A Wide Spectrum of Genetic Disorders Causing Severe Childhood Epilepsy in Taiwan: A Case Series of Ultrarare Genetic Cause and Novel Mutation Analysis in a Pilot Study



Abstract
:1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Extracting DNA from Peripheral Blood
2.3. WES
2.4. Data Analysis and Interpretation
3. Results
3.1. Demographic Data of the Enrolled Patients
3.2. Diagnostic Yield
3.3. Genotype
3.4. Refractory Seizure Cases
3.5. Recurrent Mutations
3.6. Genes Accounting for Epilepsy Demonstrated by the Age of Seizure Onset
3.7. Nonseizure Group
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Engel, J., Jr. A proposed diagnostic scheme for people with epileptic seizures and with epilepsy: Report of the ILAE Task Force on Classification and Terminology. Epilepsia 2001, 42, 796–803. [Google Scholar] [CrossRef]
- Scheffer, I.E.; Berkovic, S.; Capovilla, G.; Connolly, M.B.; French, J.; Guilhoto, L.; Hirsch, E.; Jain, S.; Mathern, G.W.; Moshé, S.L.; et al. ILAE classification of the epilepsies: Position paper of the ILAE Commission for Classification and Terminology. Epilepsia 2017, 58, 512–521. [Google Scholar] [CrossRef] [Green Version]
- El Kosseifi, C.; Cornet, M.C.; Cilio, M.R. Neonatal Developmental and Epileptic Encephalopathies. Semin. Pediatr. Neurol. 2019, 32, 100770. [Google Scholar] [CrossRef]
- Howell, K.B.; Harvey, A.S.; Archer, J.S. Epileptic encephalopathy: Use and misuse of a clinically and conceptually important concept. Epilepsia 2016, 57, 343–347. [Google Scholar] [CrossRef] [Green Version]
- McTague, A.; Howell, K.B.; Cross, J.H.; Kurian, M.A.; Scheffer, I.E. The genetic landscape of the epileptic encephalopathies of infancy and childhood. Lancet Neurol. 2016, 15, 304–316. [Google Scholar] [CrossRef]
- Nashabat, M.; Al Qahtani, X.S.; Almakdob, S.; Altwaijri, W.; Ba-Armah, D.M.; Hundallah, K.; Al Hashem, A.; Al Tala, S.; Maddirevula, S.; Alkuraya, F.S.; et al. The landscape of early infantile epileptic encephalopathy in a consanguineous population. Seizure 2019, 69, 154–172. [Google Scholar] [CrossRef]
- Nieh, S.E.; Sherr, E.H. Epileptic encephalopathies: New genes and new pathways. Neurotherapeutics 2014, 11, 796–806. [Google Scholar] [CrossRef] [Green Version]
- Khan, S.; Al Baradie, R. Epileptic encephalopathies: An overview. Epilepsy Res. Treat. 2012, 2012, 403592. [Google Scholar] [CrossRef]
- Noh, G.J.; Asher, Y.J.T.; Graham, J.M., Jr. Clinical review of genetic epileptic encephalopathies. Eur. J. Med. Genet. 2012, 55, 281–298. [Google Scholar] [CrossRef] [Green Version]
