Latin American Genes: The Great Forgotten in Rheumatoid Arthritis

 ,
,  and
and 
Abstract
:1. Introduction
2. Rheumatoid Arthritis in Latin American Populations
3. Genetics in Rheumatoid Arthritis
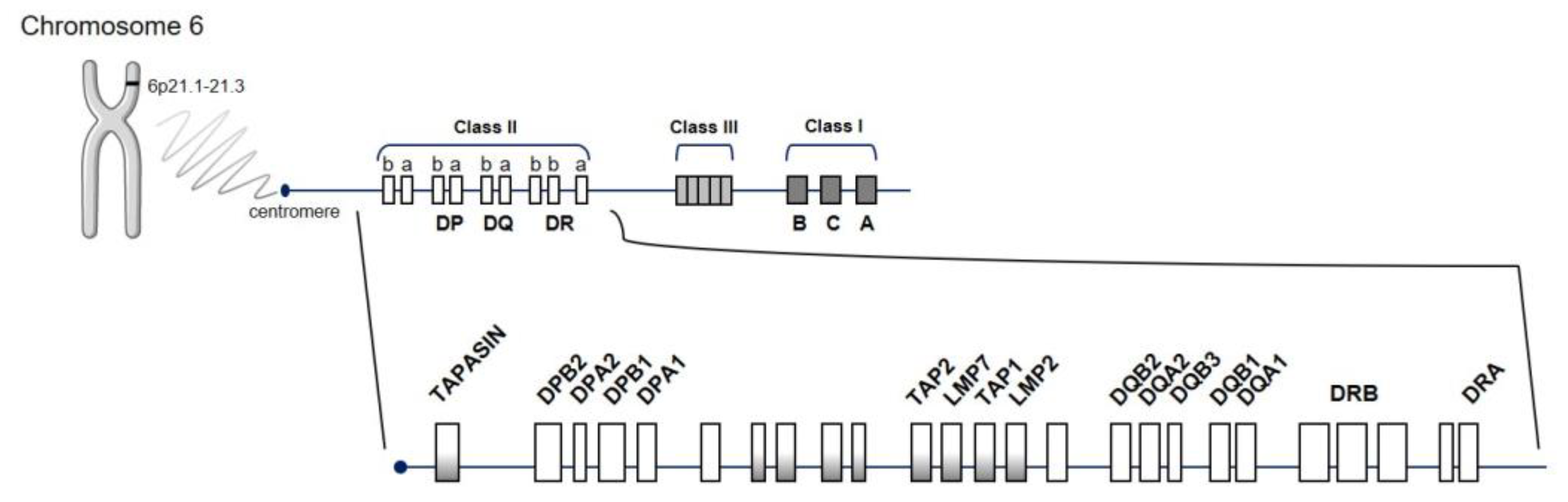
3.1. Human Leukocyte Antigen Region
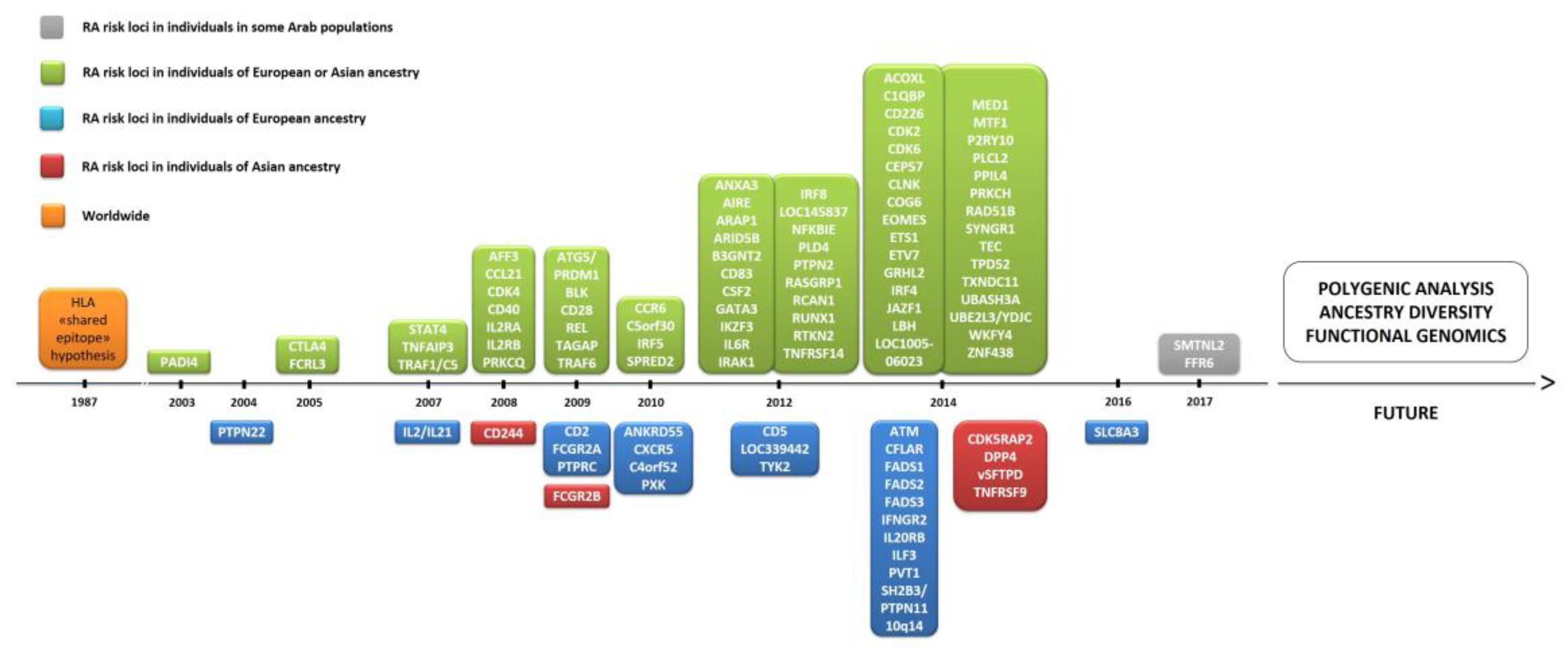
3.2. Genome-Wide Association Studies
3.2.1. Non-HLA Genetic Associations
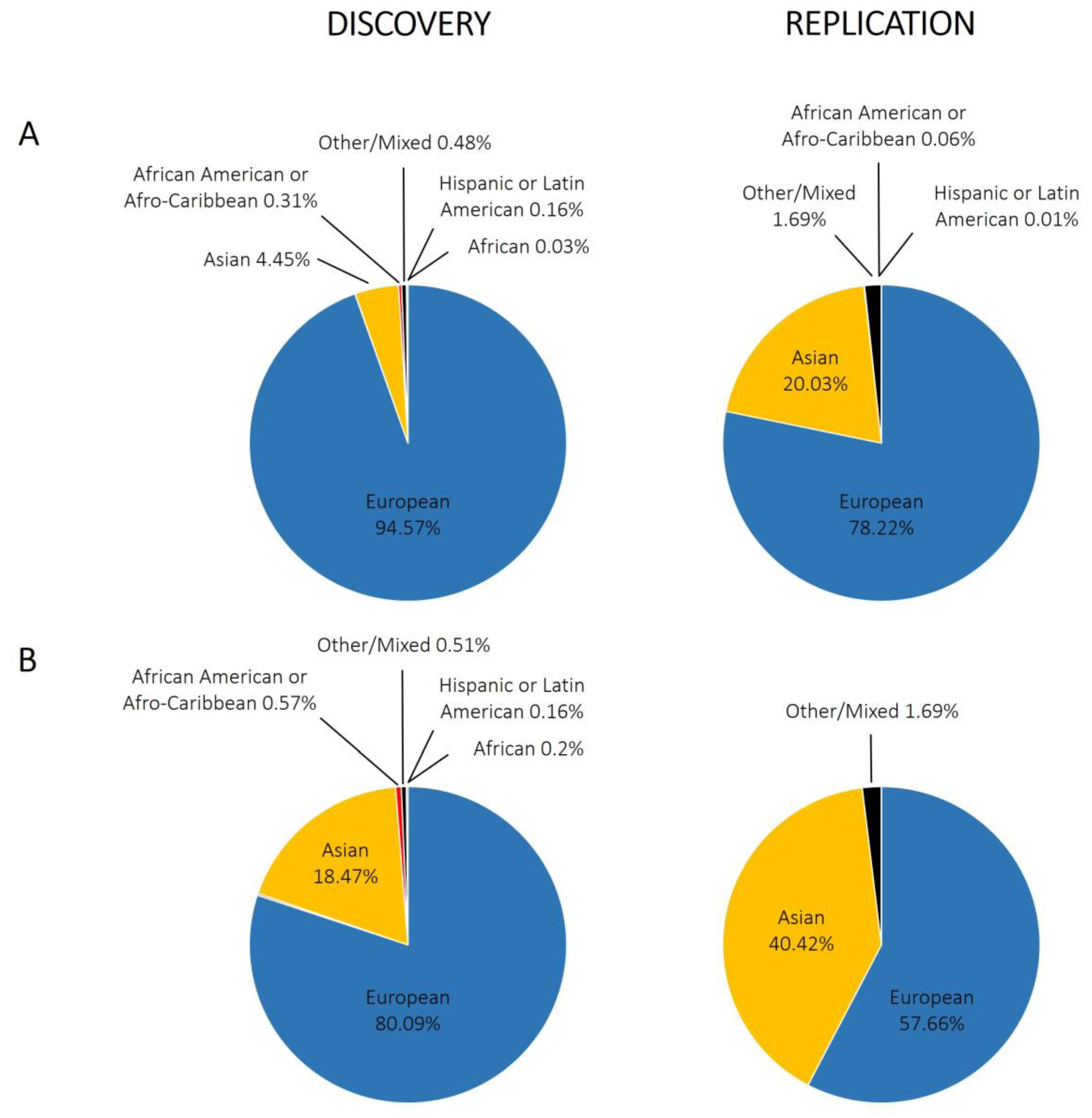
3.2.2. Ancestral Diversity and Sample Sizes
3.2.3. Population-Specific and Rare Variants
3.2.4. Implications for Variant Discovery and Medical Genetics
4. Pharmacogenetics and (Pharmaco) Epigenomics
4.1. Pharmacogenetics
4.2. Epigenomics
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Tobón, G.J.; Youinou, P.; Saraux, A. The environment, geo-epidemiology, and autoimmune disease: Rheumatoid arthritis. J. Autoimmun. 2010, 35, 10–14. [Google Scholar] [CrossRef] [PubMed]
- del Rincón, I.; Battafarano, D.F.; Arroyo, R.A.; Murphy, F.T.; Fischbach, M.; Escalante, A. Ethnic variation in the clinical manifestations of rheumatoid arthritis: Role of HLA–DRB1 alleles. Arthritis Rheum. 2003, 49, 200–208. [Google Scholar] [CrossRef] [PubMed]
- Daha, N.A.; Toes, R.E.M. Rheumatoid arthritis: Are ACPA-positive and ACPA-negative RA the same disease? Nat. Rev. Rheumatol. 2011, 7, 202–203. [Google Scholar] [CrossRef]
- Gurdasani, D.; Barroso, I.; Zeggini, E.; Sandhu, M.S. Genomics of disease risk in globally diverse populations. Nat. Rev. Genet. 2019, 20, 520–535. [Google Scholar] [CrossRef]
- Castro-Santos, P.; Díaz-Peña, R. Genetics of rheumatoid arthritis: A new boost is needed in Latin American populations. Rev. Bras. Reumatol. 2016, 56, 171–177. [Google Scholar] [CrossRef] [Green Version]
- Massardo, L.; Pons-Estel, B.A.; Wojdyla, D.; Cardiel, M.H.; Galarza-Maldonado, C.M.; Sacnun, M.P.; Soriano, E.R.; Laurindo, I.M.; Acevedo-Vásquez, E.M.; Caballero-Uribe, C.V.; et al. Early rheumatoid arthritis in Latin America: Low socioeconomic status related to high disease activity at baseline. Arthritis Care Res. 2012, 64, 1135–1143. [Google Scholar] [CrossRef]
- Peláez-Ballestas, I.; Sanin, L.H.; Moreno-Montoya, J.; Alvarez-Nemegyei, J.; Burgos-Vargas, R.; Garza-Elizondo, M.; Rodríguez-Amado, J.; Goycochea-Robles, M.V.; Madariaga, M.; Zamudio, J.; et al. Epidemiology of the rheumatic diseases in Mexico. A study of 5 regions based on the COPCORD methodology. J. Rheumatol. 2011, 38, 3–6. [Google Scholar] [CrossRef]
- Spindler, A.; Bellomio, V.; Berman, A.; Lucero, E.; Baigorria, M.; Paz, S.; Garrone, N.; Torres, A.I.; Romano, O.; Carraccio, A.; et al. Prevalence of rheumatoid arthritis in Tucuman, Argentina. J. Rheumatol. 2002, 29, 1166–1170. [Google Scholar]
- Scublinsky, D.; Venarotti, H.; Citera, G.; Messina, O.D.; Scheines, E.; Rillo, O.; Arturi, A.; Hofman, J.; Somma, L.F.; Casado, G.; et al. The prevalence of rheumatoid arthritis in argentina: A capture-recapture study in a city of Buenos Aires Province. J. Clin. Rheumatol. 2010, 16, 317–321. [Google Scholar] [CrossRef]
