
Common Treatment, Common Variant: Evolutionary Prediction of Functional Pharmacogenomic Variants

 ,
,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Samples and Data
2.2. Enrichment Testing
2.3. Machine Learning Modeling
3. Results
3.1. Annotated PGx Variation Is Negatively Impacted by Ascertainment Bias
3.2. Pharmacogenes Are Enriched for Adaptive Signatures
3.3. In Silico Model Development
3.4. Comparison with Existing Methods
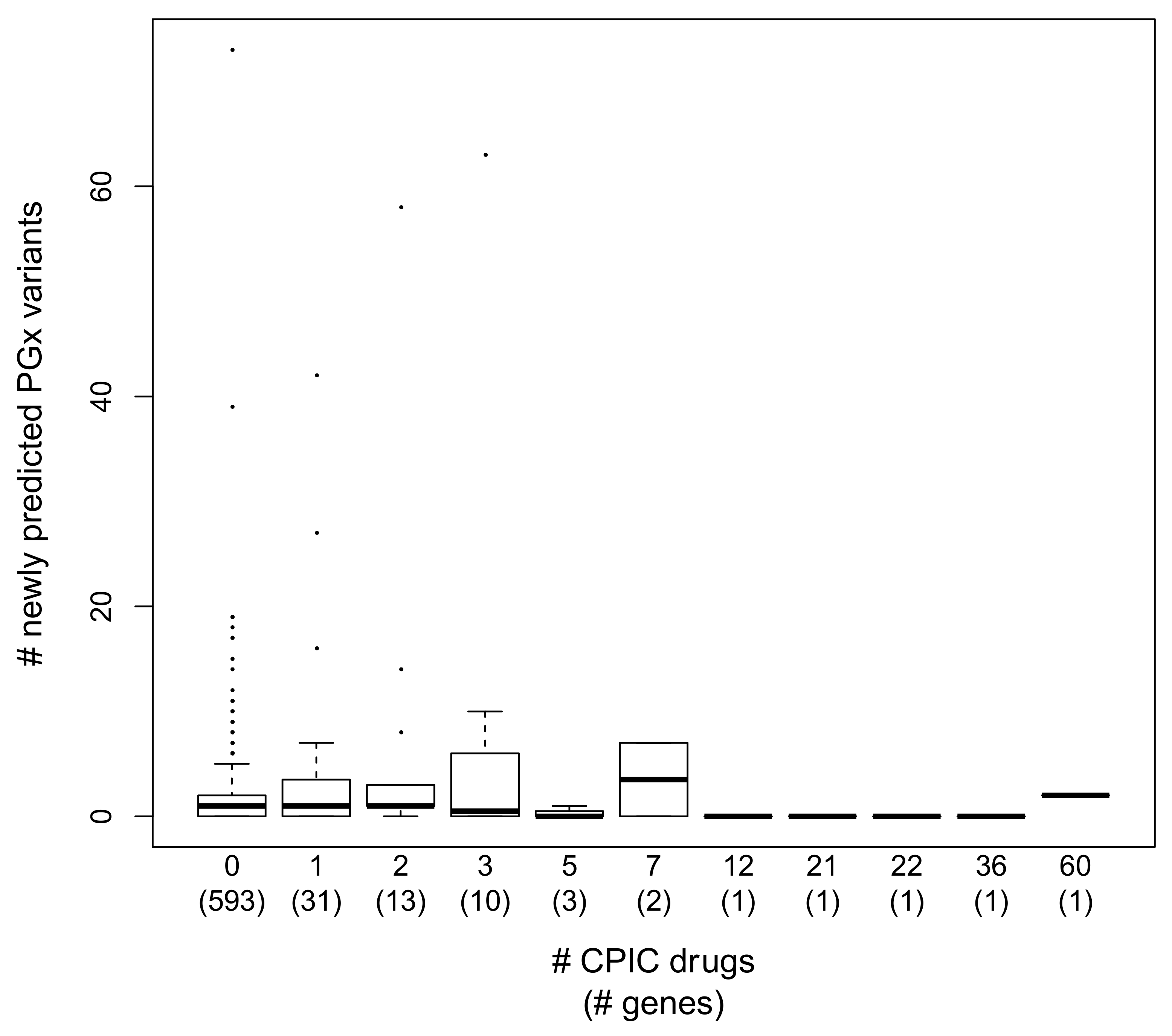
3.5. Annotation Trends in PGx Variant Prediction
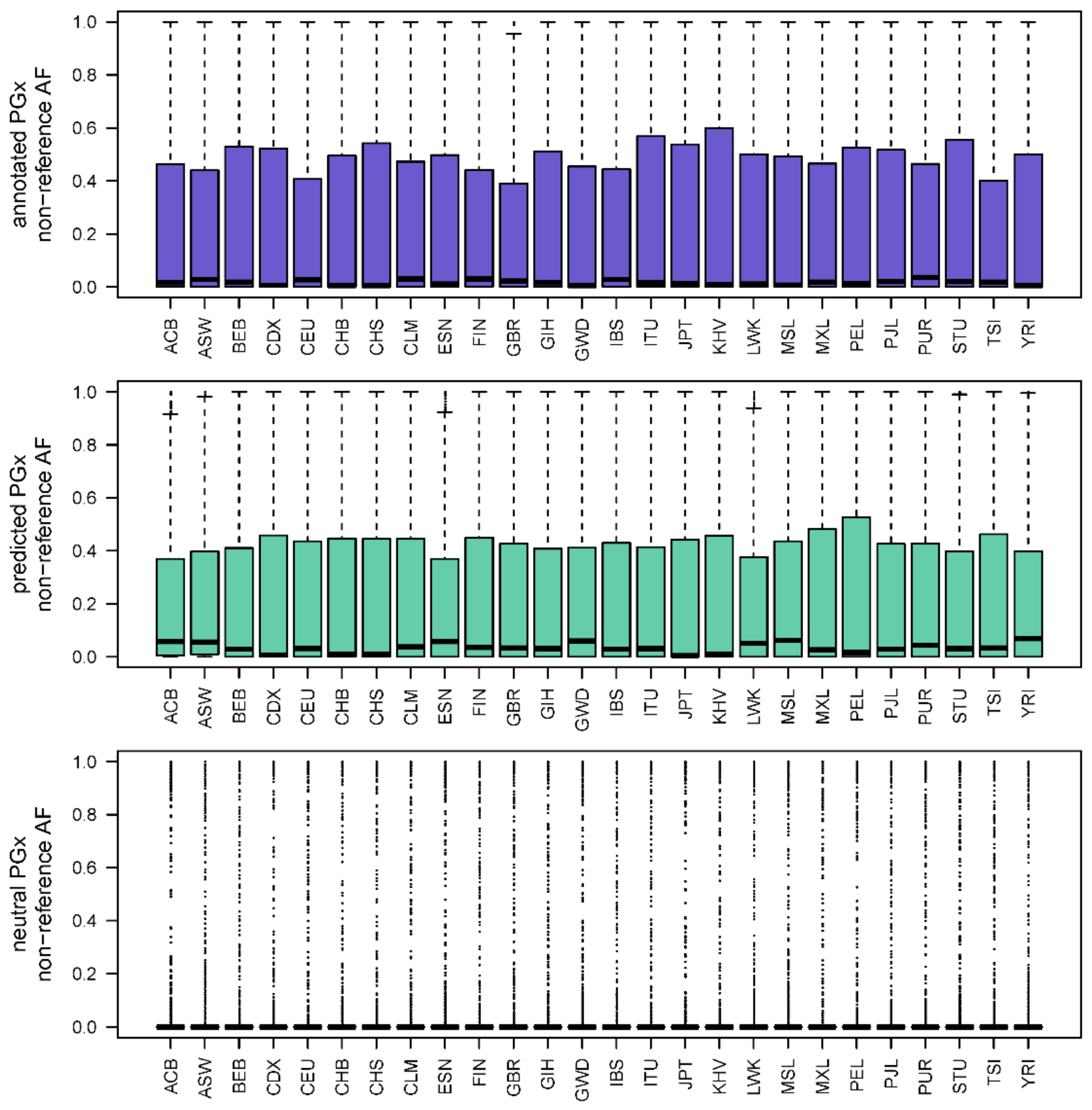
3.6. Allele Frequency Trends in PGx Variant Prediction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Crews, K.R.; Hicks, J.K.; Pui, C.H.; Relling, M.V.; Evans, W.E. Pharmacogenomics and individualized medicine: Translating science into practice. Clin. Pharmacol. Ther. 2012, 92, 467–475. [Google Scholar] [CrossRef] [Green Version]
- Relling, M.V.; Evans, W.E. Pharmacogenomics in the clinic. Nature 2015, 526, 343–350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Relling, M.V.; Krauss, R.M.; Roden, D.M.; Klein, T.E.; Fowler, D.M.; Terada, N.; Lin, L.; Riel-Mehan, M.; Do, T.P.; Kubo, M.; et al. New Pharmacogenomics Research Network: An Open Community Catalyzing Research and Translation in Precision Medicine. Clin. Pharmacol. Ther. 2017, 102, 897–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rasmussen-Torvik, L.J.; Stallings, S.C.; Gordon, A.S.; Almoguera, B.; Basford, M.A.; Bielinski, S.J.; Brautbar, A.; Brilliant, M.H.; Carrell, D.S.; Connolly, J.J.; et al. Design and anticipated outcomes of the eMERGE-PGx project: A multicenter pilot for preemptive pharmacogenomics in electronic health record systems. Clin. Pharmacol. Ther. 2014, 96, 482–489. [Google Scholar] [CrossRef] [PubMed]
- Gharani, N.; Keller, M.A.; Stack, C.B.; Hodges, L.M.; Schmidlen, T.J.; Lynch, D.E.; Gordon, E.S.; Christman, M.F. The Coriell personalized medicine collaborative pharmacogenomics appraisal, evidence scoring and interpretation system. Genome Med. 2013, 5, 93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunnenberger, H.M.; Crews, K.R.; Hoffman, J.M.; Caudle, K.E.; Broeckel, U.; Howard, S.C.; Hunkler, R.J.; Klein, T.E.; Evans, W.E.; Relling, M.V. Preemptive clinical pharmacogenetics implementation: Current programs in five US medical centers. Annu. Rev. Pharmacol. Toxicol. 2015, 55, 89–106. [Google Scholar] [CrossRef] [Green Version]
