Combination of Genome-Wide Polymorphisms and Copy Number Variations of Pharmacogenes in Koreans


Abstract
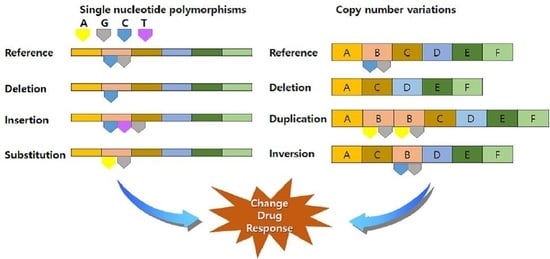
:
1. Introduction
2. Materials and Methods
2.1. Study Subjects
2.2. Pharmacogenes
2.3. Data Collection and Preprocessing
2.4. CNV Calling
2.5. Data Analysis
3. Results
3.1. Characteristics of the Study Population
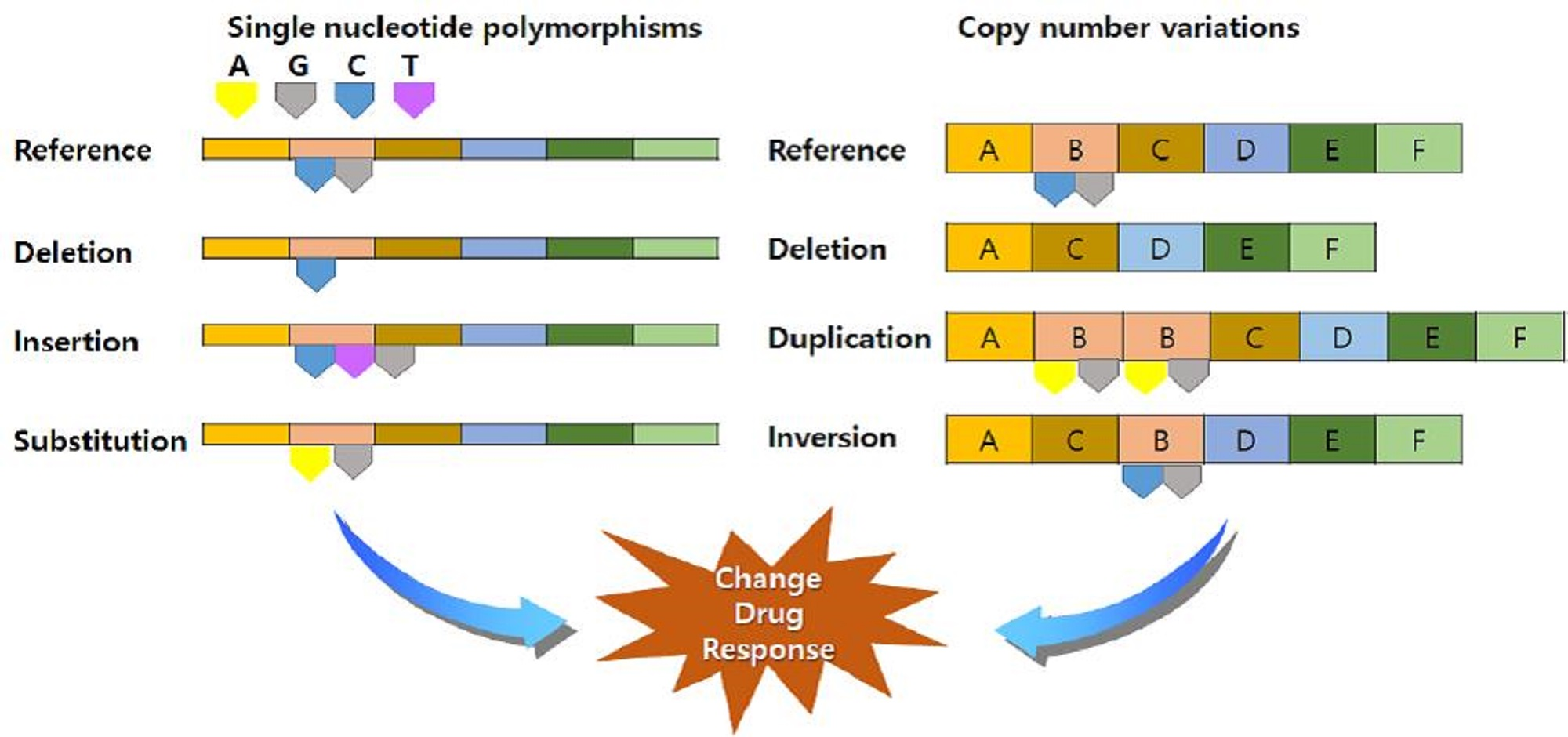
3.2. Genotype Variants
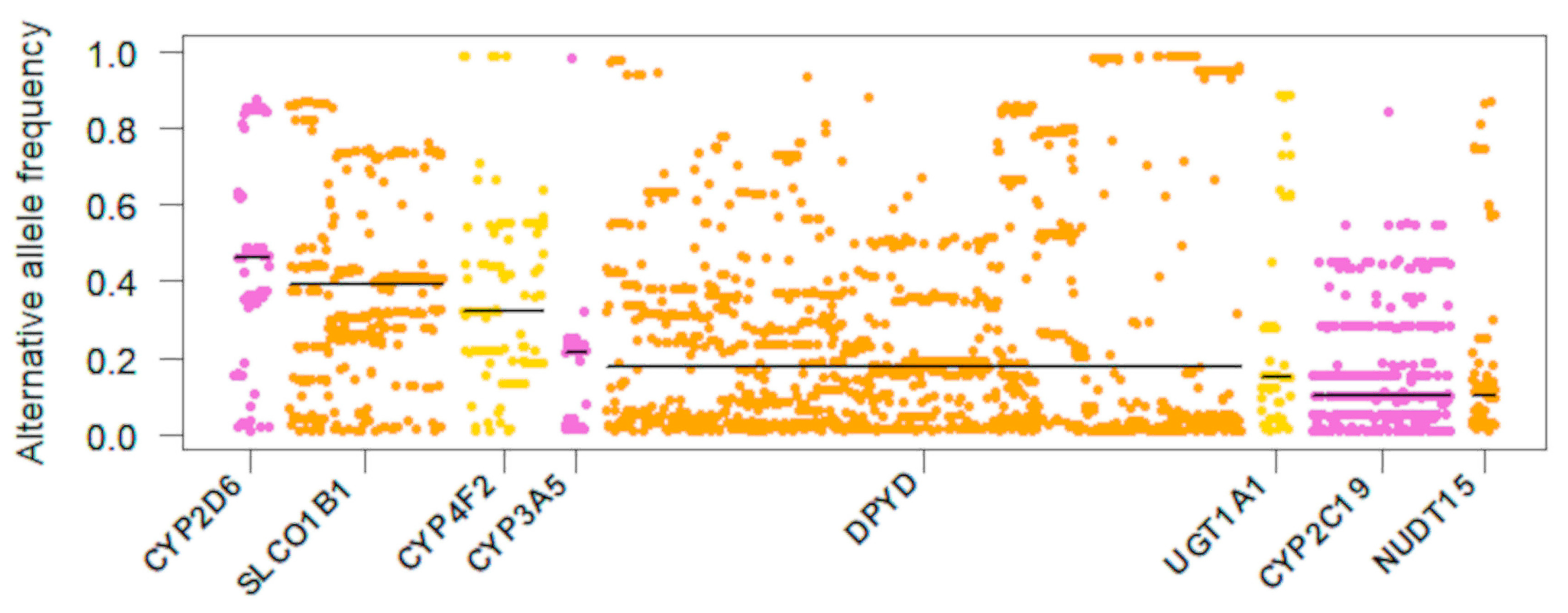
3.3. Haplotype Analysis
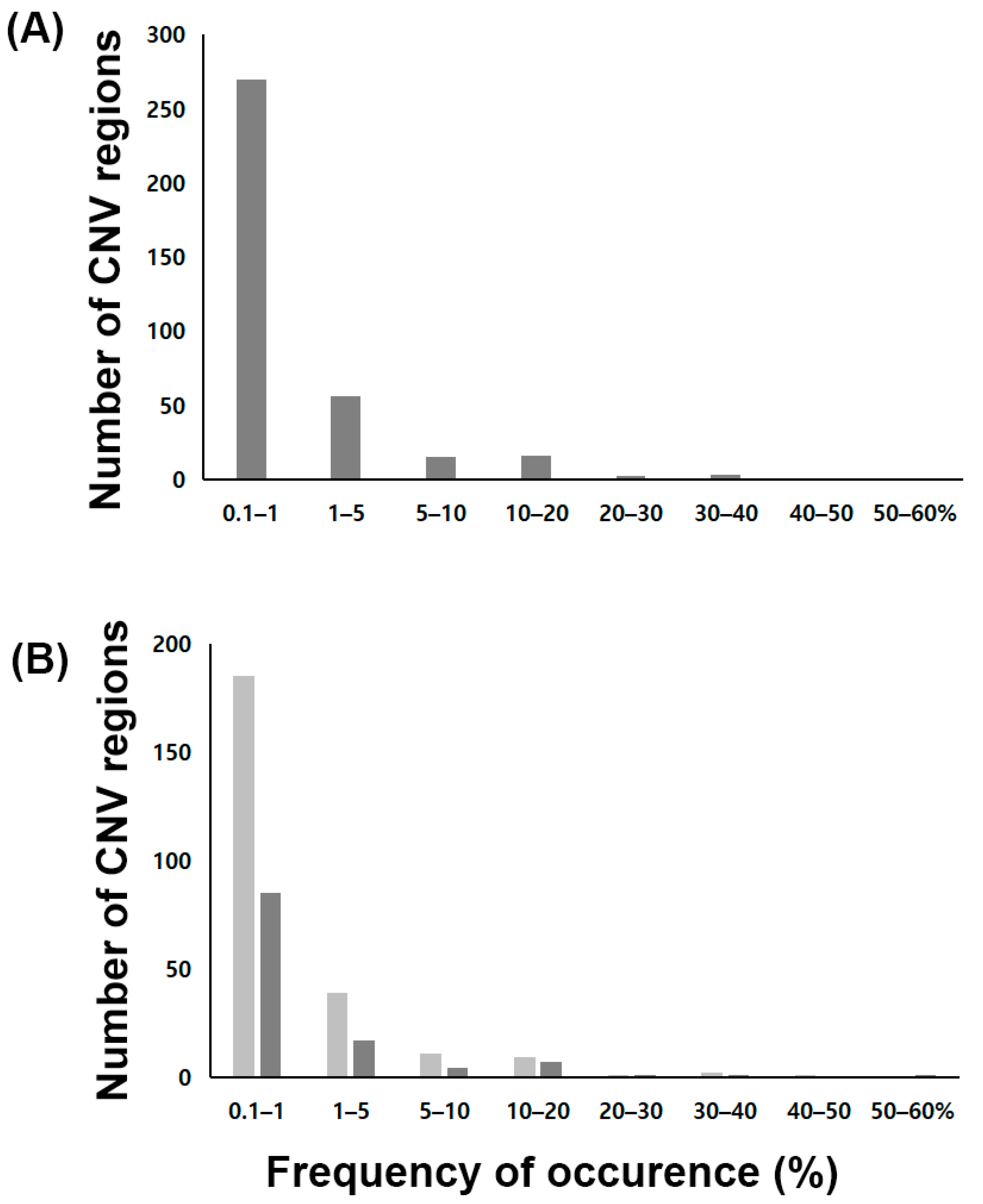
3.4. Copy Number Variation Profiling
3.5. Combination of Genotype Variants and CNVs
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Liu, R.; Yan, C.; Liu, L.; Tong, Z.; Jiang, W.; Yao, M.; Fang, W.; Chen, Z. Advantage of next-generation sequencing in dynamic monitoring of circulating tumor DNA over droplet digital PCR in cetuximab treated colorectal cancer patients. Transl. Oncol. 2019, 12, 426–431. [Google Scholar] [CrossRef] [PubMed]
- Galvan-Femenia, I.; Obon-Santacana, M.; Pineyro, D.; Guindo-Martinez, M.; Duran, X.; Carreras, A.; Pluvinet, R.; Velasco, J.; Ramos, L.; Ausso, S.; et al. Multitrait genome association analysis identifies new susceptibility genes for human anthropometric variation in the GCAT cohort. J. Med. Genet. 2018, 55, 765–778. [Google Scholar] [CrossRef] [PubMed]
- Tachmazidou, I.; Suveges, D.; Min, J.L.; Ritchie, G.R.S.; Steinberg, J.; Walter, K.; Iotchkova, V.; Schwartzentruber, J.; Huang, J.; Memari, Y.; et al. Whole-genome sequencing coupled to imputation discovers genetic signals for anthropometric traits. Am. J. Hum. Genet. 2017, 100, 865–884. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hellwege, J.N.; Velez Edwards, D.R.; Giri, A.; Qiu, C.; Park, J.; Torstenson, E.S.; Keaton, J.M.; Wilson, O.D.; Robinson-Cohen, C.; Chung, C.P.; et al. Mapping eGFR loci to the renal transcriptome and phenome in the VA Million Veteran Program. Nat. Commun. 2019, 10, 3842. [Google Scholar] [CrossRef] [Green Version]
- Din, L.; Sheikh, M.; Kosaraju, N.; Smedby, K.E.; Bernatsky, S.; Berndt, S.I.; Skibola, C.F.; Nieters, A.; Wang, S.; McKay, J.D.; et al. Genetic overlap between autoimmune diseases and non-Hodgkin lymphoma subtypes. Genet. Epidemiol. 2019, 43, 844–863. [Google Scholar] [CrossRef]
- Namjou, B.; Lingren, T.; Huang, Y.; Parameswaran, S.; Cobb, B.L.; Stanaway, I.B.; Connolly, J.J.; Mentch, F.D.; Benoit, B.; Niu, X.; et al. GWAS and enrichment analyses of non-alcoholic fatty liver disease identify new trait-associated genes and pathways across eMERGE Network. BMC Med. 2019, 17, 135. [Google Scholar] [CrossRef] [PubMed]