- Guevara-Pacheco, S.; Feicán-Alvarado, A.; Sanín, L.H.; Vintimilla-Ugalde, J.; Vintimilla-Moscoso, F.; Delgado-Pauta, J.; Lliguisaca-Segarra, A.; Dután-Erráez, H.; Guevara-Mosquera, D.; Ochoa-Robles, V.; et al. Prevalence of musculoskeletal disorders and rheumatic diseases in Cuenca, Ecuador: A WHO-ILAR COPCORD study. Rheumatol. Int. 2016, 36, 1195–1204. [Google Scholar] [CrossRef] [Green Version]
- Cuervo, F.; Santos, A.; Saldarriaga, E.; Angarita, I.; Peláez-Ballestas, I.; Giraldo, R.; Ballesteros, J.; Forero, E.; Ramírez, J.; Toro, C.; et al. Prevalencia de las enfermedades reumáticas en Colombia. Med. Bogotá 2018, 40, 94–95. [Google Scholar]
- Granados, Y.; Cedeño, L.; Rosillo, C.; Berbin, S.; Azocar, M.; Molina, M.E.; Lara, O.; Sanchez, G.; Peláez-Ballestas, I. Prevalence of musculoskeletal disorders and rheumatic diseases in an urban community in Monagas State, Venezuela: A COPCORD study. Clin. Rheumatol. 2015, 34, 871–877. [Google Scholar] [CrossRef] [PubMed]
- Gamboa, R.; Medina, M.; Acevedo, E.; Pastor, C.; Cucho, J.; Gutiérrez, C.; Ugarte, M.; Sánchez, C.; Perich, R.; Alfaro, J.; et al. Prevalencia de enfermedades reumatológicas y discapacidad en una comunidad urbano-marginal: Resultados del primer estudio COPCORD en el Perú. Rev. Peru. Reumatol. 2009, 15, 40–46. [Google Scholar]
- Rodrigues Senna, É.; De Barros, A.L.P.; Silva, E.O.; Costa, I.F.; Pereira, L.V.B.; Ciconelli, R.M.; Ferraz, M.B. Prevalence of Rheumatic Diseases in Brazil: A Study Using the COPCORD Approach. J. Rheumatol. 2004, 31, 594–597. [Google Scholar]
- Durán, J.; Massardo, L.; Llanos, C.; Iacobelli, S.; Burgos, P.; Cisternas, M.; Iruretagoyena, M.; Armstrong, M.; Aguilera, R.; Radrigán, F.; et al. The Prevalence of Rheumatoid Arthritis in Chile: A Nationwide Study Performed as Part of the National Health Survey. J. Rheumatol. 2020, 47, 951–958. [Google Scholar] [CrossRef]
- Sparks, J.A.; Costenbader, K.H. Genetics, environment, and gene-environment interactions in the development of systemic rheumatic diseases. Rheum. Dis. Clin. N. Am. 2014, 40, 637–657. [Google Scholar] [CrossRef] [Green Version]
- MacGregor, A.J.; Snieder, H.; Rigby, A.S.; Koskenvuo, M.; Kaprio, J.; Aho, K.; Silman, A.J. Characterizing the quantitative genetic contribution to rheumatoid arthritis using data from twins. Arthritis Rheum. 2000, 43, 30–37. [Google Scholar] [CrossRef]
- van der Helm-van Mil, A.H.M.; Wesoly, J.Z.; Huizinga, T.W.J. Understanding the genetic contribution to rheumatoid arthritis. Curr. Opin. Rheumatol. 2005, 17, 299–304. [Google Scholar] [CrossRef]
- van der Helm-van Mil, A.H.M.; Toes, R.E.M.; Huizinga, T.W.J. Genetic variants in the prediction of rheumatoid arthritis. Ann. Rheum. Dis. 2010, 69, 1694–1696. [Google Scholar] [CrossRef] [PubMed]
- Stastny, P. Association of the B-Cell Alloantigen DRw4 with Rheumatoid Arthritis. N. Engl. J. Med. 1978, 298, 869–871. [Google Scholar] [CrossRef]
- Gregersen, P.K.; Silver, J.; Winchester, R.J. The shared epitope hypothesis. an approach to understanding the molecular genetics of susceptibility to rheumatoid arthritis. Arthritis Rheum. 1987, 30, 1205–1213. [Google Scholar] [CrossRef] [PubMed]