- Bank, P.C.D.; Swen, J.J.; Guchelaar, H.J. Implementation of Pharmacogenomics in Everyday Clinical Settings. Adv. Pharmacol. 2018, 83, 219–246. [Google Scholar] [CrossRef] [PubMed]
- Bentley, A.R.; Callier, S.; Rotimi, C.N. Diversity and inclusion in genomic research: Why the uneven progress? J. Community Genet. 2017, 8, 255–266. [Google Scholar] [CrossRef] [Green Version]
- Martin, A.R.; Gignoux, C.R.; Walters, R.K.; Wojcik, G.L.; Neale, B.M.; Gravel, S.; Daly, M.J.; Bustamante, C.D.; Kenny, E.E. Human Demographic History Impacts Genetic Risk Prediction across Diverse Populations. Am. J. Hum. Genet. 2017, 100, 635–649. [Google Scholar] [CrossRef] [Green Version]
- Popejoy, A.B.; Fullerton, S.M. Genomics is failing on diversity. Nature 2016, 538, 161–164. [Google Scholar] [CrossRef] [Green Version]
- Scheinfeldt, L.B.; Tishkoff, S.A. Recent human adaptation: Genomic approaches, interpretation and insights. Nat. Rev. Genet. 2013, 14, 692–702. [Google Scholar] [CrossRef] [Green Version]
- Scheinfeldt, L.B.; Soi, S.; Tishkoff, S.A. Colloquium paper: Working toward a synthesis of archaeological, linguistic, and genetic data for inferring African population history. Proc. Natl. Acad. Sci. USA 2010, 107 (Suppl. 2), 8931–8938. [Google Scholar] [CrossRef] [Green Version]
- Genomes Project, C.; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef] [Green Version]
- Biswas, S.; Scheinfeldt, L.B.; Akey, J.M. Genome-wide insights into the patterns and determinants of fine-scale population structure in humans. Am. J. Hum. Genet. 2009, 84, 641–650. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scheinfeldt, L.B.; Biswas, S.; Madeoy, J.; Connelly, C.F.; Schadt, E.E.; Akey, J.M. Population genomic analysis of ALMS1 in humans reveals a surprisingly complex evolutionary history. Mol. Biol. Evol. 2009, 26, 1357–1367. [Google Scholar] [CrossRef] [Green Version]
- Choudhury, A.; Aron, S.; Botigue, L.R.; Sengupta, D.; Botha, G.; Bensellak, T.; Wells, G.; Kumuthini, J.; Shriner, D.; Fakim, Y.J.; et al. High-depth African genomes inform human migration and health. Nature 2020, 586, 741–748. [Google Scholar] [CrossRef] [PubMed]