- Chang, X.; Wang, K. wANNOVAR: Annotating genetic variants for personal genomes via the web. J. Med. Genet. 2012, 49, 433–436. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Wang, K. Genomic variant annotation and prioritization with ANNOVAR and wANNOVAR. Nat. Protoc. 2015, 10, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Landrum, M.J.; Lee, J.M.; Riley, G.R.; Jang, W.; Rubinstein, W.S.; Church, D.M.; Maglott, D.R. ClinVar: Public archive of relationships among sequence variation and human phenotype. Nucleic. Acids. Res. 2014, 42 (Database issue), D980–D985. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef] [PubMed]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amberger, J.S.; Bocchini, C.A.; Schiettecatte, F.; Scott, A.F.; Hamosh, A. OMIM.org: Online Mendelian Inheritance in Man (OMIM®), an online catalog of human genes and genetic disorders. Nucleic. Acids. Res. 2015, 43 (Database issue), D789–D798. [Google Scholar] [CrossRef] [Green Version]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical. Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Demos, M.; Guella, I.; DeGuzman, C.; McKenzie, M.B.; Buerki, S.E.; Evans, D.M.; Toyota, E.B.; Boelman, C.; Huh, L.L.; Datta, A.; et al. Diagnostic Yield and Treatment Impact of Targeted Exome Sequencing in Early-Onset Epilepsy. Front Neurol. 2019, 10, 434. [Google Scholar] [CrossRef]
- Snoeijen-Schouwenaars, F.M.; van Ool, J.S.; Verhoeven, J.S.; van Mierlo, P.; Braakman, H.M.; Smeets, E.E.; Nicolai, J.; Schoots, J.; Teunissen, M.W.; Rouhl, R.P.; et al. Diagnostic exome sequencing in 100 consecutive patients with both epilepsy and intellectual disability. Epilepsia 2019, 60, 155–164. [Google Scholar] [CrossRef] [Green Version]
- Butler, K.M.; da Silva, C.; Alexander, J.J.; Hegde, M.; Escayg, A. Diagnostic Yield from 339 Epilepsy Patients Screened on a Clinical Gene Panel. Pediatric Neurol. 2017, 77, 61–66. [Google Scholar] [CrossRef]
- Juanes, M.; Veneruzzo, G.; Loos, M.; Reyes, G.; Araoz, H.V.; Garcia, F.M.; Gomez, G.; Alonso, C.N.; Chertkoff, L.P.; Caraballo, R. Molecular diagnosis of epileptic encephalopathy of the first year of life applying a customized gene panel in a group of Argentinean patients. Epilepsy Behav. 2020, 111, 107322. [Google Scholar] [CrossRef] [PubMed]
- Ostrander, B.E.; Butterfield, R.J.; Pedersen, B.S.; Farrell, A.J.; Layer, R.M.; Ward, A.; Miller, C.; DiSera, T.; Filloux, F.M.; Candee, M.S.; et al. Whole-genome analysis for effective clinical diagnosis and gene discovery in early infantile epileptic encephalopathy. NPJ Genom. Med. 2018, 3, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Srivastava, S.; Cohen, J.S.; Vernon, H.; Barañano, K.; McClellan, R.; Jamal, L.; Naidu, S.; Fatemi, A. Clinical whole exome sequencing in child neurology practice. Ann Neurol. 2014, 76, 473–483. [Google Scholar] [CrossRef]