- Robinson, J.; Halliwell, J.A.; Hayhurst, J.D.; Flicek, P.; Parham, P.; Marsh, S.G.E. The IPD and IMGT/HLA database: Allele variant databases. Nucleic Acids Res. 2015, 43, D423–D431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wysocki, T.; Olesińska, M.; Paradowska-Gorycka, A. Current Understanding of an Emerging Role of HLA-DRB1 Gene in Rheumatoid Arthritis-From Research to Clinical Practice. Cells 2020, 9, 1127. [Google Scholar] [CrossRef]
- Dedmon, L.E. The genetics of rheumatoid arthritis. Rheumatology (Oxford, England) 2020, 59, 2661–2670. [Google Scholar] [CrossRef]
- Delgado-Vega, A.M.; Anaya, J.-M. Meta-analysis of HLA-DRB1 polymorphism in Latin American patients with rheumatoid arthritis. Autoimmun. Rev. 2007, 6, 402–408. [Google Scholar] [CrossRef]
- Herráez, D.L.; Martínez-Bueno, M.; Riba, L.; De La Torre, I.G.; Sacnún, M.; Goñi, M.; Berbotto, G.A.; Paira, S.; Musuruana, J.L.; Graf, C.E.; et al. Rheumatoid arthritis in latin americans enriched for amerindian ancestry is associated with loci in chromosomes 1, 12, and 13, and the HLA Class II region. Arthritis Rheum. 2013, 65, 1457–1467. [Google Scholar] [CrossRef]
- Mills, M.C.; Rahal, C. A scientometric review of genome-wide association studies. Commun. Biol. 2019, 2, 9. [Google Scholar] [CrossRef] [Green Version]
- Amariuta, T.; Luo, Y.; Knevel, R.; Okada, Y.; Raychaudhuri, S. Advances in genetics toward identifying pathogenic cell states of rheumatoid arthritis. Immunol. Rev. 2020, 294, 188–204. [Google Scholar] [CrossRef]
- Suzuki, A.; Yamada, R.; Chang, X.; Tokuhiro, S.; Sawada, T.; Suzuki, M.; Nagasaki, M.; Nakayama-Hamada, M.; Kawaida, R.; Ono, M.; et al. Functional haplotypes of PADI4, encoding citrullinating enzyme peptidylarginine deiminase 4, are associated with rheumatoid arthritis. Nat. Genet. 2003, 34, 395–402. [Google Scholar] [CrossRef]
- Plenge, R.M.; Padyukov, L.; Remmers, E.F.; Purcell, S.; Lee, A.T.; Karlson, E.W.; Wolfe, F.; Kastner, D.L.; Alfredsson, L.; Altshuler, D.; et al. Replication of Putative Candidate-Gene Associations with Rheumatoid Arthritis in >4,000 Samples from North America and Sweden: Association of Susceptibility with PTPN22, CTLA4, and PADI4. Am. J. Hum. Genet. 2005, 77, 1044–1060. [Google Scholar] [CrossRef] [Green Version]
- Márquez, A.; Kerick, M.; Zhernakova, A.; Gutierrez-Achury, J.; Chen, W.-M.; Onengut-Gumuscu, S.; González-Álvaro, I.; Rodriguez-Rodriguez, L.; Rios-Fernández, R.; González-Gay, M.A.; et al. Meta-analysis of Immunochip data of four autoimmune diseases reveals novel single-disease and cross-phenotype associations. Genome Med. 2018, 10, 97. [Google Scholar] [CrossRef] [PubMed]
- Okada, Y.; Wu, D.; Trynka, G.; Raj, T.; Terao, C.; Ikari, K.; Kochi, Y.; Ohmura, K.; Suzuki, A.; Yoshida, S.; et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature 2013, 506, 376–381. [Google Scholar] [CrossRef] [PubMed]
- Tam, V.; Patel, N.; Turcotte, M.; Bossé, Y.; Paré, G.; Meyre, D. Benefits and limitations of genome-wide association studies. Nat. Rev. Genet. 2019, 20, 467–484. [Google Scholar] [CrossRef] [PubMed]