- Popejoy, A.B. Diversity in precision medicine and pharmacogenetics: Methodological and conceptual considerations for broadening participation. Pharmgenom. Pers. Med. 2019, 12, 257–271. [Google Scholar] [CrossRef] [Green Version]
- Massey, J.; Plant, D.; Hyrich, K.; Morgan, A.W.; Wilson, A.G.; Spiliopoulou, A.; Colombo, M.; McKeigue, P.; Isaacs, J.; Cordell, H.; et al. Genome-wide association study of response to tumour necrosis factor inhibitor therapy in rheumatoid arthritis. Pharmacogenom. J. 2018, 18, 657–664. [Google Scholar] [CrossRef] [PubMed]
- Fabbri, C.; Tansey, K.E.; Perlis, R.H.; Hauser, J.; Henigsberg, N.; Maier, W.; Mors, O.; Placentino, A.; Rietschel, M.; Souery, D.; et al. New insights into the pharmacogenomics of antidepressant response from the GENDEP and STAR*D studies: Rare variant analysis and high-density imputation. Pharmacogenom. J. 2018, 18, 413–421. [Google Scholar] [CrossRef] [Green Version]
- Yu, H.; Yan, H.; Wang, L.; Li, J.; Tan, L.; Deng, W.; Chen, Q.; Yang, G.; Zhang, F.; Lu, T.; et al. Five novel loci associated with antipsychotic treatment response in patients with schizophrenia: A genome-wide association study. Lancet Psychiatry 2018, 5, 327–338. [Google Scholar] [CrossRef]
- Walker, G.J.; Harrison, J.W.; Heap, G.A.; Voskuil, M.D.; Andersen, V.; Anderson, C.A.; Ananthakrishnan, A.N.; Barrett, J.C.; Beaugerie, L.; Bewshea, C.M.; et al. Association of genetic variants in NUDT15 with thiopurine-induced myelosuppression in patients with inflammatory bowel disease. JAMA 2019, 321, 773–785. [Google Scholar] [CrossRef] [Green Version]
- Carr, D.F.; Francis, B.; Jorgensen, A.L.; Zhang, E.; Chinoy, H.; Heckbert, S.R.; Bis, J.C.; Brody, J.A.; Floyd, J.S.; Psaty, B.M.; et al. Genomewide association study of statin-induced myopathy in patients recruited using the UK clinical practice research datalink. Clin. Pharmacol. Ther. 2019, 106, 1353–1361. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nicoletti, P.; Barrett, S.; McEvoy, L.; Daly, A.K.; Aithal, G.; Lucena, M.I.; Andrade, R.J.; Wadelius, M.; Hallberg, P.; Stephens, C.; et al. Shared genetic risk factors across carbamazepine-induced hypersensitivity reactions. Clin. Pharmacol. Ther. 2019, 106, 1028–1036. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, S.; Zhou, Z.; Zhou, J.; Chen, S.Q. Pharmacogenomics of drug metabolizing enzymes and transporters: Relevance to precision medicine. Genom. Proteom. Bioinform. 2016, 14, 298–313. [Google Scholar] [CrossRef] [Green Version]