- Dixon-Salazar, T.J.; Silhavy, J.L.; Udpa, N.; Schroth, J.; Bielas, S.; Schaffer, A.E.; Olvera, J.; Bafna, V.; Zaki, M.S.; Abdel-Salam, G.H.; et al. Exome sequencing can improve diagnosis and alter patient management. Sci. Transl. Med. 2012, 4, 138ra178. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuperberg, M.; Lev, D.; Blumkin, L.; Zerem, A.; Ginsberg, M.; Linder, I.; Carmi, N.; Kivity, S.; Lerman-Sagie, T.; Leshinsky-Silver, E. Utility of Whole Exome Sequencing for Genetic Diagnosis of Previously Undiagnosed Pediatric Neurology Patients. J. Child Neurol. 2016, 31, 1534–1539. [Google Scholar] [CrossRef]
- Du, W.; Bautista, J.F.; Yang, H.; Diez-Sampedro, A.; You, S.A.; Wang, L.; Kotagal, P.; Lüders, H.O.; Shi, J.; Cui, J. Calcium-sensitive potassium channelopathy in human epilepsy and paroxysmal movement disorder. Nat. Genet. 2005, 37, 733–738. [Google Scholar] [CrossRef]
- Lee, I.C.; Hong, S.Y. Treatment of intractable seizure in Wolf-Hirschhorn syndrome with bromide. Brain Dev. 2017, 39, 633. [Google Scholar] [CrossRef]
- Marini, C.; Scheffer, I.E.; Nabbout, R.; Mei, D.; Cox, K.; Dibbens, L.M.; McMahon, J.M.; Iona, X.; Carpintero, R.S.; Elia, M.; et al. SCN1A duplications and deletions detected in Dravet syndrome: Implications for molecular diagnosis. Epilepsia 2009, 50, 1670–1678. [Google Scholar] [CrossRef]
- Lin, W.D.; Chang, K.P.; Wang, C.H.; Chen, S.J.; Fan, P.C.; Weng, W.C.; Lin, W.C.; Tsai, Y.; Tsai, C.H.; Chou, I.C.; et al. Molecular aspects of Dravet syndrome patients in Taiwan. Clin. Chim. Acta 2013, 421, 34–40. [Google Scholar] [CrossRef]
- Claes, L.; Del-Favero, J.; Ceulemans, B.; Lagae, L.; Van Broeckhoven, C.; De Jonghe, P. De novo mutations in the sodium-channel gene SCN1A cause severe myoclonic epilepsy of infancy. Am. J. Hum. Genet. 2001, 68, 1327–1332. [Google Scholar] [CrossRef] [Green Version]
- Vadlamudi, L.; Dibbens, L.M.; Lawrence, K.M.; Iona, X.; McMahon, J.M.; Murrell, W.; Mackay-Sim, A.; Scheffer, I.E.; Berkovic, S.F. Timing of de novo mutagenesis—A twin study of sodium-channel mutations. N. Engl. J. Med. 2010, 363, 1335–1340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kanai, K.; Hirose, S.; Oguni, H.; Fukuma, G.; Shirasaka, Y.; Miyajima, T.; Wada, K.; Iwasa, H.; Yasumoto, S.; Matsuo, M.; et al. Effect of localization of missense mutations in SCN1A on epilepsy phenotype severity. Neurology 2004, 63, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Ceulemans, B.P.; Claes, L.R.; Lagae, L.G. Clinical correlations of mutations in the SCN1A gene: From febrile seizures to severe myoclonic epilepsy in infancy. Pediatric Neurol. 2004, 30, 236–243. [Google Scholar] [CrossRef] [PubMed]