- Jansen, P.R.; Watanabe, K.; Stringer, S.; Skene, N.; Bryois, J.; Hammerschlag, A.R.; de Leeuw, C.A.; Benjamins, J.S.; Muñoz-Manchado, A.B.; Nagel, M.; et al. Genome-wide analysis of insomnia in 1,331,010 individuals identifies new risk loci and functional pathways. Nat. Genet. 2019, 51, 394–403. [Google Scholar] [CrossRef] [Green Version]
- Mills, M.C.; Rahal, C. The GWAS Diversity Monitor tracks diversity by disease in real time. Nat. Genet. 2020, 52, 242–243. [Google Scholar] [CrossRef]
- Sans, M. Admixture studies in Latin America: From the 20th to the 21st century. Hum. Biol. 2000, 72, 155–177. [Google Scholar]
- Darvasi, A.; Shifman, S. The beauty of admixture. Nat. Genet. 2005, 37, 118–119. [Google Scholar] [CrossRef]
- Bergström, A.; McCarthy, S.A.; Hui, R.; Almarri, M.A.; Ayub, Q.; Danecek, P.; Chen, Y.; Felkel, S.; Hallast, P.; Kamm, J.; et al. Insights into human genetic variation and population history from 929 diverse genomes. Science 2020, 367, 5012. [Google Scholar] [CrossRef]
- Bodmer, W.; Bonilla, C. Common and rare variants in multifactorial susceptibility to common diseases. Nat. Genet. 2008, 40, 695–701. [Google Scholar] [CrossRef] [Green Version]
- Kryukov, G.V.; Pennacchio, L.A.; Sunyaev, S.R. Most rare missense alleles are deleterious in humans: Implications for complex disease and association studies. Am. J. Hum. Genet. 2007, 80, 727–739. [Google Scholar] [CrossRef] [Green Version]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [Green Version]
- Bowes, J.; Lawrence, R.; Eyre, S.; Panoutsopoulou, K.; Orozco, G.; Elliott, K.S.; Ke, X.; Morris, A.P.; Thomson, W.; Worthington, J.; et al. Rare variation at the TNFAIP3 locus and susceptibility to rheumatoid arthritis. Hum. Genet. 2010, 128, 627–633. [Google Scholar] [CrossRef] [Green Version]
- Bang, S.-Y.; Na, Y.-J.; Kim, K.; Joo, Y.B.; Park, Y.; Lee, J.; Lee, S.-Y.; Ansari, A.A.; Jung, J.; Rhee, H.; et al. Targeted exon sequencing fails to identify rare coding variants with large effect in rheumatoid arthritis. Arthritis Res. Ther. 2014, 16, 447. [Google Scholar] [CrossRef] [Green Version]
- Diogo, D.; Kurreeman, F.; Stahl, E.A.; Liao, K.P.; Gupta, N.; Greenberg, J.D.; Rivas, M.A.; Hickey, B.; Flannick, J.; Thomson, B.; et al. Rare, Low-Frequency, and Common Variants in the Protein-Coding Sequence of Biological Candidate Genes from GWASs Contribute to Risk of Rheumatoid Arthritis. Am. J. Hum. Genet. 2013, 92, 15–27. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Leung, E.L.H.; Pan, H.; Yao, X.; Huang, Q.; Wu, M.; Xu, T.; Wang, Y.; Cai, J.; Li, R.; et al. Identification of potential genetic causal variants for rheumatoid arthritis by whole-exome sequencing. Oncotarget 2017, 8, 111119–111129. [Google Scholar] [CrossRef] [PubMed]
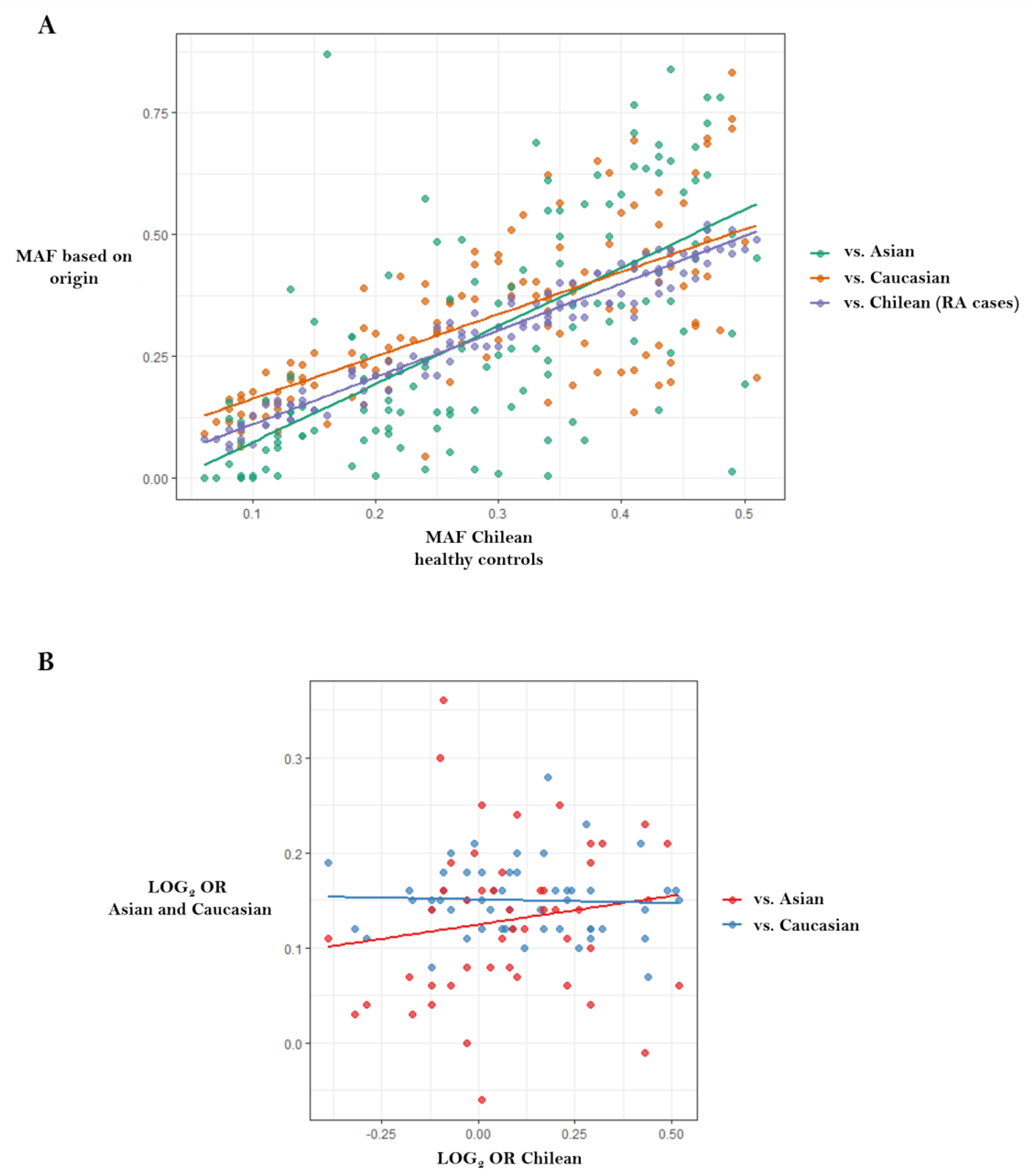
- Castro-Santos, P.; Verdugo, R.A.; Alonso-Arias, R.; Gutiérrez, M.A.; Suazo, J.; Aguillón, J.C.; Olloquequi, J.; Pinochet, C.; Lucia, A.; Quiñones, L.A.; et al. Association analysis in a Latin American population revealed ethnic differences in rheumatoid arthritis-associated SNPs in Caucasian and Asian populations. Sci. Rep. 2020, 10, 7879. [Google Scholar] [CrossRef] [PubMed]
- Frazer, K.A.; Murray, S.S.; Schork, N.J.; Topol, E.J. Human genetic variation and its contribution to complex traits. Nat. Rev. Genet. 2009, 10, 241–251. [Google Scholar] [CrossRef]