- Zarrei, M.; MacDonald, J.R.; Merico, D.; Scherer, S.W. A copy number variation map of the human genome. Nat. Rev. Genet. 2015, 16, 172–183. [Google Scholar] [CrossRef]
- Pang, A.W.; MacDonald, J.R.; Pinto, D.; Wei, J.; Rafiq, M.A.; Conrad, D.F.; Park, H.; Hurles, M.E.; Lee, C.; Venter, J.C.; et al. Towards a comprehensive structural variation map of an individual human genome. Genome. Biol. 2010, 11, R52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sudmant, P.H.; Mallick, S.; Nelson, B.J.; Hormozdiari, F.; Krumm, N.; Huddleston, J.; Coe, B.P.; Baker, C.; Nordenfelt, S.; Bamshad, M.; et al. Global diversity, population stratification, and selection of human copy-number variation. Science 2015, 349, aab3761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arcella, A.; Limanaqi, F.; Ferese, R.; Biagioni, F.; Oliva, M.A.; Storto, M.; Fanelli, M.; Gambardella, S.; Fornai, F. Dissecting molecular features of gliomas: Genetic loci and validated biomarkers. Int. J. Mol. Sci. 2020, 21, 685. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gentile, G.; La Cognata, V.; Cavallaro, S. The contribution of CNVs to the most common aging-related neurodegenerative diseases. Aging Clin. Exp. Res. 2020. [Google Scholar] [CrossRef] [PubMed]
- Sullivan, P.F.; Owen, M.J. Increasing the clinical psychiatric knowledge base about pathogenic copy number variation. Am. J. Psychiatry 2020, 177, 204–209. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lauer, S.; Gresham, D. An evolving view of copy number variants. Curr. Genet. 2019, 65, 1287–1295. [Google Scholar] [CrossRef]
- Kim, J.; Lee, S.Y.; Lee, K.A. Copy number variation and gene rearrangements in CYP2D6 genotyping using multiplex ligation-dependent probe amplification in Koreans. Pharmacogenomics 2012, 13, 963–973. [Google Scholar] [CrossRef]
- Qiao, W.; Martis, S.; Mendiratta, G.; Shi, L.; Botton, M.R.; Yang, Y.; Gaedigk, A.; Vijzelaar, R.; Edelmann, L.; Kornreich, R.; et al. Integrated CYP2D6 interrogation for multiethnic copy number and tandem allele detection. Pharmacogenomics 2019, 20, 9–20. [Google Scholar] [CrossRef] [PubMed]
- Santos, M.; Niemi, M.; Hiratsuka, M.; Kumondai, M.; Ingelman-Sundberg, M.; Lauschke, V.M.; Rodriguez-Antona, C. Novel copy-number variations in pharmacogenes contribute to interindividual differences in drug pharmacokinetics. Genet. Med. 2018, 20, 622–629. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Han, J.; Shon, J.; Hwang, J.Y.; Park, Y.J. Effects of Coffee Intake on Dyslipidemia Risk According to Genetic Variants in the ADORA Gene Family among Korean Adults. Nutrients 2020, 12, 493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kwon, Y.J.; Hong, K.W.; Park, B.J.; Jung, D.H. Serotonin receptor 3B polymorphisms are associated with type 2 diabetes: The Korean Genome and Epidemiology Study. Diabetes Res. Clin. Pract. 2019, 153, 76–85. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.J.; Kim, J.; Kwock, C.K. Association of Genetic Variation in the Epithelial Sodium Channel Gene with Urinary Sodium Excretion and Blood Pressure. Nutrients 2018, 10, 612. [Google Scholar] [CrossRef] [Green Version]
- Kim, Y.; Han, B.G. Cohort Profile: The Korean Genome and Epidemiology Study (KoGES) Consortium. Int. J. Epidemiol. 2017, 46, 1350. [Google Scholar] [CrossRef]
- Thorn, C.F.; Klein, T.E.; Altman, R.B. PharmGKB: The Pharmacogenomics Knowledge Base. Methods Mol. Biol. 2013, 1015, 311–320. [Google Scholar]
- Relling, M.V.; Klein, T.E. CPIC: Clinical Pharmacogenetics Implementation Consortium of the Pharmacogenomics Research Network. Clin. Pharmacol. Ther. 2011, 89, 464–467. [Google Scholar] [CrossRef] [PubMed]
- Tutton, R. Pharmacogenomic biomarkers in drug labels: What do they tell us? Pharmacogenomics 2014, 15, 297–304. [Google Scholar] [CrossRef] [PubMed]
- Moon, S.; Kim, Y.J.; Han, S.; Hwang, M.Y.; Shin, D.M.; Park, M.Y.; Lu, Y.; Yoon, K.; Jang, H.M.; Kim, Y.K.; et al. The Korea Biobank array: Design and identification of coding variants associated with blood biochemical traits. Sci. Rep. 2019, 9, 1382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delaneau, O.; Marchini, J.; Zagury, J.F. A linear complexity phasing method for thousands of genomes. Nat. Methods 2011, 9, 179–181. [Google Scholar] [CrossRef]
- Marchini, J.; Howie, B.; Myers, S.; McVean, G.; Donnelly, P. A new multipoint method for genome-wide association studies by imputation of genotypes. Nat. Genet. 2007, 39, 906–913. [Google Scholar] [CrossRef]
- Durinck, S.; Moreau, Y.; Kasprzyk, A.; Davis, S.; De Moor, B.; Brazma, A.; Huber, W. BioMart and Bioconductor: A powerful link between biological databases and microarray data analysis. Bioinformatics 2005, 21, 3439–3440. [Google Scholar] [CrossRef] [Green Version]
- Moon, S.; Kim, Y.J.; Hong, C.B.; Kim, D.J.; Lee, J.Y.; Kim, B.J. Data-driven approach to detect common copy-number variations and frequency profiles in a population-based Korean cohort. Eur. J. Hum. Genet. 2011, 19, 1167–1172. [Google Scholar] [CrossRef]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic. Acids. Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Pique-Regi, R.; Caceres, A.; Gonzalez, J.R. R-Gada: A fast and flexible pipeline for copy number analysis in association studies. BMC Bioinform. 2010, 11, 380. [Google Scholar] [CrossRef] [Green Version]