- Baker, J.L.; Shriner, D.; Bentley, A.R.; Rotimi, C.N. Pharmacogenomic implications of the evolutionary history of infectious diseases in Africa. Pharmacogenom. J. 2017, 17, 112–120. [Google Scholar] [CrossRef] [PubMed]
- Voora, D.; Koboldt, D.C.; King, C.R.; Lenzini, P.A.; Eby, C.S.; Porche-Sorbet, R.; Deych, E.; Crankshaw, M.; Milligan, P.E.; McLeod, H.L.; et al. A polymorphism in the VKORC1 regulator calumenin predicts higher warfarin dose requirements in African Americans. Clin. Pharmacol. Ther. 2010, 87, 445–451. [Google Scholar] [CrossRef] [Green Version]
- Shahabi, P.; Scheinfeldt, L.B.; Lynch, D.E.; Schmidlen, T.J.; Perreault, S.; Keller, M.A.; Kasper, R.; Wawak, L.; Jarvis, J.P.; Gerry, N.P.; et al. An expanded pharmacogenomics warfarin dosing table with utility in generalised dosing guidance. Thromb. Haemost. 2016, 116, 337–348. [Google Scholar] [CrossRef] [Green Version]
- Kaye, J.B.; Schultz, L.E.; Steiner, H.E.; Kittles, R.A.; Cavallari, L.H.; Karnes, J.H. Warfarin Pharmacogenomics in Diverse Populations. Pharmacotherapy 2017, 37, 1150–1163. [Google Scholar] [CrossRef]
- Kimmel, S.E.; French, B.; Kasner, S.E.; Johnson, J.A.; Anderson, J.L.; Gage, B.F.; Rosenberg, Y.D.; Eby, C.S.; Madigan, R.A.; McBane, R.B.; et al. A pharmacogenetic versus a clinical algorithm for warfarin dosing. N. Engl. J. Med. 2013, 369, 2283–2293. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.C.; Henikoff, S. Predicting deleterious amino acid substitutions. Genome Res. 2001, 11, 863–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [Green Version]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2019, 47, D886–D894. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Tamura, K.; Sanderford, M.; Gray, V.E.; Kumar, S. A Molecular Evolutionary Reference for the Human Variome. Mol. Biol. Evol. 2016, 33, 245–254. [Google Scholar] [CrossRef] [Green Version]
- Li, B.; Seligman, C.; Thusberg, J.; Miller, J.L.; Auer, J.; Whirl-Carrillo, M.; Capriotti, E.; Klein, T.E.; Mooney, S.D. In silico comparative characterization of pharmacogenomic missense variants. BMC Genom. 2014, 15 (Suppl. 4), S4. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Mkrtchian, S.; Kumondai, M.; Hiratsuka, M.; Lauschke, V.M. An optimized prediction framework to assess the functional impact of pharmacogenetic variants. Pharmacogenom. J. 2019, 19, 115–126. [Google Scholar] [CrossRef]