- Caulfield, T.; Fullerton, S.M.; Ali-Khan, S.E.; Arbour, L.; Burchard, E.G.; Cooper, R.S.; Hardy, B.J.; Harry, S.; Hyde-Lay, R.; Kahn, J.; et al. Race and ancestry in biomedical research: Exploring the challenges. Genome Med. 2009, 1, 8. [Google Scholar] [CrossRef] [Green Version]
- Stahl, E.A.; Wegmann, D.; Trynka, G.; Gutierrez-Achury, J.; Do, R.; Voight, B.F.; Kraft, P.; Chen, R.; Kallberg, H.J.; Kurreeman, F.A.S.; et al. Bayesian inference analyses of the polygenic architecture of rheumatoid arthritis. Nat. Genet. 2012, 44, 483–489. [Google Scholar] [CrossRef]
- Khera, A.V.; Chaffin, M.; Aragam, K.G.; Haas, M.E.; Roselli, C.; Choi, S.H.; Natarajan, P.; Lander, E.S.; Lubitz, S.A.; Ellinor, P.T.; et al. Genome-wide polygenic scores for common diseases identify individuals with risk equivalent to monogenic mutations. Nat. Genet. 2018, 50, 1219–1224. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.R.; Kanai, M.; Kamatani, Y.; Okada, Y.; Neale, B.M.; Daly, M.J. Clinical use of current polygenic risk scores may exacerbate health disparities. Nat. Genet. 2019, 51, 584–591. [Google Scholar] [CrossRef] [PubMed]
- Ling, S.F.; Bluett, J. Pharmacogenetics of methotrexate response in rheumatoid arthritis: An update. Pharmacogenomics 2019, 21, 3–6. [Google Scholar] [CrossRef]
- Bluett, J.; Sergeant, J.C.; MacGregor, A.J.; Chipping, J.R.; Marshall, T.; Symmons, D.P.M.; Verstappen, S.M.M. Risk factors for oral methotrexate failure in patients with inflammatory polyarthritis: Results from a UK prospective cohort study. Arthritis Res. Ther. 2018, 20, 50. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.C. Swollen to tender joint count ratio: A novel combination of routine measures to assess pain and treatment response in rheumatoid arthritis. Arthritis Care Res. 2014, 66, 171–172. [Google Scholar] [CrossRef] [PubMed]
- Wolfe, F.; Michaud, K.; Pincus, T. Preliminary evaluation of a visual analog function scale for use in rheumatoid arthritis. J. Rheumatol. 2005, 32, 1261–1266. [Google Scholar]
- Oliver, J.; Plant, D.; Webster, A.P.; Barton, A. Genetic and genomic markers of anti-TNF treatment response in rheumatoid arthritis. Biomark. Med. 2015, 9, 499–512. [Google Scholar] [CrossRef] [PubMed]
- Pallio, G.; Mannino, F.; Irrera, N.; Eid, A.H.; Squadrito, F.; Bitto, A. Polymorphisms Involved in Response to Biological Agents Used in Rheumatoid Arthritis. Biomolecules 2020, 10, 1203. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Sun, L.; Zhang, X. Integration of microbiome and epigenome to decipher the pathogenesis of autoimmune diseases. J. Autoimmun. 2017, 83, 31–42. [Google Scholar] [CrossRef]
- Takami, N.; Osawa, K.; Miura, Y.; Komai, K.; Taniguchi, M.; Shiraishi, M.; Sato, K.; Iguchi, T.; Shiozawa, K.; Hashiramoto, A.; et al. Hypermethylated promoter region of DR3, the death receptor 3 gene, in rheumatoid arthritis synovial cells. Arthritis Rheum. 2006, 54, 779–787. [Google Scholar] [CrossRef]
- Nile, C.J.; Read, R.C.; Akil, M.; Duff, G.W.; Wilson, A.G. Methylation status of a single CpG site in the IL6 promoter is related to IL6 messenger RNA levels and rheumatoid arthritis. Arthritis Rheum. 2008, 58, 2686–2693. [Google Scholar] [CrossRef]
