1. Introduction
Modern medicine is at an inflexion point [
1], whereby advances in computational methods, wearable sensing technology and open access to Big Data are reshaping the ways in which we inform basic science and rapidly translate our knowledge to actionable treatments. Psychiatry is one of those medical fields that is rapidly evolving, while adapting traditional models to help advance the main goal of helping patients improve their quality of life. Along those lines, computational psychiatry [
2], a nascent subfield within psychiatry, is merging methods from Computational Neuroscience with clinical approaches through successful collaborations. These new developments are bound to open new frontiers in therapeutic treatments. Further, as part of a more general effort in the medical field, precision medicine (PM) [
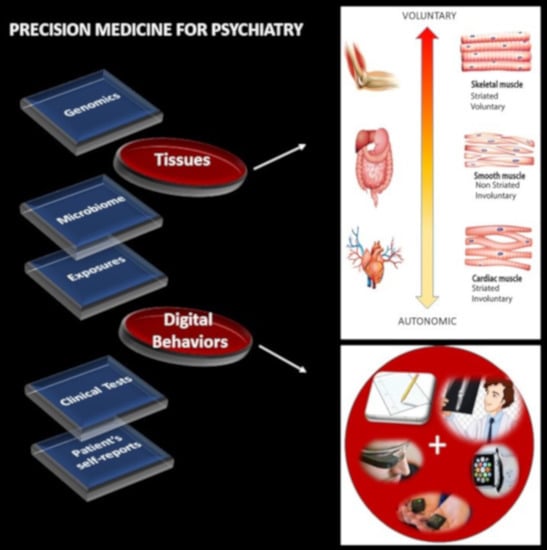
1] has emerged as a new platform to combine expertise from multiple layers of the knowledge network in order to ultimately design personalized targeted treatments (
Figure 1A). Integrating the personalized concept of PM with the new advances in computational psychiatry could give us a new way to approach mental illness and help patients cope with lifelong changing needs.
The task ahead is challenging because there is no proper roadmap to connect the layers of the knowledge network in PM and produce personalized diagnoses and measures of treatment outcomes that truly separate disease progression from treatment effectiveness according to age and development. Part of the problem is that most brain science has focused on experimental assays and methods that curtail natural movements. As such, our knowledge about the dynamics of natural behaviors is very limited, particularly in reference to those aspects of behavior that remain hidden to the naked eye of the clinician trained to observe specific expected behavioral landmarks of a psychiatric disorder conceived exclusively as a mental illness. In so doing, the clinically trained eye may miss important information that is perhaps common across different disorders of the nervous systems and rather relevant to help improve the patient’s quality of life. For example, motor coordination and volitional control are critical ingredients of autonomy in any natural behavior underlying activities of daily living. Yet, these are not considered part of the diagnostics criteria for mental illnesses such as autism, schizophrenia and depression, as per the Diagnostics Statistical Manual (DSM-5) [
3] (and see
Supplementary Materials).
Research on the underlying neurobiology of mental illnesses has revealed their associated genetics [
4] and/or helped characterize patterns of brain activity in response to external stimuli [
5] (while curtailing naturalistic bodily motions to avoid instrumentation artifacts in imaging data or in EEG, MEG, etc.). This central approach to brain science has left us with a paucity of information about the possible contributions to mental illness from the peripheral nervous systems, and from vital organs important for autonomous living. The peripheral activity, however, continuously feeds back to the brain via afferent (body-to-brain) channels and is, in turn, dynamically updated through efferent (brain-to-body) activity, self-generated by the system itself. This recursive loop, whereby re-entrant information that is partly self-produced by the organism and partly influenced by external environmental conditions, would provide important clues about truly evolving dynamics and stochastic (variability) across all-natural behaviors. Approaching the problem through this lens could bring a new quantifiable layer of granularity to basic research. This would include the design of age-appropriate metrics reflecting the development of the organism as it ages and as it copes with a disorder [
6,
7]. The micro- and macro-motion data from the nervous systems biorhythms is the low hanging fruit that we can easily attain by leveraging the wearable sensors revolution. Further, because these quantifiable digitized activities and signals therein are partly self-generated, self-monitored and self-corrected by and within the nervous systems, this quantitative approach has the potential to take us from a purely correlational science to a science that is based on causal relations between nervous system activities and external/contextual stimuli. In this sense, the new proposed approach to mental illness is amenable to intervene and modify the system with well-informed, near-optimal means capable of improving its performance.
Micro- and macro-motions that underly all aspects of human behavior depend on the intactness of fundamental tissues, many of which have already been characterized in genomics according to cell types [
8]. Here, we propose to combine micro-level underlying aspects of behavior with the current genomics knowledge to inform psychiatry of possible ways to improve quantification of nervous system activities. Ultimately, we seek to compile this information to help build accommodations and support for the patient population, while reconceptualizing mental illness as a physically quantifiable disorder of
the nervous systems.
The nervous systems already offer a taxonomy of function and control that is phylogenetically ordered and well organized along several axes. Some of these axes are accessible today with non-invasive means and, as such, we can obtain signals and build computational models to understand mechanisms and translate them to actionable societal solutions. One possible orderly structure is suggested in
Figure 1B, where we propose to map levels of neuromotor control (voluntary, involuntary, and autonomic) to fundamental types of muscles (skeletal, smooth, and cardiac) linked to commonly sampled tissues in genomic datasets. Combining information about gene expression on tissues that involve key components of the central nervous systems (the brain and the spinal cord), key organs for vital bodily functions (including smooth muscle lining internal organs), muscle-skeletal tissues and nerves, and cardiac tissues (for autonomic heart functioning), we explore the effects of removing disease-associated genes, on the overall remaining genome expression on these tissues. As a first step in this exercise, we reasoned that the genes associated with a given disorder ought to be important in the functioning of certain systems, which in turn depend on certain tissues. We also reasoned that such stochastic variations and combinations could be measured relative to the presence of all genes and to the absence of genes across neurological or non-neurological conditions.
