.; Beermann, M.L.; King, O.D.; Miller, J.B.; Kunkel, L.M.; Emerson, C.P., Jr.; et al. Facioscapulohumeral muscular dystrophy family studies of DUX4 expression: Evidence for disease modifiers and a quantitative model of pathogenesis. Hum. Mol. Genet. 2012, 21, 4419–4430. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krom, Y.D.; Dumonceaux, J.; Mamchaoui, K.; den Hamer, B.; Mariot, V.; Negroni, E.; Geng, L.N.; Martin, N.; Tawil, R.; Tapscott, S.J.; et al. Generation of isogenic D4Z4 contracted and noncontracted immortal muscle cell clones from a mosaic patient: A cellular model for FSHD. Am. J. Pathol. 2012, 181, 1387–1401. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Broucqsault, N.; Morere, J.; Gaillard, M.C.; Dumonceaux, J.; Torrents, J.; Salort-Campana, E.; Maues de Paula, A.; Bartoli, M.; Fernandez, C.; Chesnais, A.L.; et al. Dysregulation of 4q35- and muscle-specific genes in fetuses with a short D4Z4 array linked to Facio-Scapulo-Humeral Dystrophy. Hum. Mol. Genet. 2013, 22, 4206–4214. [Google Scholar] [CrossRef] [Green Version]
- Ferreboeuf, M.; Mariot, V.; Bessieres, B.; Vasiljevic, A.; Attie-Bitach, T.; Collardeau, S.; Morere, J.; Roche, S.; Magdinier, F.; Robin-Ducellier, J.; et al. DUX4 and DUX4 downstream target genes are expressed in fetal FSHD muscles. Hum. Mol. Genet. 2014, 23, 171–181. [Google Scholar] [CrossRef] [Green Version]
- Marsollier, A.C.; Ciszewski, L.; Mariot, V.; Popplewell, L.; Voit, T.; Dickson, G.; Dumonceaux, J. Antisense targeting of 3’ end elements involved in DUX4 mRNA processing is an efficient therapeutic strategy for facioscapulohumeral dystrophy: A new gene-silencing approach. Hum. Mol. Genet. 2016, 25, 10. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Brockman, J.M.; Dass, B.; Hutchins, L.N.; Singh, P.; McCarrey, J.R.; MacDonald, C.C.; Graber, J.H. Systematic variation in mRNA 3’-processing signals during mouse spermatogenesis. Nucleic Acids Res. 2007, 35, 234–246. [Google Scholar] [CrossRef] [Green Version]
- Ji, Z.; Lee, J.Y.; Pan, Z.; Jiang, B.; Tian, B. Progressive lengthening of 3’ untranslated regions of mRNAs by alternative polyadenylation during mouse embryonic development. Proc. Natl. Acad. Sci. USA 2009, 106, 7028–7033. [Google Scholar] [CrossRef] [Green Version]
- Tassin, A.; Laoudj-Chenivesse, D.; Vanderplanck, C.; Barro, M.; Charron, S.; Ansseau, E.; Chen, Y.W.; Mercier, J.; Coppee, F.; Belayew, A. DUX4 expression in FSHD muscle cells: How could such a rare protein cause a myopathy? J. Cell Mol. Med. 2012, 17, 76–89. [Google Scholar] [CrossRef]
- Ferreboeuf, M.; Mariot, V.; Furling, D.; Butler-Browne, G.; Mouly, V.; Dumonceaux, J. Nuclear protein spreading: Implication for pathophysiology of neuromuscular diseases. Hum. Mol. Genet. 2014, 23, 4125–4133. [Google Scholar] [CrossRef] [Green Version]
- Rickard, A.M.; Petek, L.M.; Miller, D.G. Endogenous DUX4 expression in FSHD myotubes is sufficient to cause cell death and disrupts RNA splicing and cell migration pathways. Hum. Mol. Genet. 2015, 24, 5901–5914. [Google Scholar] [CrossRef] [PubMed]