- Karouzakis, E.; Rengel, Y.; Jüngel, A.; Kolling, C.; Gay, R.E.; Michel, B.A.; Tak, P.P.; Gay, S.; Neidhart, M.; Ospelt, C. DNA methylation regulates the expression of CXCL12 in rheumatoid arthritis synovial fibroblasts. Genes Immun. 2011, 12, 643–652. [Google Scholar] [CrossRef] [Green Version]
- Nakano, K.; Whitaker, J.W.; Boyle, D.L.; Wang, W.; Firestein, G.S. DNA methylome signature in rheumatoid arthritis. Ann. Rheum. Dis. 2013, 72, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Aryee, M.J.; Padyukov, L.; Fallin, M.D.; Hesselberg, E.; Runarsson, A.; Reinius, L.; Acevedo, N.; Taub, M.; Ronninger, M.; et al. Epigenome-wide association data implicate DNA methylation as an intermediary of genetic risk in rheumatoid arthritis. Nat. Biotechnol. 2013, 31, 142–147. [Google Scholar] [CrossRef]
- de la Rica, L.; Urquiza, J.M.; Gómez-Cabrero, D.; Islam, A.B.M.M.K.; López-Bigas, N.; Tegnér, J.; Toes, R.E.M.; Ballestar, E. Identification of novel markers in rheumatoid arthritis through integrated analysis of DNA methylation and microRNA expression. J. Autoimmun. 2013, 41, 6–16. [Google Scholar] [CrossRef]
- Glossop, J.R.; Emes, R.D.; Nixon, N.B.; Haworth, K.E.; Packham, J.C.; Dawes, P.T.; Fryer, A.A.; Mattey, D.L.; Farrell, W.E. Genome-wide DNA methylation profiling in rheumatoid arthritis identifies disease-associated methylation changes that are distinct to individual T- and B-lymphocyte populations. Epigenetics 2014, 9, 1228–1237. [Google Scholar] [CrossRef] [Green Version]
- Julià, A.; Absher, D.; López-Lasanta, M.; Palau, N.; Pluma, A.; Waite Jones, L.; Glossop, J.R.; Farrell, W.E.; Myers, R.M.; Marsal, S. Epigenome-wide association study of rheumatoid arthritis identifies differentially methylated loci in B cells. Hum. Mol. Genet. 2017, 26, 2803–2811. [Google Scholar] [CrossRef]
- Zhu, H.; Wu, L.F.; Mo, X.B.; Lu, X.; Tang, H.; Zhu, X.W.; Xia, W.; Guo, Y.F.; Wang, M.J.; Zeng, K.Q.; et al. Rheumatoid arthritis-associated DNA methylation sites in peripheral blood mononuclear cells. Ann. Rheum. Dis. 2019, 78, 36–42. [Google Scholar] [CrossRef]
- Nemtsova, M.V.; Zaletaev, D.V.; Bure, I.V.; Mikhaylenko, D.S.; Kuznetsova, E.B.; Alekseeva, E.A.; Beloukhova, M.I.; Deviatkin, A.A.; Lukashev, A.N.; Zamyatnin, A.A. Epigenetic changes in the pathogenesis of rheumatoid arthritis. Front. Genet. 2019, 10, 570. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Laird, P.W. Principles and challenges of genome-wide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Gowers, I.R.; Walters, K.; Kiss-Toth, E.; Read, R.C.; Duff, G.W.; Wilson, A.G. Age-related loss of CpG methylation in the tumour necrosis factor promoter. Cytokine 2011, 56, 792–797. [Google Scholar] [CrossRef] [Green Version]