What is the tissue distribution of gene expression in neuropsychiatric disorders such as autism, schizophrenia, and depression in relation to well-characterized neurological conditions? Is there convergence in the remaining gene expression on the tissues upon removal of the genes associated with that disease? Furthermore, how would the gene expression change across the tissues in non-neurological conditions such as various forms of cancer, immunodeficiencies, endocrine system deficiencies and so forth? How would it change in acquired disorders such as Post Traumatic Stress Syndrome (PTSD), currently diagnosed through observation?
Take autism for example. Autism is an umbrella term for a very heterogeneous set of neurodevelopmental disorders, but no gold-standard criteria include core neurological symptoms that could help us create early accommodations and support for the nascent nervous systems of the infant (during pre-cognitive stages of neurodevelopment). The rule of thumb is to assume that the child has odd, socially inappropriate behaviors and that they should be modified through operant and cognitive conditioning techniques—often translated from lab animals to human babies, without any type of collaboration with other fields studying infant development. Current methods of diagnoses and treatments in autism are not based on normative neurodevelopmental data charts to understand age-dependent departures from typical neurodevelopment. Without any systematic way to build age-appropriate metrics in order to capture highly non-linear, stochastic patterns and rates of change in the (rather accelerated) infant neurodevelopment, entire generations of infants, children and adolescents have been exposed to such means of behavioral treatments and no information can tie these back to the underlying genomic pool of this population.
In schizophrenia, delusions, avolition and catatonia are at the core of the disorder, but as in autism above, no criteria in the DSM highlight the profound somatic sensory-motor issues that have been found in patients [
9]—even without the use of psychotropic medications known to alter motility. Interestingly, historical accounts of psychiatry (in pre-Freudian times) show the reliance on motor aspects of the behaviors that defined several mental illness from a neurological perspective [
10].
Depression is also currently treated purely as a mental illness, but it may be important to understand potential contributions to various forms of depression, from the peripheral nervous systems and from the body in general. Genetic information may give us a way to link tissues affected in these neuropsychiatric conditions with those affected in neurological conditions, for which treatments and interventions of various forms may be effective. These may be in the form of drugs, or in the form of physical, mindfulness and occupational therapies aimed at helping support the person’s bodily autonomy and overall increase the chances for independent living.
We here offer a new lens to help balance psychiatric with neurological criteria derived from genomic information specific to each disorder. In a first (crude) step of many to come, we start by comparing well-known neuropsychiatric and neurological conditions, the results from eliminating the genes associated with each disorder and quantifying the degree of convergence in the maximally affected tissues, in relation to those resulting from eliminating the genes associated with non-neurological conditions. We focus our discussion on possible ways to continue this path of inquiry and highlight current caveats for future improved iterations of the proposed methods.
2. Materials and Methods
We combine the datasets from genes associated with mental illnesses with well-known neurological disorders and with illnesses that are not directly associated with the nervous systems. We also include genes associated with manifestations of acquired Post-Traumatic Stress Syndrome Disorder (PTSD). Among mental illnesses defined by the DSM-5, we include autism, schizophrenia and mental depression of different types, (e.g., general, bipolar and unipolar). Among neurological conditions, we include ataxias (e.g., cerebellar, spinocerebellar, progressive, and gait) and Parkinson’s disease. Among non-neurological disorders, we include colon cancer, breast cancer, diabetes, congenital heart disease, hematologic neoplasm, and various autoimmune disorders (lupus systemic erythematosus, psoriasis, and irritable bowel syndrome).
We use the genes, gene expression, and tissues from the Genotype-Tissue Expression project, GTEx pPortal human RNA-seq (Transcripts Per Million (TPM), see
Appendix A for note in TPM) as reference specifically using the files denoted in
Appendix B. In autism, we use the gene scoring module of the Simons Foundation Autism Research Initiative (SFARI) scored according to evidence from the literature. We also use ataxia genes, the X genes and the FX genes taken from various literature reviews [
11,
12]. Furthermore, we use genes associated with mitochondrial disorders [
13] and genes identified in Parkinson’s disease, taken from [
14,
15,
16,
17,
18,
19]. Besides the autism SFARI genes and the genes reported in literature reviews, we take the genes associated with autism, schizophrenia and depression reported in
https://www.disgenet.org/home/ along with other genes from the above-mentioned non-neurological disorders. The latter will inform us of fundamental differences in gene expression between these diseases and those which affect neuromotor control and basic functioning, as mediated by interactions between the brain and the peripheral nervous systems (including the autonomic nervous system).
The SFARI autism categories that we used were those reported as of 03-04-2020. Quoting from their site:
CATEGORY 1
Genes in this category are all found on the SPARK gene list. Each of these genes has been clearly implicated in Autism Spectrum Disorders, ASD—typically by the presence of at least three de novo likely-gene-disrupting mutations being reported in the literature—and such mutations identified in the sequencing of the SPARK cohort are typically returned to the participants. Some of these genes meet the most rigorous threshold of genome-wide significance; all at least meet a threshold false discovery rate of <0.1.
CATEGORY 2
Genes with two reported de novo likely-gene-disrupting mutations.
A gene uniquely implicated by a genome-wide association study, either reaching genome-wide significance or, if not, consistently replicated and accompanied by evidence that the risk variant has a functional effect.
CATEGORY 3
Genes with a single reported de novo likely-gene-disrupting mutation.
Evidence from a significant but unreplicated association study, or a series of rare inherited mutations for which there is not a rigorous statistical comparison with controls.
SYNDROMIC
The syndromic category includes mutations that are associated with a substantial degree of increased risk and consistently linked to additional characteristics not required for an ASD diagnosis. If there is independent evidence implicating a gene in idiopathic ASD, it will be listed as “#S” (e.g., 2S, 3S). If there is no such independent evidence, the gene will be listed simply as “S”.
The GTEx dataset is as the 06-05-2017 v8 release. For every gene in autism, ataxia, X, FX, mitochondrial diseases, Parkinson’s disease, and the non-neurological diseases, we first confirmed the presence of the gene in the GTEx dataset and then incorporated it into the analyses.
The genes from the DisGeNet portal were found by interrogation of their dataset under disease type and saving the outcome to excel files containing all pertinent information. All sample files used in our analyses are provided in
Supplementary Materials.
2.1. Count Normalization
The GTEx matrix of RNA-seq genes along the rows (56,146) × the tissues (54) along the columns was transposed (54 × 56,246), such that we expressed each tissue as a function of the gene expression denoted by the count (TPM). Each individual count value was then normalized using Equation (1).