- Scheinfeldt, L.B.; Soi, S.; Lambert, C.; Ko, W.Y.; Coulibaly, A.; Ranciaro, A.; Thompson, S.; Hirbo, J.; Beggs, W.; Ibrahim, M.; et al. Genomic evidence for shared common ancestry of East African hunting-gathering populations and insights into local adaptation. Proc. Natl. Acad. Sci. USA 2019, 116, 4166–4175. [Google Scholar] [CrossRef] [Green Version]
- Patel, R.; Scheinfeldt, L.B.; Sanderford, M.D.; Lanham, T.R.; Tamura, K.; Platt, A.; Glicksberg, B.S.; Xu, K.; Dudley, J.T.; Kumar, S. Adaptive Landscape of Protein Variation in Human Exomes. Mol. Biol. Evol. 2018, 35, 2015–2025. [Google Scholar] [CrossRef] [Green Version]
- Fumagalli, M.; Sironi, M.; Pozzoli, U.; Ferrer-Admetlla, A.; Pattini, L.; Nielsen, R. Signatures of environmental genetic adaptation pinpoint pathogens as the main selective pressure through human evolution. PLoS Genet. 2011, 7, e1002355. [Google Scholar] [CrossRef]
- Grossman, S.R.; Andersen, K.G.; Shlyakhter, I.; Tabrizi, S.; Winnicki, S.; Yen, A.; Park, D.J.; Griesemer, D.; Karlsson, E.K.; Wong, S.H.; et al. Identifying recent adaptations in large-scale genomic data. Cell 2013, 152, 703–713. [Google Scholar] [CrossRef] [Green Version]
- McDonagh, E.M.; Whirl-Carrillo, M.; Garten, Y.; Altman, R.B.; Klein, T.E. From pharmacogenomic knowledge acquisition to clinical applications: The PharmGKB as a clinical pharmacogenomic biomarker resource. Biomark. Med. 2011, 5, 795–806. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The human genome browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, W.J.; Eizirik, E.; O’Brien, S.J.; Madsen, O.; Scally, M.; Douady, C.J.; Teeling, E.; Ryder, O.A.; Stanhope, M.J.; de Jong, W.W.; et al. Resolution of the early placental mammal radiation using Bayesian phylogenetics. Science 2001, 294, 2348–2351. [Google Scholar] [CrossRef]
- Karim, S.; NourEldin, H.F.; Abusamra, H.; Salem, N.; Alhathli, E.; Dudley, J.; Sanderford, M.; Scheinfeldt, L.B.; Chaudhary, A.G.; Al-Qahtani, M.H.; et al. e-GRASP: An integrated evolutionary and GRASP resource for exploring disease associations. BMC Genom. 2016, 17, 770. [Google Scholar] [CrossRef] [Green Version]
- Kircher, M.; Witten, D.M.; Jain, P.; O’Roak, B.J.; Cooper, G.M.; Shendure, J. A general framework for estimating the relative pathogenicity of human genetic variants. Nat. Genet. 2014, 46, 310–315. [Google Scholar] [CrossRef] [Green Version]