- Lim, B.C.; Hwang, H.; Chae, J.H.; Choi, J.E.; Hwang, Y.S.; Kang, S.H.; Ki, C.S.; Kim, K.J. SCN1A mutational analysis in Korean patients with Dravet syndrome. Seizure 2011, 20, 789–794. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldberg-Stern, H.; Aharoni, S.; Afawi, Z.; Bennett, O.; Appenzeller, S.; Pendziwiat, M.; Kuhlenbäumer, G.; Basel-Vanagaite, L.; Shuper, A.; Korczyn, A.D.; et al. Broad phenotypic heterogeneity due to a novel SCN1A mutation in a family with genetic epilepsy with febrile seizures plus. J. Child Neurol. 2014, 29, 221–226. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.S.; Pan, Z.; Shi, W.; Brown, B.S.; Wymore, R.S.; Cohen, I.S.; Dixon, J.E.; McKinnon, D. KCNQ2 and KCNQ3 potassium channel subunits: Molecular correlates of the M-channel. Science 1998, 282, 1890–1893. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weckhuysen, S.; Mandelstam, S.; Suls, A.; Audenaert, D.; Deconinck, T.; Claes, L.R.; Deprez, L.; Smets, K.; Hristova, D.; Yordanova, I.; et al. KCNQ2 encephalopathy: Emerging phenotype of a neonatal epileptic encephalopathy. Ann Neurol. 2012, 71, 15–25. [Google Scholar] [CrossRef]
- Dedek, K.; Fusco, L.; Teloy, N.; Steinlein, O.K. Neonatal convulsions and epileptic encephalopathy in an Italian family with a missense mutation in the fifth transmembrane region of KCNQ2. Epilepsy Res. 2003, 54, 21–27. [Google Scholar] [CrossRef]
- Shashi, V.; McConkie-Rosell, A.; Rosell, B.; Schoch, K.; Vellore, K.; McDonald, M.; Jiang, Y.H.; Xie, P.; Need, A.; Goldstein, D.B. The utility of the traditional medical genetics diagnostic evaluation in the context of next-generation sequencing for undiagnosed genetic disorders. Genet. Med. 2014, 16, 176–182. [Google Scholar] [CrossRef] [Green Version]
- Majumdar, K. Differential operator in seizure detection. Comput. Biol. Med. 2012, 42, 70–74. [Google Scholar] [CrossRef]





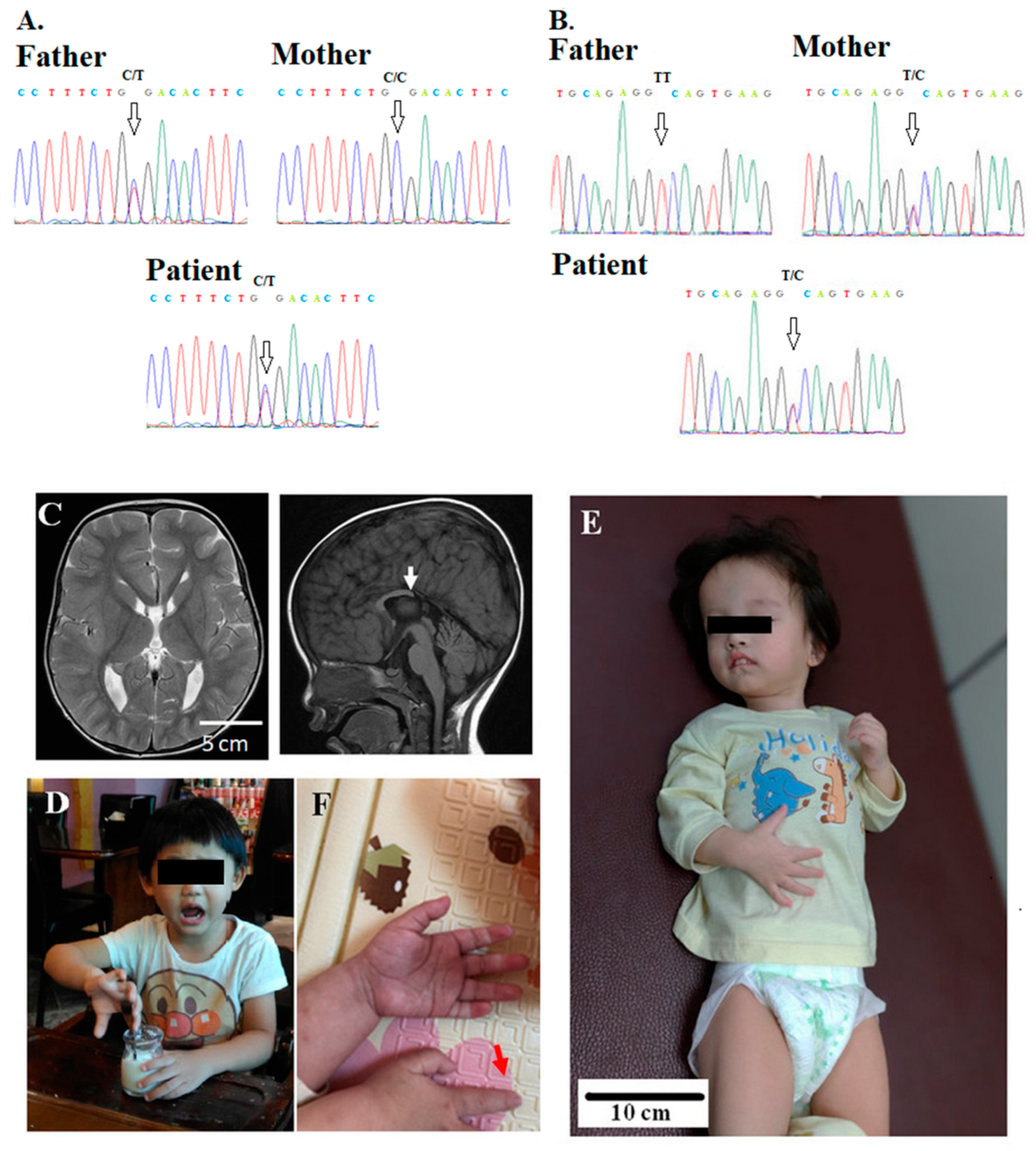{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Data (N) | Identified Genetic Group (N = 24) | Nonidentified Genetic Group (N = 30) | |
|---|---|---|---|
| Symptoms | NS | ||
| Seizures (45) | 20 (44%) | 25 (56%) | |
| No seizure (9) | 4 (44%) | 5 (56%) | |
| Age at first seizure onset | NS | ||
| 0–1 month (10) | 5 (50%) | 5 (50%) | |
| 1 month–2 years (31) | 13 (42%) | 18 (58%) | |
| >2 years (4) | 2 (50%) | 2 (50%) | |
| No seizure (8) | 4 (44%) | 5 (56%) | |
| Sex | p < 0.05 ※ | ||
| Male (30) | 17 (57%) | 13 (43%) | |
| Female (24) | 7 (29%) | 17 (71%) | |
| Number of antiepileptic drugs at the time of study | NS | ||
| 0 (8) | 4 (44%) | 5 (56%) | |
| 1 (10) | 2 (20%) | 8 (80%) | |
| 2 (11) | 5 (49%) | 6 (51%) | |
| >3 or =3 (25) | 13 (54%) | 11 (46%) |
| Gene Name | Genotype | Inheritance Pattern | Phenotype | Age of Diagnosis/ Age of First Seizure | Sex | NCBI ClinVar | Seizure Types | Neurodevelopmental Outcomes | |
|---|---|---|---|---|---|---|---|---|---|
| P 1 | PACS1 (NM_018026.4) | c.607C > T p.(Arg203Trp) | AD, de novo | DEE, spinal tethered cord | 12 years/ Newborn | F | Pathogenic | Focal seizures | Walk by ambulance, cognition delay at 14 years |