- Moon, S.; Kim, Y.J.; Kim, Y.K.; Kim, D.J.; Lee, J.Y.; Go, M.J.; Shin, Y.A.; Hong, C.B.; Kim, B.J. Genome-wide survey of copy number variants associated with blood pressure and body mass index in a Korean population. Genom. Inform. 2011, 9, 152–160. [Google Scholar] [CrossRef]
- Bambury, R.M.; Bhatt, A.S.; Riester, M.; Pedamallu, C.S.; Duke, F.; Bellmunt, J.; Stack, E.C.; Werner, L.; Park, R.; Iyer, G.; et al. DNA copy number analysis of metastatic urothelial carcinoma with comparison to primary tumors. BMC Cancer 2015, 15, 242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lindgren, D.; Sjodahl, G.; Lauss, M.; Staaf, J.; Chebil, G.; Lovgren, K.; Gudjonsson, S.; Liedberg, F.; Patschan, O.; Mansson, W.; et al. Integrated genomic and gene expression profiling identifies two major genomic circuits in urothelial carcinoma. PLoS ONE 2012, 7, e38863. [Google Scholar] [CrossRef]
- Chang, C.C.; Chow, C.C.; Tellier, L.C.; Vattikuti, S.; Purcell, S.M.; Lee, J.J. Second-generation PLINK: Rising to the challenge of larger and richer datasets. Gigascience 2015, 4, 7. [Google Scholar] [CrossRef]
- Barrett, J.C.; Fry, B.; Maller, J.; Daly, M.J. Haploview: Analysis and visualization of LD and haplotype maps. Bioinformatics 2005, 21, 263–265. [Google Scholar] [CrossRef] [Green Version]
- Owen, R.P.; Gong, L.; Sagreiya, H.; Klein, T.E.; Altman, R.B. VKORC1 pharmacogenomics summary. Pharm. Genom. 2010, 20, 642–644. [Google Scholar] [CrossRef] [Green Version]
- Sakuyama, K.; Sasaki, T.; Ujiie, S.; Obata, K.; Mizugaki, M.; Ishikawa, M.; Hiratsuka, M. Functional characterization of 17 CYP2D6 allelic variants (CYP2D6.2, 10, 14A-B, 18, 27, 36, 39, 47-51, 53-55, and 57). Drug Metab. Dispos. 2008, 36, 2460–2467. [Google Scholar] [CrossRef]
- Kim, K.A.; Song, W.K.; Kim, K.R.; Park, J.Y. Assessment of CYP2C19 genetic polymorphisms in a Korean population using a simultaneous multiplex pyrosequencing method to simultaneously detect the CYP2C19*2, CYP2C19*3, and CYP2C19*17 alleles. J. Clin. Pharm. Ther. 2010, 35, 697–703. [Google Scholar] [CrossRef]
- Scott, S.A.; Sangkuhl, K.; Stein, C.M.; Hulot, J.S.; Mega, J.L.; Roden, D.M.; Klein, T.E.; Sabatine, M.S.; Johnson, J.A.; Shuldiner, A.R.; et al. Clinical Pharmacogenetics Implementation Consortium guidelines for CYP2C19 genotype and clopidogrel therapy: 2013 update. Clin. Pharmacol. Ther. 2013, 94, 317–323. [Google Scholar] [CrossRef]