- Tao, W.; Concepcion, A.N.; Vianen, M.; Marijnissen, A.C.A.; Lafeber, F.P.G.J.; Radstake, T.R.D.J.; Pandit, A. Multi-omics and machine learning accurately predicts clinical response to Adalimumab and Etanercept therapy in patients with rheumatoid arthritis. Arthritis Rheumatol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Raychaudhuri, S.; Sandor, C.; Stahl, E.A.; Freudenberg, J.; Lee, H.S.; Jia, X.; Alfredsson, L.; Padyukov, L.; Klareskog, L.; Worthington, J.; et al. Five amino acids in three HLA proteins explain most of the association between MHC and seropositive rheumatoid arthritis. Nat. Genet. 2012, 44, 291–296. [Google Scholar] [CrossRef] [PubMed]
- Knevel, R.; Huizinga, T.W.J.; Kurreeman, F. Genomic Influences on Susceptibility and Severity of Rheumatoid Arthritis. Rheum. Dis. Clin. N. Am. 2017, 43, 347–361. [Google Scholar] [CrossRef]
- Maehlen, M.T.; Olsen, I.C.; Andreassen, B.K.; Viken, M.K.; Jiang, X.; Alfredsson, L.; Källberg, H.; Brynedal, B.; Kurreeman, F.; Daha, N.; et al. Genetic risk scores and number of autoantibodies in patients with rheumatoid arthritis. Ann. Rheum. Dis. 2015, 74, 762–768. [Google Scholar] [CrossRef]
- Yarwood, A.; Han, B.; Raychaudhuri, S.; Bowes, J.; Lunt, M.; Pappas, D.A.; Kremer, J.; Greenberg, J.D.; Plenge, R.; Worthington, J.; et al. A weighted genetic risk score using all known susceptibility variants to estimate rheumatoid arthritis risk. Ann. Rheum. Dis. 2015, 74, 170–176. [Google Scholar] [CrossRef]
- Sparks, J.A.; Chen, C.Y.; Jiang, X.; Askling, J.; Hiraki, L.T.; Malspeis, S.; Klareskog, L.; Alfredsson, L.; Costenbader, K.H.; Karlson, E.W. Improved performance of epidemiologic and genetic risk models for rheumatoid arthritis serologic phenotypes using family history. Ann. Rheum. Dis. 2015, 74, 1522–1529. [Google Scholar] [CrossRef] [Green Version]
- Knevel, R.; le Cessie, S.; Terao, C.C.; Slowikowski, K.; Cui, J.; Huizinga, T.W.J.; Costenbader, K.H.; Liao, K.P.; Karlson, E.W.; Raychaudhuri, S. Using genetics to prioritize diagnoses for rheumatology outpatients with inflammatory arthritis. Sci. Transl. Med. 2020, 12, eaay1548. [Google Scholar] [CrossRef]
- Johnson, S.B.; Slade, I.; Giubilini, A.; Graham, M. Rethinking the ethical principles of genomic medicine services. Eur. J. Hum. Genet. 2020, 28, 147–154. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | HLA-DRB1 Allele(s) | Sequence (70–74 Position) |
|---|---|---|
| Caucasoid | * 04:01/* 04:04, * 04:08 | QKRAA/QRRAA |
| Asian | * 04:05 | QRRAA |
| Native Americans | * 14:02 | QRRAA |
| African Americans | * 01:01, * 04:05/* 10:01 | QRRAA/RRRAA |
| Israeli Jews | * 01:01, * 01:02 | QRRAA |
| Latin Americans | * 04:01/* 04:04, * 04:05 | QKRAA/QRRAA |
| Study | Gene (s)/Region (s) Implicated | Main Results |
|---|---|---|
| [59] | Human leukocyte antigen (HLA)-DR-3 | DR-3 gene was differentially methylated in patients with RA. As a result, the expression of DR-3 protein was downregulated in synovium, thereby providing higher resistance to apoptosis in these cells |
| [60] | Interleukin 6 (IL6) | Lower methylation and subsequent higher expression of IL6 in peripheral blood mononuclear cells in patients with RA |
| [61] | C-X-C motif chemokine 12 (CXCL12) | CXCL12 gene was hypomethylated in patients with RA and the levels of CXCL12 were subsequently higher in these patients than in those with osteoarthritis, promoting activation of matrix metalloproteinases and joint destruction |