Here,
counti is the count value of the gene
i,
AvrgGlobalCount is the overall average of the matrix of values taken along the columns and the rows.
MaxGlobalCount is the maximum count value, also taken globally across the matrix values.
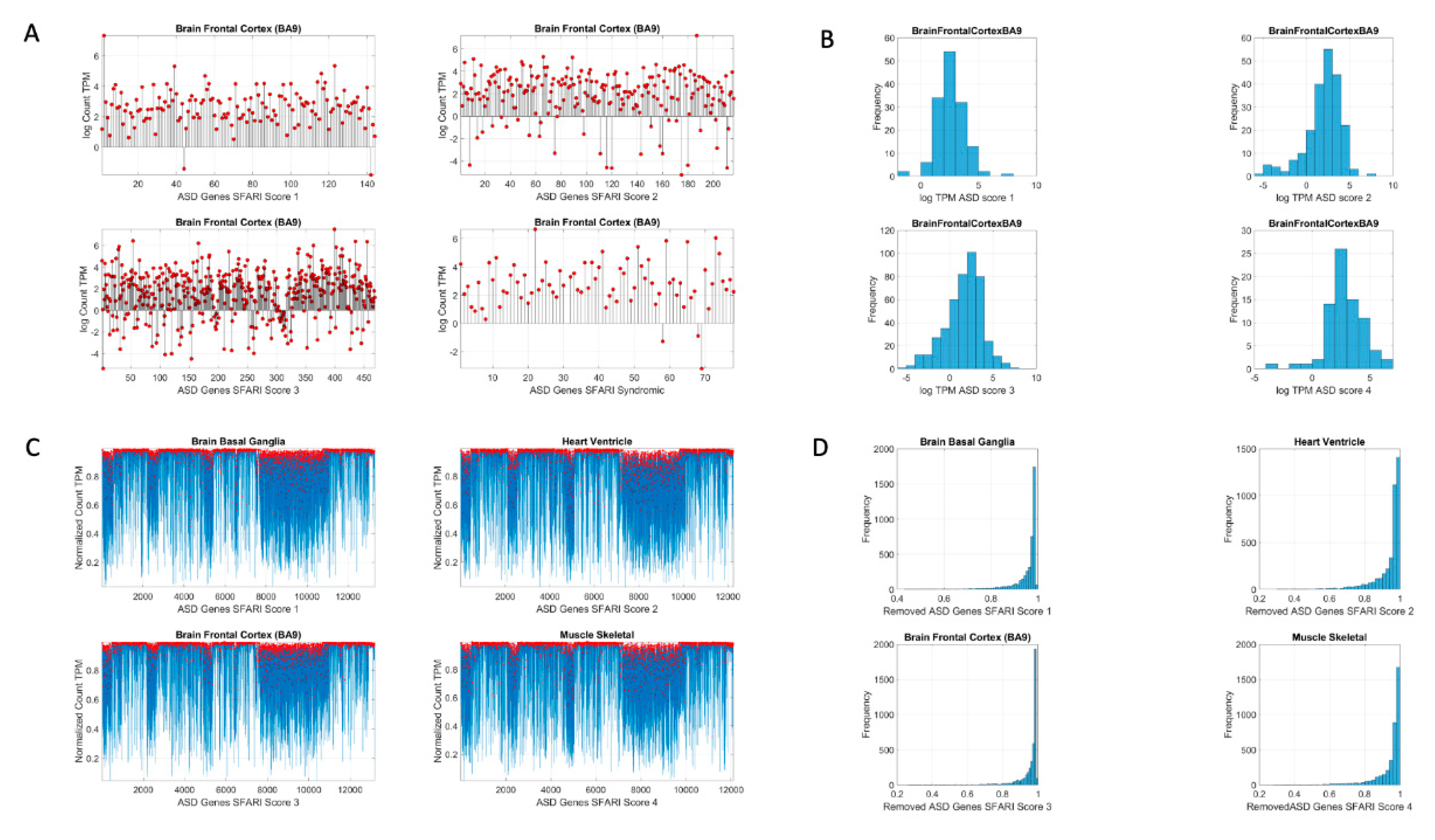
Figure 2 shows the original count numbers (
Figure 2A) and the normalized version (coined micro-movement spikes (MMS)) in
Figure 2B.
Figure 2C shows the MMS derived from the fluctuations in counts normalized by Equation (1), while
Figure 2D shows the histograms of the peaks (marked in red dots) for different tissues and genes scored by the SFARI.
2.2. Gene Removal
For each of the disorders of interest in
Appendix B Table A1 (mental illness and neurological) and
Table A2 (non-neurological), we remove the genes associated with each condition from the human GTEx dataset. These disease-gene associations are as reported in the various databases (the SFARI, DisGeNet
https://www.disgenet.org/home/ and the literature meta reviews). We then treat the resulting count series as a random process. We use the exponential distribution to characterize it and to assess the differential expression across the tissues relative to non-removal in the original human genome.
The question that we ask is: given the known neurological phenotypes, is there convergence between the most-affected tissues upon associated gene removal and the changes in the tissues that will be obtained by the removal of genes associated with mental illnesses? Furthermore, is there convergence with the outcome from removing the genes associated with non-neurological illnesses?
Appendix B Table A1 and
Table A2 show the number of genes removed in each respective case as well as the source of the reported genes associated with each condition/disease.
2.3. Stochastic Analyses
Since the count values for each tissue can be conceived as a random series of numbers, we use maximum likelihood estimation (MLE) to model the numbers representing the counts, as generated by the exponential distribution using Equation (2)
Here,
x represents the normalized count value (as per Equation (1)) and
y represents the value from the exponential distribution. We seek the value of the rate parameter
λ to model this random counting process, which we use to represent the gene expression in the tissue. To that end, we estimate the likelihood
where the series of counts
xi, with
i ranging from 1 to
n, represent the normalized counts (according to Equation (1)) across all genes for one tissue.
Appendix B shows the steps to find
λ. Further, this is computed for each of the 54 tissues. We then rank the departure of
λ (resulting from gene removal) from the
λ obtained for the full human genome (see below). This is explained in
Figure 3.