- Carpenter, D.; Ringrose, C.; Leo, V.; Morris, A.; Robinson, R.L.; Halsall, P.J.; Hopkins, P.M.; Shaw, M.A. The role of CACNA1S in predisposition to malignant hyperthermia. BMC Med. Genet. 2009, 10, 104. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.H.; Choi, J.H.; Namkung, W.; Hanrahan, J.W.; Chang, J.; Song, S.Y.; Park, S.W.; Kim, D.S.; Yoon, J.H.; Suh, Y.; et al. A haplotype-based molecular analysis of CFTR mutations associated with respiratory and pancreatic diseases. Hum. Mol. Genet. 2003, 12, 2321–2332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guan, S.; Huang, M.; Li, X.; Chen, X.; Chan, E.; Zhou, S.F. Intra- and inter-ethnic differences in the allele frequencies of cytochrome P450 2B6 gene in Chinese. Pharm. Res. 2006, 23, 1983–1990. [Google Scholar] [CrossRef]
- Gadel, S.; Crafford, A.; Regina, K.; Kharasch, E.D. Methadone N-demethylation by the common CYP2B6 allelic variant CYP2B6.6. Drug Metab. Dispos. 2013, 41, 709–713. [Google Scholar] [CrossRef] [Green Version]
- Paganotti, G.M.; Russo, G.; Sobze, M.S.; Mayaka, G.B.; Muthoga, C.W.; Tawe, L.; Martinelli, A.; Romano, R.; Vullo, V. CYP2B6 poor metaboliser alleles involved in efavirenz and nevirapine metabolism: CYP2B6*9 and CYP2B6*18 distribution in HIV-exposed subjects from Dschang, Western Cameroon. Infect. Genet. Evol. 2015, 35, 122–126. [Google Scholar] [CrossRef]
- Ishmukhametova, A.; Chen, J.M.; Bernard, R.; de Massy, B.; Baudat, F.; Boyer, A.; Mechin, D.; Thorel, D.; Chabrol, B.; Vincent, M.C.; et al. Dissecting the structure and mechanism of a complex duplication-triplication rearrangement in the DMD gene. Hum. Mutat. 2013, 34, 1080–1084. [Google Scholar] [CrossRef]
- Iskandar, K.; Dwianingsih, E.K.; Pratiwi, L.; Kalim, A.S.; Mardhiah, H.; Putranti, A.H.; Nurputra, D.K.; Triono, A.; Herini, E.S.; Malueka, R.G.; et al. The analysis of DMD gene deletions by multiplex PCR in Indonesian DMD/BMD patients: The era of personalized medicine. BMC Res. Notes 2019, 12, 704. [Google Scholar] [CrossRef] [Green Version]
- Cho, A.; Seong, M.W.; Lim, B.C.; Lee, H.J.; Byeon, J.H.; Kim, S.S.; Kim, S.Y.; Choi, S.A.; Wong, A.L.; Lee, J.; et al. Consecutive Analysis of Mutation Spectrum in the Dystrophin Gene of 507 Korean Boys with Duchenne/Becker Muscular Dystrophy in a Single Center. Muscle Nerve 2017, 55, 727–734. [Google Scholar] [CrossRef]