- Kuhn, M. Building Predictive Models in R Using the caret Package. J. Stat. Softw. 2008, 28, 1–26. [Google Scholar] [CrossRef] [Green Version]
- Liaw, A.; Wiener, M. Classification and Regression by randomForest. R News 2002, 2, 18–22. [Google Scholar]
- Chen, T.; Guestrin, C. XGBoost: A Scalable Tree Boosting System. In Proceedings of the 22nd ACM SIGKDD International Conference on Knowledge Discovery and Data Mining, New York, NY, USA, 13 August 2016; Association for Computing Machinery: New York, NY, USA, 2016; pp. 785–794. [Google Scholar] [CrossRef] [Green Version]
- Torgo, L. Data Mining with R, Learning with Case Studies; Chapman and Hall/CRC: Boca Raton, FL, USA, 2010. [Google Scholar]
- Wright, G.E.B.; Carleton, B.; Hayden, M.R.; Ross, C.J.D. The global spectrum of protein-coding pharmacogenomic diversity. Pharmacogenom. J. 2018, 18, 187–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, J.; Zhang, L.; Zhou, H.; Stoneking, M.; Tang, K. Global patterns of genetic diversity and signals of natural selection for human ADME genes. Hum. Mol. Genet. 2011, 20, 528–540. [Google Scholar] [CrossRef] [Green Version]
- Gerek, N.Z.; Liu, L.; Gerold, K.; Biparva, P.; Thomas, E.D.; Kumar, S. Evolutionary Diagnosis of non-synonymous variants involved in differential drug response. BMC Med. Genomics 2015, 8 (Suppl. 1), S6. [Google Scholar] [CrossRef] [Green Version]
- Ng, P.C.; Henikoff, S. SIFT: Predicting amino acid changes that affect protein function. Nucleic Acids Res. 2003, 31, 3812–3814. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nickerson, D.A.; Tobe, V.O.; Taylor, S.L. PolyPhred: Automating the detection and genotyping of single nucleotide substitutions using fluorescence-based resequencing. Nucleic Acids Res. 1997, 25, 2745–2751. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, S.; Akey, J.M. Genomic insights into positive selection. Trends Genet. 2006, 22, 437–446. [Google Scholar] [CrossRef] [PubMed]
- Devarajan, S.; Moon, I.; Ho, M.F.; Larson, N.B.; Neavin, D.R.; Moyer, A.M.; Black, J.L.; Bielinski, S.J.; Scherer, S.E.; Wang, L.; et al. Pharmacogenomic Next-Generation DNA Sequencing: Lessons from the Identification and Functional Characterization of Variants of Unknown Significance in CYP2C9 and CYP2C19. Drug Metab. Dispos. 2019, 47, 425–435. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Description | Label | Sample Size |