| P 2 | TBCID 24 (NM_001199 107.1) | c.1499C > T (p.Ala500Val)/c.229_240del (p.77_80del) | AR | DEE | 2 years /10 months | M | Pathogenic/Pathogenic | Multiple focal, general, nonconvulsive status epilepticus | Attention deficit. cognition delay |
| P 3 | FBN1 (NM_000138. 4) | c.794_795insGTAT (p.Val266Tyr fs * 6) | AD | Marfan syndrome | 16 years /- | F | Pathogenic | No seizure | Normal |
| P 4 | KCNQ2 (NM_172107.3) | c.740C > T (p.Ser247Leu) | AD, de novo | DEE | 10 months /Day 3 | M | Pathogenic | General tonic, apnea | No language, hypotonia at 3 years |
| P 5 | KCNQ2 (NM_172107.3) | c.853C > A (p.Pro285Thr) | AD, de novo | DEE | 2 months /Day 3 | F | Novel | General tonic | No language, hypotonia at 1 year |
| P 6 | OSGEP (NM_017807.4) | c.740G > A (p.Arg247Gln)/ c.740G > A (p.Arg247Gln) | AR | Lissencephaly, seizures | 3 months /3 weeks | M | Pathogenic/Pathogenic | Focal | Severe, died at 6 months |
| P 7 | SCN2A (NM_021007.2) | c.4958delT (p.Leu1653Ter) | AD, de novo | Autism | 4 years/- | M | Pathogenic | No seizure | Severe autism, no language at 4 years |
| P 8 | 16p13.11 microdeletion | AD | Frequent seizures | 12 years/10 years | F | Pathogenic | General tonic | Attention deficit | |
| P 9 ※ | 4p16.3p16.1(68,345–7,739,782)X1, 17q25.1q25.3(73,608,322–81,041,938)X3 | AD, de novo | DEE | 1 year/ 6 months | M | Pathogenic | General | Global DD | |
| P 10 | IRF2BPL (NM_024496.3) | c.562C > T (p.Arg188Ter) | AD, de novo | DEE and regressive encephalopathy | 12 years /11 years | F | Pathogenic | General | Profound ID |
| P 11 | PIGA: (NM.002641) | c.356G > A (p.Arg119Gln) | X-linked | DEE | 0.5 month /Day 3 | M | Pathogenic | Apnea, cyanosis, absence, atonic, spasms | Severe global DD |
| P 12 | PPP1CB | c.548A > C (p.Glu183Ala) | AD, de novo | DEE, dysmorphism | 4 months/ 2 months | M | Pathogenic | Apnea, eye gazed deviation, myoclonic | Severe global DD |
| P 13 | TBCID 24 (NM_001199 107.1) | c.119G > A (p.Arg40His)/ c.1499C > T (p.Ala500Val) | AR | DEE | 8 months /6 months | M | Pathogenic /Pathogenic | Multifocal, myoclonus | Global DD |
| P 14 | UBE3A (NM_130838) | c.C219A (p.Thr73Ter) | AD, maternal imprinting | Regressive encephalopathy | 14 months /11 months | M | Novel | General tonic | Profound ID |
| P 15 | SCN1A (NM_001165963) | c.362delC (p.Ala121fs) | AD, de novo | DEE | 5 months /4 months | M | Novel | Focal clonic, general tonic | Profound ID |
| P 16 | SCN1A (NM_001165963) | c.3918 + 1 G > - | AD, de novo | DEE | 8 months /5 months | F | Novel | Focal clonic, general tonic | Profound ID |
| P 17 $ | MECP2 (NM_004992.3) | rsa Xq28 MECP2 (exons3-4) x1 | X-linked | Regressive encephalopathy | 14 months /8 months | F | Pathogenic | General tonic | Profound ID |
| P 18 | DMD (NM_004006.2) | c.C5287T (p.Arg1763Ter) | X-linked | Muscle dystrophy | 3 years /- | M | Pathogenic | No seizure | Motor delay |