- Spire-Vayron de la Moureyre, C.; Debuysere, H.; Fazio, F.; Sergent, E.; Bernard, C.; Sabbagh, N.; Marez, D.; Lo Guidice, J.M.; D’Halluin, J.C. Characterization of a variable number tandem repeat region in the thiopurine S-methyltransferase gene promoter. Pharmacogenetics 1999, 9, 189–198. [Google Scholar]
- Urbancic, D.; Smid, A.; Stocco, G.; Decorti, G.; Mlinaric-Rascan, I.; Karas Kuzelicki, N. Novel motif of variable number of tandem repeats in TPMT promoter region and evolutionary association of variable number of tandem repeats with TPMT*3 alleles. Pharmacogenomics 2018, 19, 1311–1322. [Google Scholar] [CrossRef]
- Green, D.J.; Duong, S.Q.; Burckart, G.J.; Sissung, T.; Price, D.K.; Figg, W.D., Jr.; Brooks, M.M.; Chinnock, R.; Canter, C.; Addonizio, L.; et al. Association Between Thiopurine S-Methyltransferase (TPMT) Genetic Variants and Infection in Pediatric Heart Transplant Recipients Treated With Azathioprine: A Multi-Institutional Analysis. J. Pediatr. Pharmacol. Ther. 2018, 23, 106–110. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.S.; Kim, W.Y.; Jang, Y.J.; Shin, J.G. Duplex pyrosequencing of the TPMT*3C and TPMT*6 alleles in Korean and Vietnamese populations. Clin. Chim. Acta 2008, 398, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Yang, S.K.; Hong, M.; Baek, J.; Choi, H.; Zhao, W.; Jung, Y.; Haritunians, T.; Ye, B.D.; Kim, K.J.; Park, S.H.; et al. A common missense variant in NUDT15 confers susceptibility to thiopurine-induced leukopenia. Nat. Genet. 2014, 46, 1017–1020. [Google Scholar] [CrossRef] [PubMed]
- Vijzelaar, R.; Botton, M.R.; Stolk, L.; Martis, S.; Desnick, R.J.; Scott, S.A. Multi-ethnic SULT1A1 copy number profiling with multiplex ligation-dependent probe amplification. Pharmacogenomics 2018, 19, 761–770. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guthrie, P.A.; Gaunt, T.R.; Abdollahi, M.R.; Rodriguez, S.; Lawlor, D.A.; Smith, G.D.; Day, I.N. Amplification ratio control system for copy number variation genotyping. Nucleic. Acids. Res. 2011, 39, e54. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haraksingh, R.R.; Snyder, M.P. Impacts of variation in the human genome on gene regulation. J. Mol. Biol. 2013, 425, 3970–3977. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Characteristics | SNV | CNV | Combination of SNV with CNV |
|---|---|---|---|
| Number of patients, n | 69,027 | 947 | 614 |
| Age, years | 54.08 ± 8.31 | 54.05 ± 9.08 | 52.82 ± 8.80 |
| Gender | |||
| male | 25,004 (36.22) | 474 (50.05) | 311 (50.65) |
| female | 44,023 (63.78) | 473 (49.95) | 303 (49.35) |