2.4. Stochastic Analyses—Visualization of Change Relative to the Normative Data of the Full Human Genome
Using MLE, we also obtain for each of the 54 tissues, the frequency histograms of the normalized counts across all genes and fit the continuous gamma family of probability distributions with shape (a) and scale (b) values in order to obtain the gamma moments and plot them on a parameter space. We do this to visualize the spread of the tissues and their shift upon gene removal. To that end, we plot the mean, the variance, and the skewness across the x, y and z axes, respectively. We plot the size of the marker representing the tissue proportional to the kurtosis value, and we color the marker based on the change relative to the original genome count (i.e., containing all the genes, without removal).
To measure the stochastic shift between the tissues from the full genome and those upon removal of the genes identified with each known neurological condition, we take the absolute difference between the MLE λ for the full GTEx genome and that for the genome upon removal of the genes associated with each condition, disorder or disease, as shown in
Figure 3.
We median rank the Δλ for each tissue, sorting Δλ in ascending order across the 54 tissues. Then we create four median-ranked blocks and plot the maximally affected block of tissues (
Figure 3A). The highest-ranked group is then compared across all conditions—mental illness vs. those neurologically defined vs. those non-neurologically defined. We annotate the neurological functions that such tissues are known to maximally disrupt. Further, we determine whether there is convergence between the tissue outcome in mental illnesses, upon removing the associated genes from the human genome-tissue model, and the outcome upon removing those genes tied to the other known disorders of the nervous systems. We repeat this interrogation process using the genes associated with disorders in the DisGeNet portal, including autism, schizophrenia, depression, the neurological disorders in
Appendix B Table A1, and the non-neurological disorders in
Appendix B Table A2.
To assess tissue outcome upon removal of genes associated with various non-neurological diseases, we follow these procedures and compare these to the above results. These diseases include colon cancer, breast cancer, psoriasis, diabetes, congenital heart disease, hematologic neoplast and systemic lupus.
Appendix B Table A2 describes the number of genes associated with each of these diseases and the sources.
We also examine other mental illnesses described by the DSM. These include schizophrenia, depression, unipolar depression, and bipolar depression. We determine whether there are tissues that overlap with those affected in the neurological disorders. Lastly, we examine mitochondria-related disorders and PTSD using these methods. We reasoned that these may be disorders that have potentially affected tissues across a broader range of functions, including those from the brain and other bodily organs.
3. Results
3.1. Autism, Ataxia and FX Have Convergence in Maximally Affected Tissues by the Removal of Associated Genes
The maximally affected tissues upon gene removal, according genes stochastic expression (count in Transcripts Per Million (TPM)), are depicted in
Appendix B Table A3. Tissue gene expression was modelled by the exponential distribution
, with
x as the gene combination expressed in the tissues, and λ as the exponential rate parameter. The Δλ between the neurotypical template case from the GTEx portal (containing all genes) and the modeled disorder case (upon removal of the SFARI genes) provides a sense of the departure from the normative case. This difference, taken for the removal of the SFARI genes, is depicted in
Figure 4A, with samples of maximally affected tissues in
Figure 4B that are known to be critical for motor control, regulation, adaptation/learning, and coordination.
The Δλ median ranking quantified the difference between neurotypical tissue gene expression vs. tissue gene expression upon removal of the genes corresponding to the disorders in question, with four groups ordered by the size of Δλ. This λ quantity was first obtained relative to the neurotypical population tissues, i.e., including all the counts (gene expression) from all genes, in order to model an exponential process. The computation of λ using maximum likelihood estimation (MLE) is explained in
Appendix A. It does not assume any order of the counts, but rather seeks to identify the resulting λ for each tissue, treating the gene count (expression) as a random, memoryless stochastic process. Typically, the exponential distribution is used to model times between events, but here we used it to model the fluctuations in the values of the counts across the genes, as they randomly fluctuate their expression across each of the 54 tissues reported in the GTEx portal. We note this to underscore that the results spontaneously self-emerge from the random combination of the genes involved (with and without removal), rather than from the clinical criteria used to denote gene relevance to autism, or the evidence from the literature used to determine their association. There is in fact no scoring of such relevance for the genes associated with the other neurological disorders under consideration (e.g., ataxias and Parkinson’s disease) or for the autistic disorders reported in the DisGenNet portal. Using those genes instead of the SFARI genes reveals tissues in
Appendix B Table A4, where we report the convergence.
Removal of the SFARI genes, ranked by change in gene expression, reveals brain tissues linked to the CNS, in brain subcortical tissues linked to motor control (basal ganglia, striatum), memory (hypocampus), emotions (amygdala) and regulation (hypothalamus); and the spinal cord. This is also generally the case for the removal of the DisGeNet genes associated with autistic disorders and with FX and ataxia. Congruent with the outcome from the removal of the SFARI genes from the GTEx genome, the DisGeNet gene removal also affected the tissues associated with CNS function. Important tissues for systemic organ functioning such as those containing smooth muscles, cardiac and skeletal muscles in the taxonomy proposed in
Figure 1B were also affected (commonly) across these disorders.
Figure 4 shows a summary of the results, visualizing the stochastic shifts.
Appendix B Table A3 and
Table A4 show the tissues ranked in descending order and color coded according to CNS (brain and spinal cord in blue), heart related (pink), muscle-skeletal (green), and peripheral vital organs (gray). Most tissues in autism and the neurological disorders are from the CNS, followed by PNS-related tissues in the heart and muscle-skeletal and with vital organs towards the end of the Δλ ranking.
Supplementary Figures S1–S3 show these results separately for each neurological condition. We note in
Supplementary Materials that removal of the SFARI autism syndromic genes from the GTEx genome reveals maximal differences in tissues of organs with smooth and cardiac muscles linked to involuntary and autonomic function in the proposed taxonomy of
Figure 1B.
Removal of the overlapping SFARI genes and neurological disorders also reveal brain tissues linked to motor control, memory, emotions, and regulation. This is depicted in
Figure 4. Given the congruence between the tissues maximally affected by removing the SFARI autism genes from the GTEx database and those from the neurological conditions, we next ascertain the extent to which these genes overlap with those used from ataxias in the literature. To that end, we divide them into the autosomal dominant, the autosomal recessive and the X-chromosome genes.