| P 19 | SMARCA4 (NM_001128849) | c.3595G > A (p.Val1199Met) | AD, de novo | Seizures, encephalopathy | 3 months /2 months | M | Novel | General tonic | Severe global DD |
| P 20 | GRIN1 (NM_007327.3) | c.C2414T (p.Pro805Leu) | AD, de novo | DEE | 4 months /2 months | M | Pathogenic | Infantile spasms | Global DD |
| P 21 | SZT2 (NM_015284) | c.1496 + 2T > C/c.9055T > C (p.Arg3019Ter) | AR | DEE | 3 years /6 months | M | Novel/Novel | Focal motor | Severe hypotonia, gastrostomy |
| P 22 | SUOX (NM_001032386.2) | c.650G > A (p.Arg217Gln)/ c.258dupT (p.Lys87Ter) | AR | Leigh-like, regression | 8 months /4 months | M | Pathogenic /novel | Apnea, cyanosis, opisthotonos | Severe DD, hypertonia |
| P 23 | SPTBN2 (NM_006946. 2) | c.5515C > A (p.Gln1839Lys) | AD | Ataxia, vertigo | 14 years /- | M | Novel | No seizure | Frequent ataxia |
| P 24 | LAMA2 NM_000426.3 | c.1583dupA (p.Ser529GlufsTer19) /c.6931A > T (p.Lys2311Ter) | AR | Hypotonia, seizures | 2 years /1.5 months | M | Novel/Novel | Focal | Severe global DD |
| Gene Name | Genotype (N) | Phenotype | NCBI ClinVar | Critical Functional | Global | East Asia | Taiwan Biobank. | CADD | Poly | SIFT | ACMG | ||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Domain | MAF | MAF | Predict | Phen2 | Score | ||||||||
| Patient 15 | SCN1A (NM_001165963) | c.362delC (p.Ala121fs) | Frameshift | DEE | Novel | 0 | 0 | 0 | D | D | D | AD, de novo, (PVS1, PM2, PP3) | |
| Patient 14 | UBE3A (NM_130838) | c.C219A (p.Thr73Ter) | Nonsense | Encephalopathy and regression | Novel | 0 | 0 | 0 | D | D | D | AD from mother (PVS1, PM2, PP3) | |
| Patient 16 | SCN1A NM_001165963) | c.3918 + 1 G > - | Splicing | DEE | Novel | 0 | 0 | 0 | D | D | D | AD, de novo (PVS1, PM2, PP3) | |
| Patient 19 | SMARCA4 (NM_001128849) | c.3595G > A (p.Val1199Met) | Missense | Seizure, severe global delay | Novel | Helicase C-terminal’ | 0 | 0 | 0 | D | D | D | AD, de novo (PS2, PM1, PM2, PP3) |
| Patient 21 | SZT2 (NM_015284) | c.1496 + 2T > C | Splicing | DEE | Novel | 0 | 0 | 0 | D | D | D | AR (PVS1, PM2, PP3) | |
| Patient 21 | SZT2 (NM_015284) | c.9055T > C (p.Arg3019Ter) | Nonsense | DEE | Novel | 0 | 0 | 0 | D | D | D | AR (PVS1, PM2, PP3) | |
| Patient 22 | SUOX (NM_001032386.2) | c.258dupT (p.Lys87Ter) | Indel | Leigh-like, regression | Novel | 0 | 0 | 0 | D | D | D | AR (PVS1, PM2, PP3) | |
| Patient 23 | SPTBN2 (NM_006946. 2) | c.5515C > A (p.Gln1839Lys) | Missense | Spinocerebellar ataxia 5 | Novel | Ankyrin binding domain | 0 | 0 | 0 | D | D | T | AD from mother (PM1 + PM2 + PP1 + PP3 + PP4) |
| Patient 24 | LAMA2 NM_000426.3 | c.1583dupA(p.Ser529GlufsTer19) | Indel | CMD, seizures | Novel | 0 | 0 | 0 | D | D | D | AR (PVS1, PM2, PP3) | |