| Gene | Position | Gain Frequency (%) | Loss Frequency (%) |
|---|---|---|---|
| ABCB1 | 7: 87133179−87342639 | 0.11 | 15.31 |
| ALK | 2: 29415640−30144477 | 6.12 | 1.06 |
| ALOX5 | 10: 45869624−45941567 | 6.65 | 1.58 |
| BCR | 11: 23522552−23660224 | 0.11 | 19.01 |
| BRCA | 17: 41196312−41277500 | 2.22 | 2.64 |
| COMT | 19: 19929263−19957498 | 7.07 | 0.32 |
| CYP2A6 | 19: 41349443−41356352 | 1.27 | 1.48 |
| CYP4F2 | 19: 15988834−16008884 | 3.80 | 0.42 |
| DMD | X: 31137345−33229673 | 64.52 | 20.27 |
| EGFR | 7: 55086725−55275031 | 2.32 | 41.39 |
| ESR1 | 6: 152128814−152424408 | 0 | 1.48 |
| G6PD | X: 153759606−153775233 | 17.21 | 0.42 |
| HLA-B | 6: 31237743−31324989 | 0.42 | 36.54 |
| HLA-DRB1 | 6: 32489683−32557613 | 0.32 | 40.65 |
| KIT | 4: 55524095−55606881 | 21.12 | 2.22 |
| OTC | X: 38211736−38280703 | 57.76 | 0.42 |
| PDGFRA | 4: 55095264−55164412 | 0.11 | 21.44 |
| RYR1 | 19: 38924340−39078204 | 5.07 | 2.11 |
| SMN2 | 5: 70220768−70248842 | 2.11 | 0.11 |
| SULT1A1 | 16: 28616908−28620649 | 7.71 | 19.75 |
| TPMT | 6: 18128545−18155374 | 0 | 51.80 |
| Gene Allele | Subjects (N) | Frequency (%) |
|---|---|---|
| CYP4F2*1*1 | 258 | 42.02 |
| CYP4F2*1*2 | 1 | 0.16 |
| CYP4F2*1*3 | 150 | 24.43 |
| CYP4F2*3*3 | 22 | 3.58 |
| CYP4F2*2*3 | 114 | 18.57 |
| CYP4F2*1*2-*3*3 | 31 | 5.05 |
| CYP4F2*2*2-*3*3 | 13 | 2.12 |
| CYP4F2*1*1 gain | 9 | 1.47 |
| CYP4F2*1*3 gain | 6 | 0.98 |
| CYP4F2*2*3 gain | 6 | 0.98 |
| CYP4F2*3*3 gain | 1 | 0.16 |
| CYP4F2*1*1 loss | 2 | 0.33 |
| CYP4F2*2*3 loss | 1 | 0.16 |
| TPMT*1*1 | 287 | 46.74 |
| TPMT*1*3C | 8 | 1.30 |
| TPMT*1*1 loss | 308 | 50.16 |
| TPMT*1*3C loss | 11 | 1.79 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, N.; Oh, J.M.; Kim, I.-W. Combination of Genome-Wide Polymorphisms and Copy Number Variations of Pharmacogenes in Koreans. J. Pers. Med. 2021, 11, 33. https://doi.org/10.3390/jpm11010033
Han N, Oh JM, Kim I-W. Combination of Genome-Wide Polymorphisms and Copy Number Variations of Pharmacogenes in Koreans. Journal of Personalized Medicine. 2021; 11(1):33. https://doi.org/10.3390/jpm11010033
Chicago/Turabian StyleHan, Nayoung, Jung Mi Oh, and In-Wha Kim. 2021. "Combination of Genome-Wide Polymorphisms and Copy Number Variations of Pharmacogenes in Koreans" Journal of Personalized Medicine 11, no. 1: 33. https://doi.org/10.3390/jpm11010033
APA StyleHan, N., Oh, J. M., & Kim, I.-W. (2021). Combination of Genome-Wide Polymorphisms and Copy Number Variations of Pharmacogenes in Koreans. Journal of Personalized Medicine, 11(1), 33. https://doi.org/10.3390/jpm11010033