| [62] | Genome-wide studies comparing stromal fibroblast-like synoviocytes in patients with RA or osteoarthritis | Identification of ~2000 loci differentially methylated, including genes involved in immune response, migration, and cellular adhesion |
| [63] | Identification of 2 methylation clusters in the major histocompatibility complex (MHC) region associated with epigenetic risk for RA | The DNA methylation study sorted CD14+ monocytes of patients with RA and controls, finding 9 differential methylated sites located in the MHC region and suggesting that monocytes are more proximal to the pathogenic cell type |
| [64] | Interleukin 6 receptor (IL6R), calpain 8 (CAPN8), homeobox protein Hox-A11 (HOXA11), dipeptidyl-peptidase 4 (DPP4), and homeobox protein Hox-C4 (HOXC4) | The study showed hypomethylation of IL6R, CAPN8, and HOXA11, and hypermethylation of DPP4 and HOXC4, respectively, in the synovial fibroblasts of patients with RA |
| [65] | Dual specificity phosphatase 22 (DUSP22) and polypeptide N-acetylgalactosaminyltransferase 9 (GALNT9) | Multiple sites within DUSP22 and GALNT9 genes were consistently hypermethylated and hypomethylated, respectively, in T-lymphocytes from patients with RA |
| [66] | T-cell surface glycoprotein CD1c (CD1C), TNF superfamily member 10 (TNFSF10), parvin gamma (PARVG), nidogen 1 (NID1), dehydrogenase/reductase 12 (DHRS12), inositol-tetrakisphosphate 1-kinase (ITPK1), acyl-CoA synthetase family member 3 (ACSF3), and TNF receptor superfamily member 13C (TNFRSF13C) | Although there were differentially methylated genes in patients and control groups, the study showed similar patterns of epigenetic changes in B-lymphocytes from patients with RA or systemic lupus erythematosus |
| [67] | Poly (ADP-ribose) polymerase family member 9 (PARP9) | The study identified an interferon-inducible gene interaction network. The significance of PARP9 gene methylation and the resulting change in gene expression in the pathogenesis of RA was demonstrated. In addition, the ability of PARP9 gene to positively regulate interleukin 2, which stimulates various cells of the immune response, was revealed. |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Díaz-Peña, R.; Quiñones, L.A.; Castro-Santos, P.; Durán, J.; Lucia, A. Latin American Genes: The Great Forgotten in Rheumatoid Arthritis. J. Pers. Med. 2020, 10, 196. https://doi.org/10.3390/jpm10040196
Díaz-Peña R, Quiñones LA, Castro-Santos P, Durán J, Lucia A. Latin American Genes: The Great Forgotten in Rheumatoid Arthritis. Journal of Personalized Medicine. 2020; 10(4):196. https://doi.org/10.3390/jpm10040196
Chicago/Turabian StyleDíaz-Peña, Roberto, Luis A. Quiñones, Patricia Castro-Santos, Josefina Durán, and Alejandro Lucia. 2020. "Latin American Genes: The Great Forgotten in Rheumatoid Arthritis" Journal of Personalized Medicine 10, no. 4: 196. https://doi.org/10.3390/jpm10040196
APA StyleDíaz-Peña, R., Quiñones, L. A., Castro-Santos, P., Durán, J., & Lucia, A. (2020). Latin American Genes: The Great Forgotten in Rheumatoid Arthritis. Journal of Personalized Medicine, 10(4), 196. https://doi.org/10.3390/jpm10040196