- Gabellini, D.; Green, M.R.; Tupler, R. Inappropriate gene activation in FSHD: A repressor complex binds a chromosomal repeat deleted in dystrophic muscle. Cell 2002, 110, 339–348. [Google Scholar] [CrossRef] [Green Version]
- Vanderplanck, C.; Ansseau, E.; Charron, S.; Stricwant, N.; Tassin, A.; Laoudj-Chenivesse, D.; Wilton, S.D.; Coppee, F.; Belayew, A. The FSHD Atrophic Myotube Phenotype Is Caused by DUX4 Expression. PLoS ONE 2011, 6, e26820. [Google Scholar] [CrossRef] [PubMed]
- Wallace, L.M.; Liu, J.; Domire, J.S.; Garwick-Coppens, S.E.; Guckes, S.M.; Mendell, J.R.; Flanigan, K.M.; Harper, S.Q. RNA Interference Inhibits DUX4-induced Muscle Toxicity In Vivo: Implications for a Targeted FSHD Therapy. Mol. Ther. 2012, 20, 1417–1423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, J.C.; King, O.D.; Zhang, Y.; Clayton, N.P.; Spencer, C.; Wentworth, B.M.; Emerson, C.P., Jr.; Wagner, K.R. Morpholino-mediated Knockdown of DUX4 Toward Facioscapulohumeral Muscular Dystrophy Therapeutics. Mol. Ther. 2016, 24, 1405–1411. [Google Scholar] [CrossRef] [Green Version]
- Marsollier, A.C.; Joubert, R.; Mariot, V.; Dumonceaux, J. Targeting the Polyadenylation Signal of Pre-mRNA: A New Gene Silencing Approach for Facioscapulohumeral Dystrophy. Int. J. Mol. Sci. 2018, 19, 1347. [Google Scholar] [CrossRef] [Green Version]
- Bosnakovski, D.; da Silva, M.T.; Sunny, S.T.; Ener, E.T.; Toso, E.A.; Yuan, C.; Cui, Z.; Walters, M.A.; Jadhav, A.; Kyba, M. A novel P300 inhibitor reverses DUX4-mediated global histone H3 hyperacetylation, target gene expression, and cell death. Sci. Adv. 2019, 5, eaaw7781. [Google Scholar] [CrossRef] [Green Version]
- DeSimone, A.M.; Leszyk, J.; Wagner, K.; Emerson, C.P., Jr. Identification of the hyaluronic acid pathway as a therapeutic target for facioscapulohumeral muscular dystrophy. Sci. Adv. 2019, 5, eaaw7099. [Google Scholar] [CrossRef] [Green Version]
- Klingler, C.; Ashley, J.; Shi, K.; Stiefvater, A.; Kyba, M.; Sinnreich, M.; Aihara, H.; Kinter, J. DNA aptamers against the DUX4 protein reveal novel therapeutic implications for FSHD. FASEB J. 2020, 34, 4573–4590. [Google Scholar] [CrossRef] [Green Version]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sidlauskaite, E.; Le Gall, L.; Mariot, V.; Dumonceaux, J. DUX4 Expression in FSHD Muscles: Focus on Its mRNA Regulation. J. Pers. Med. 2020, 10, 73. https://doi.org/10.3390/jpm10030073
Sidlauskaite E, Le Gall L, Mariot V, Dumonceaux J. DUX4 Expression in FSHD Muscles: Focus on Its mRNA Regulation. Journal of Personalized Medicine. 2020; 10(3):73. https://doi.org/10.3390/jpm10030073
Chicago/Turabian StyleSidlauskaite, Eva, Laura Le Gall, Virginie Mariot, and Julie Dumonceaux. 2020. "DUX4 Expression in FSHD Muscles: Focus on Its mRNA Regulation" Journal of Personalized Medicine 10, no. 3: 73. https://doi.org/10.3390/jpm10030073
APA StyleSidlauskaite, E., Le Gall, L., Mariot, V., & Dumonceaux, J. (2020). DUX4 Expression in FSHD Muscles: Focus on Its mRNA Regulation. Journal of Personalized Medicine, 10(3), 73. https://doi.org/10.3390/jpm10030073