Figure 5A shows the result upon removal of overlapping genes between the SFARI autism set and ataxias (dominant and recessive and X-chromosome sets) from the literature.
Appendix B Table A3, Column 3 lists the tissues, while
Appendix B Table A5 also has the scoring from the SFARI autism genes.
Supplementary Materials Table S2 lists the phenotypic information of the disorders associated with these genes, as described by the clinical literature.
Figure 5B lists the PD gene that overlaps with the SFARI autism genes, also depicted in
Appendix B Table A5 along with the score ranking from the SFARI portal.
We note that removing this subset of 14 overlapping genes from the SFARI autism set (
Appendix B Table A5) does not change the primary result, whereby the most-affected tissues upon removal of the SFARI autism set from the GTEx dataset are those associated with subcortical brain structures critical for motor control, adaptation/learning, regulation, coordination and autonomic function as well as memory and emotion. This is shown in
Figure 5A,B and in the third column of
Appendix B Table A3. In
Figure 4D, we also plot the top-ranked tissues affected by the removal of the FX genes reported in the DisGeNet portal from the GTEx dataset.
Supplementary Figures S19–S22 further provide details on X-chromosome genes in
Figure 5A implicated in autism according to the SFARI genes portal.
The result that convergence in the ranked descending order of the CNS (brain and spinal cord tissues), followed by heart-related tissues, muscle-skeletal tissue and lastly peripheral vital organs for systemic functioning in the SFARI autism and well-known neurological disorders from the literature is also congruent with the results using the genes associated with these conditions in the DisGeNet portal. There, we interrogated autistic disorders, ataxias and fragile X, confirming the overlap in genes, their expression on the 54 tissues of the GTEx database and the orderly levels of tissues maximally affected by the removal of the associated genes. We grouped the tissues by CNS, heart, muscle-skeletal and peripheral vital organs to follow the proposed taxonomy of
Figure 1B.
In the remaining sections of this paper, we consistently use this tissue grouping to simplify visualization of the
Appendix B tables and data presentation.
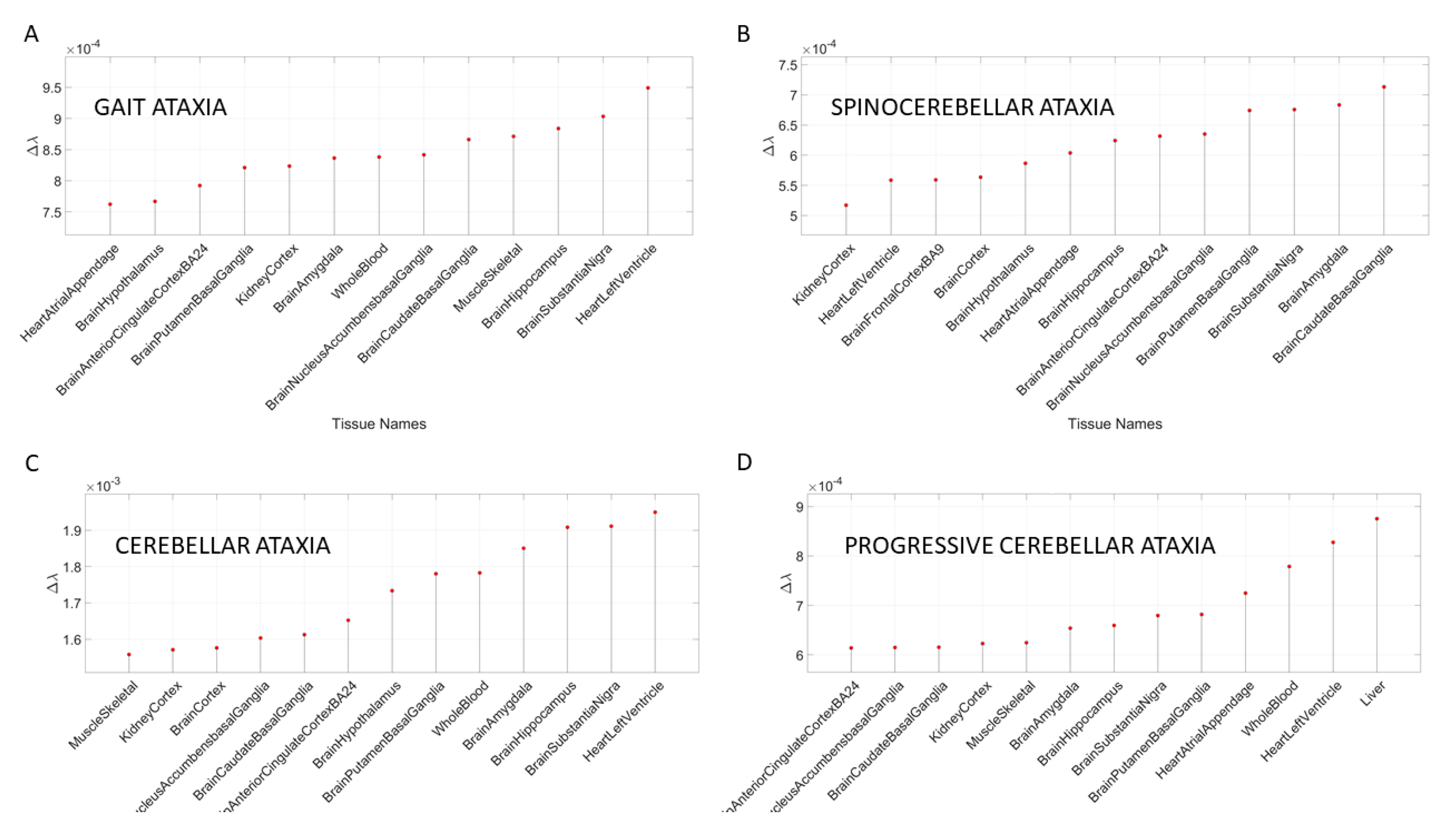
Figure 6 shows the results for different types of ataxias and FX, while
Appendix B Table A6 summarizes the top-ranked affected tissues in different types of ataxias. These are color coded according to the tissue grouping, approximating the taxonomy proposed in
Figure 1B.