| Patient 24 | LAMA2 NM_000426.3 | /c.6931A > T (p.Lys2311Ter) | Nonsense | CMD, seizures | Novel | 0 | 0 | 0 | D | D | D | AR (PVS1, PM2, PP3) |
| Reference | The Study | [18] | [19] | [20] | [21] | [22] | [23] |
|---|---|---|---|---|---|---|---|
| Method | WES | WES | WES | 110-gene panel | 47-gene panel | WGS | WES |
| Cases (N) | 54 | 360 | 100 | 339 | 17 | 14 | 78 |
| Ethnicity or region | Taiwan | Canada | Caucasian (2 patients had African ethnicity), Netherlands | United States | Argentinean | United States | United States |
| Age | 43.7 ± 62.0 months 41 (76%) seizure onset <2 years | Seizure onset was 18 months (range 0.03–60 months) | 24.1 ± 16.2 years (ranged 2.8–67.6 years) | Age ranged 2.5 months to 74 years | Average age 4 months (range: 0–10 months) | Seizure onset before first month | 8.6 ± 5.8 years (ranged 1.6–26.3 years) |
| Inclusion criteria | 45 epilepsy (35 with AEDs ≥2; 10 with 1 AED and regression) + 9 nonepileptic with regressive symptoms | Seizure onset at ≤5 years | Unexplained epilepsy and borderline intelligence disability | Epileptic patients | EE with age of onset under 12 months | EIEE | Neurodevelopmental disabilities |
| Male/Female | 30/24 | 154/206 | 55/45 | About equal | 8/9 | 5/9 | 41/37 |
| Yield | 44% | 33% | 25% | 18% | ∼50% | 100% | 41% |
| Male/Female in identified genetic group | 17 (57%)/ 7 (29%) | 23/154 (15%)/ 36/206 (19%); In 53 epilepsy diagnosis < 6 months, 12/27 (44.4%) in males and 9/26 (35%) in females | 12 (22%)/ 13 (29%) | NA | 4 (50%)/ 4 (44%) | 5 (100%)/ 9 (100%) | NA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, S.-Y.; Yang, J.-J.; Li, S.-Y.; Lee, I.-C. A Wide Spectrum of Genetic Disorders Causing Severe Childhood Epilepsy in Taiwan: A Case Series of Ultrarare Genetic Cause and Novel Mutation Analysis in a Pilot Study. J. Pers. Med. 2020, 10, 281. https://doi.org/10.3390/jpm10040281
Hong S-Y, Yang J-J, Li S-Y, Lee I-C. A Wide Spectrum of Genetic Disorders Causing Severe Childhood Epilepsy in Taiwan: A Case Series of Ultrarare Genetic Cause and Novel Mutation Analysis in a Pilot Study. Journal of Personalized Medicine. 2020; 10(4):281. https://doi.org/10.3390/jpm10040281
Chicago/Turabian StyleHong, Syuan-Yu, Jiann-Jou Yang, Shuan-Yow Li, and Inn-Chi Lee. 2020. "A Wide Spectrum of Genetic Disorders Causing Severe Childhood Epilepsy in Taiwan: A Case Series of Ultrarare Genetic Cause and Novel Mutation Analysis in a Pilot Study" Journal of Personalized Medicine 10, no. 4: 281. https://doi.org/10.3390/jpm10040281
APA StyleHong, S. -Y., Yang, J. -J., Li, S. -Y., & Lee, I. -C. (2020). A Wide Spectrum of Genetic Disorders Causing Severe Childhood Epilepsy in Taiwan: A Case Series of Ultrarare Genetic Cause and Novel Mutation Analysis in a Pilot Study. Journal of Personalized Medicine, 10(4), 281. https://doi.org/10.3390/jpm10040281