3.2. Removal of Genes Associated with Schizophrenia and Multiple Forms of Mental Depression Reveals Convergence with Neurological Disorders
The removal of the DisGeNet genes associated with mental illnesses such as schizophrenia, depression, bipolar depression and unipolar depression from the normative GTEx genome resulted in convergence of maximally affected tissues involved in the CNS, especially those brain regions necessary for neuromotor control, memory, and emotion. This is depicted in
Appendix B Table A7 and
Figure 7. Several of these tissues were also found to be affected upon removal of the SFARI genes and the genes associated in DisGeNet with autistic disorders. Furthermore, these are maximally affected tissues in the well-known neurological conditions depicted in
Figure 4 and
Figure 5 and
Appendix B Table A3,
Table A4 and
Table A5.
3.3. Removal of Genes Associated with Non-Neurological Disorders Reveals Other Non-CNS Tissues
In addition to the examination of mental illnesses and neurological disorders, we also interrogated the GTEx genome upon removal of genes associated with various non-neurological disorders. These included various forms of cancers, inflammatory and autoimmune disorders and other tissues related to the heart, the circulatory and the endocrine systems.
Appendix B Table A8 and
Table A9 summarize the results of this interrogation and
Figure 8 and
Figure 9 show the Δλ-ranking graphs.
The results of the maximally affected tissues upon the removal of the genes associated with these non-neurological disorders revealed a very different picture than those upon removal of the genes associated with the mental illnesses (autism, schizophrenia and the depressions) and those associated with the known neurological conditions (the various forms of ataxia, FX and Parkinson’s disease). Namely, the CNS-related tissues were less affected in these non-neurological diseases than those related to the PNS (muscle-skeletal and ANS heart), and those linked to peripheral bodily organs were the most visibly affected. The exception was diabetes, maximally affected tissues in peripheral organs, but also CNS and PNS tissues in the tail of the top Δλ-ranked tissues. We next interrogate the genome in relation to mitochondrial disorders of several kinds and acquired PTSD.
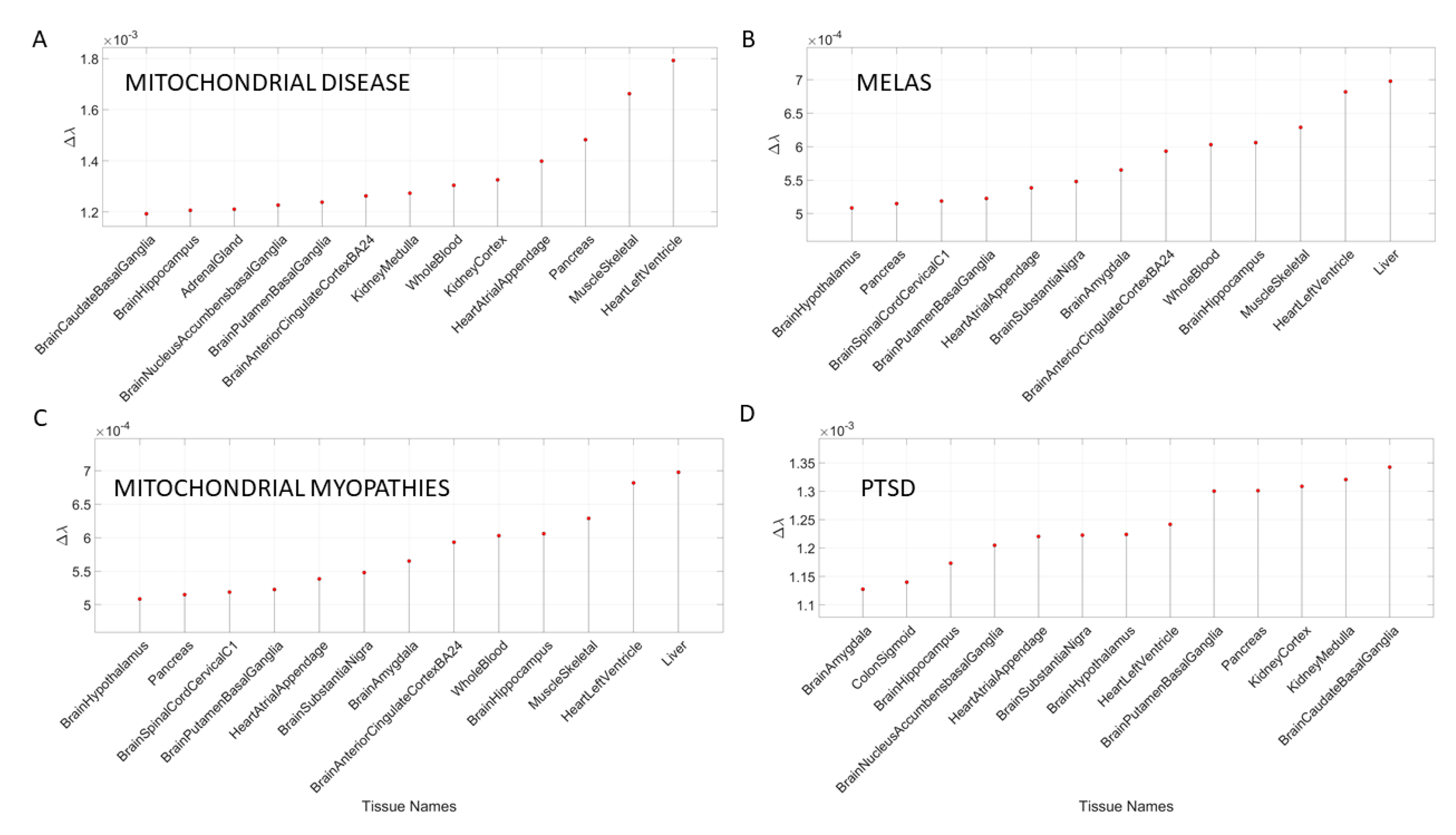
3.4. Removal of Genes Associated with Mitochondrial Diseases Reveals that Heart-Related Tissues Are Maximally Affected but PTSD Is Mixed
Removal of genes associated with mitochondrial disorders of various types from the GTEx genome, according to the genes in the DisGeNet portal, reveal a mixture of tissues associated with peripheral vital organs for systemic functions, heart-related and muscle-skeletal- and CNS-related tissues. The top half of the highest-ranked tissues in mitochondrial disease shows affected tissues related to the heart, muscle-skeletal and peripheral organs, while the bottom half shows more involvement of brain-related tissues in subcortical regions of motor control. In contrast, mitochondrial myopathies show a predominance of CNS-related tissues, including the brain and spinal cord, with top Δλ-ranked tissues related to the heart and muscle-skeletal tissues. Mitochondrial encephalopathy, lactic acidosis, and stroke-like episodes (MELAS) show a predominance of tissues associated with peripheral vital organs for systemic function and heart-related tissues. Only two brain regions for motor control and emotion are present in the bottom-ranked tissues of the most-affected tissues.
The case of acquired PTSD also reveals a mixture of tissues from brain, heart, and peripheral organs. There, we see maximally affected tissues linked to subcortical regions of the brain involved in motor control, adaptation, learning, and coordination intermixed with tissues linked to peripheral bodily organs (like the kidneys) and the autonomic systems’ heart. Furthermore, we also see tissues linked to the hypothalamus, a regulatory brain structure.
Figure 10 shows the graphs of the Δλ difference, which was median ranked, as in the previous cases, for these disorders.
We summarize the results across all 54 tissues (in alphabetical order from left to right) in
Figure 11. Here, a color map depicts the values of Δλ normalized for each disease (along the rows) across the tissues (columns) by dividing by the maximum Δλ value of each row. The patterns reveal that the maximally affected tissues (upon genes removal) are common to both neurological disorders and mental illnesses. They correspond to the brain tissues involved in motor control, adaptation, and learning (basal ganglia, striatum, substantia nigra), tissues in involved in emotion (amygdala), memory (hippocampus) and systemic regulation (hypothalamus). They also reveal that whole blood tissue is not as affected in the mental illnesses as in the neurological disorders (marking a point of divergence that warrants further investigation). Heart-related tissues and muscle-skeletal tissues are also shared between these mental illnesses and neurological disorders when the genes specific to each disorder are removed from the GTEx genome. Interestingly, the pancreas is an example of a peripheral bodily organ with tissues that are commonly affected across most of the disorders and diseases interrogated here. Yet they have lower weight the neurological disorders compared to the non-neurological diseases under examination.
The non-neurological diseases reveal less involvement of the CNS-related tissues but highly overlap with the heart and muscle-skeletal tissues. Tissues linked to the kidneys, liver and pancreas are also maximally affected by genes’ removal in these diseases. Colon cancer shows an interesting pattern whereby the pancreas reveals maximal normalized Δλ values.
Figure 12 summarizes the patterns in binary form by turning ON (yellow) values above 0.8 considered high and OFF those below (blue). This cut off is chosen to further highlight overlap and differences across diseases based on high Δλ.
The patterns revealed by the high values of the normalized Δλ quantity, show convergence of maximally affected brain tissues in mental illnesses with the neurological disorders but not with the non-neurological disorders (except for diabetes which does affect some brain regions.) The mitochondrial diseases do not show the same intensity of the CNS-related Δλ values as the mental illnesses and the neurological disorders, but they do share the heart and muscle-skeletal patterns with all the examined diseases and disorders. This is interesting, given that some of the children with various forms of mitochondrial disorders may receive diagnoses of autism. In summary, there is clear overlap between mental illness and neurological disorders, suggesting involvement of the central nervous systems in both. We also see major contributions from the peripheral nervous systems, particularly the heart, the muscle-skeletal tissues and, to a lesser degree, tissues of peripheral organs. The latter are most affected in the non-neurological diseases.
Figure 13 shows the most-affected tissue in each disease/disorder (also depicted in
Appendix B Table A11).
4. Discussion
This work interrogated the human genome by removing genes associated with various diseases and comparing the outcome from the remaining gene expression on 54 tissues commonly examined in the GTEx portal. These tissues involve parts of the central nervous systems (the brain and the spinal cord) and parts of the peripheral nervous systems (the muscle-skeletal tissue and the heart tissues as part of the autonomic nervous system), which we grouped in
Figure 1B. Other tissues are from peripheral vital organs for systemic bodily functions (whole blood, pancreas, liver, kidneys, lungs, etc.).
We compared mental illnesses such as autism, schizophrenia, and various types of mental depression (including unipolar and bipolar), with well-known neurological disorders such as different types of ataxias, fragile X and Parkinson’s disease. We found convergence between the tissues maximally affected by the removal of disease-associated genes across these mental illnesses and neurological disorders. CNS-related tissues in subcortical regions of the brain related to motor control, motor learning, motor coordination, and motor adaptation, as well as memory and emotion, were predominantly maximally affected by the corresponding gene removal across both the mental illnesses and the neurological disorders. This convergence demonstrates overlap between psychiatric and neurological conditions with specific involvement of motor, memory, emotional and regulatory axes. In autism and FX, we obtained congruent results on maximally affected tissues. The results were consistent using removal of the genes from the SFARI autism database and using the genes upon querying the DisGeNet portal. In addition to the genes reported by querying DisGeNet, we also used the genes reported in the literature for schizophrenia and depression, and for Parkinson’s disease. We found congruence in all cases.
To further test our hypothesis that these mental illnesses are disorders of the nervous systems and that removing the gene pool associated with them gives rise to overlapping tissues related to CNS functioning, we also queried DisGeNet about other non-neurological diseases. We found that in such cases, the predominance of maximally affected tissues was on tissues associated with peripheral vital bodily organs related to the disease, such as pancreas, kidney, liver, and colon transverse in colon cancer. Furthermore, several of these diseases had maximally affected heart-related tissues and whole blood. Other cases also revealed a predominance of peripheral organs. We lastly, interrogated the genome in relation to mitochondrial diseases and acquired PTSD. In these cases, we hypothesized and confirmed a mixture of tissues related to peripheral organs (for mitochondrial diseases) and the CNS (for PTSD).
In the case of mitochondrial diseases, the heart-related tissues were revealed as the most affected along with muscle-skeletal tissue. Furthermore CNS-related tissues were more affected by gene removal in mitochondrial myopathies (i.e., the amygdala and the anterior cingulate cortex), as compared to MELAS or mitochondrial disease. The common thread across all three types of mitochondrial-related disorders was the heart-related tissues. The case of acquired PTSD showed a mixture of CNS-related tissues, tissues related to bodily peripheral organs, and heart-related tissues. The kidney and pancreas were also affected in PTSD. When we examined the maximum Δλ for each disorder/disease under examination, we found that the basal ganglia was maximally affected in autism, unipolar depression, spinocerebellar ataxia, and PTSD), while the heart left ventricle tissue was maximally affected in depression, ataxia, cerebellar ataxia, gait ataxia and fragile X tremor ataxia syndrome (FXTAS). This result indicates overlap between the psychiatric mental illnesses and the neurological disorders. It also shows the importance of examining mental illnesses in a more systemic way that includes the autonomic nervous system of the PNS.
This test on non-neurological illnesses served as a control to show that removal of the genes associated with each disorder did have specificity with the disorder and yet a very different outcome when comparing the mental illnesses to the non-neurological disorders. Among the top tissues affected across non-neurological diseases, the pancreas was maximally affected by the removal of disease-associated genes in breast and colon cancer, while the liver was maximally affected in diabetes. The heart left ventricle was maximally affected across autoimmune disorders such as psoriasis, lupus systemic erythematosus, and irritable bowel disease (IBD). The heart was also maximally affected in hematologic neoplast, congenital heart disease, mitochondrial disease and MELAS, in contrast to the mitochondrial myopathies which showed the liver as the maximally affected tissue.
This exercise demonstrated that despite the stochastic nature of gene expression, upon removal and random recombination, there is convergence across psychiatric and neurological disorders, thus potentially rendering both as disorders of the nervous systems. In both cases, we found a strong prevalence of the CNS, but also found important differences in tissues from the PNS, including the heart and the muscle-skeletal tissues involved in both mental illnesses and neurological disorders. Because of these convergences, and the fact that there are treatments and accommodations to help persons with neurological disorders, it may be possible to leverage some of those types of bodily-based supports to help persons with mental illnesses. Behaviors that are described by observation to define mental illnesses can now be connected with underlying tissues involved in voluntary, involuntary, and autonomic function across the CNS and the PNS and mapped to the genome, thus closing the present gap between behaviors and genomics in the precision medicine knowledge network. In this sense, the present methods offer a new way to interrogate the genome and link tissues with behavioral phenotyping.
A surprising finding here is the potential contributions of peripheral structures and organs to mental illness. Tissues of the autonomic nervous systems were maximally impacted by the removal of the genes associated with these mental illnesses, as was the muscle-skeletal tissue among the top-ranked illnesses. Tissues associated with subcortical brain regions necessary for motor control, learning, adaptation, and coordination (basal ganglia and striatum) were highly impacted by the removal of the genes in both mental illnesses and neurological disorders, along with those tissues important for memory (hippocampus), emotion (amygdala) and regulation (hypothalamus). Surprisingly, we did not see cerebellum-related tissues among the most affected by the removal of the genes (even in ataxias) where we do know that the cerebellum plays a large role [
20,
21,
22,
23]. This was also the case in autism, where we know the cerebellum has been implicated [
24,
25].
Lastly, the autoimmune disorders that we examined had very different brain tissue patterns from the mental illnesses and neurological conditions but shared the heart-related tissues and the muscle-skeletal tissue. In this sense, the contributions from the peripheral systems to mental illnesses and to autoimmune disorders seem important. However, blood tissue marked a departure of neurological disorders from mental illnesses, as it was maximally affected in neurological disorders but not in the mental illnesses. Overall, these gene removals revealed surprising results that invite rethinking how we may want to describe, diagnose, and treat mental illnesses in general.
Caveats and Future Directions
Although we found evidence that the mental illnesses and neurological disorders have remarkable overlap in the types of brain tissues that are maximally affected by the removal of their corresponding associated genes, we recognize that gene removal is a crude way to interrogate the human genome and its expression of the 54 tissues of the GTEx database. Future work will aim at developing more sophisticated methods to explore gene overexpression and to build simulations of the use of these methods in, e.g., combination with dynamic transcriptome evolution during neuronal differentiation in the development of cell lines from induced pluripotent stem cells. This will be important to move beyond a static approach and be able to assess asynchronous gene expression behaviors over time when cell lines differentiate into neuronal types. Full transcriptome dynamic interrogation longitudinally, over time, is now possible using these stochastic analyses in combination with the various data repositories featuring disease-associated genes.
The present work merely scratches the surface on possible new ways to interrogate the human genome in relation to diseases of all types (not just mental or neurological) in order to possibly build comparative models of outcomes
in tissues that can be related to behavioral phenotypic manifestations of the clinical disorder. In this sense, the work presented here can help bridge the gap between behavioral description of a mental illness, or a neurological disorder, and its genetic underpinnings via the affected tissues. Combining this approach with the new wave of digital biomarkers that describe human behavior digitally at a microscopic level [
9,
26,
27,
28,
29], using objective means and a finer level of granularity beyond naked eye detection, could help us redefine many psychiatric disorders and medical conditions under the precision medicine paradigm.
5. Conclusions
We here offer a new roadmap to reframe psychiatry using the precision medicine paradigm. The new stochastic approach can initiate the steps to connect behavioral phenotypic description from clinical observation and digital characterizations therein, with the underlying neurobiology of mental illnesses. Borrowing knowledge from neurology and brain science, it will be possible to shift psychiatry from an art to a quantitative objective science under the tenets of precision medicine by integrating all layers of the knowledge network. This would help design personalized targeted treatments utilizing the person’s genome, localizing the most-affected tissues defining central nervous system functions and distinguishing those from tissues related to vital organs for systemic functions. This new approach could potentially mark the beginning of a transformative era in mental health.
6. Patents
E.B.T. holds the US Patent “Methods and Systems for the Diagnoses and Treatments of Nervous Systems Disorders” combined in this paper as micro-movement spikes (MMS) data type and continuous gamma probability distribution family empirical estimation.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}