|---|---|---|
| African Caribbean in Barbados | ACB | 96 |
| Esan in Nigeria | ESN | 99 |
| Gambian in Western Division, Mandinka | GWD | 113 |
| Luhya in Webuye, Kenya | LWK | 99 |
| Mende in Sierra Leone | MSL | 85 |
| People with African Ancestry in Southwest USA | ASW | 61 |
| Yoruba in Ibadan, Nigeria | YRI | 108 |
| Colombians in Medellin, Colombia | CLM | 94 |
| People with Mexican Ancestry in Los Angeles, CA, USA | MXL | 64 |
| Peruvians in Lima, Peru | PEL | 85 |
| Puerto Ricans in Puerto Rico | PUR | 104 |
| Chinese Dai in Xishuangbanna, China | CDX | 93 |
| Han Chinese in Beijing, China | CHB | 103 |
| Japanese in Tokyo, Japan | JPT | 104 |
| Kinh in Ho Chi Minh City, Vietnam | KHV | 99 |
| Southern Han Chinese | CHS | 105 |
| British in England and Scotland | GBR | 91 |
| Finnish in Finland | FIN | 99 |
| Iberian Populations in Spain | IBS | 107 |
| Toscani in Italia | TSI | 107 |
| Utah residents (CEPH) with Northern and Western European ancestry | CEU | 99 |
| Bengali in Bangladesh | BEB | 86 |
| Gujarati Indians in Houston, TX, USA | GIH | 103 |
| Indian Telugu in the UK | ITU | 102 |
| Punjabi in Lahore, Pakistan | PJL | 96 |
| Sri Lankan Tamil in the UK | STU | 102 |
| Statistic | Model | Minimum | 1st Quartile | Median | Mean | 3rd Quartile | Maximum |
|---|---|---|---|---|---|---|---|
| ROC | Random Forest | 0.80 | 0.84 | 0.85 | 0.85 | 0.87 | 0.90 |
| LogitBoost | 0.83 | 0.86 | 0.87 | 0.87 | 0.89 | 0.92 | |
| XGBoost | 0.88 | 0.90 | 0.91 | 0.91 | 0.92 | 0.94 | |
| Sensitivity | Random Forest | 0.96 | 0.97 | 0.97 | 0.97 | 0.98 | 0.98 |
| LogitBoost | 0.90 | 0.92 | 0.93 | 0.93 | 0.94 | 0.96 | |
| XGBoost | 0.93 | 0.94 | 0.95 | 0.95 | 0.95 | 0.96 | |
| Specificity | Random Forest | 0.31 | 0.40 | 0.45 | 0.45 | 0.50 | 0.57 |
| LogitBoost | 0.53 | 0.62 | 0.69 | 0.68 | 0.72 | 0.82 | |
| XGBoost | 0.61 | 0.67 | 0.70 | 0.69 | 0.72 | 0.78 |
| Model | Prediction | Not Annotated in PharmGKB | PharmGKB PGx |
|---|---|---|---|
| Random Forest | neutral | 11,076 | 105 |
| PGx | 326 | 98 | |
| LogitBoost | neutral | 10,877 | 539 |
| PGx | 525 | 125 | |
| XGBoost | neutral | 10,716 | 63 |
| PGx | 686 | 140 |
| Variable | Overall Variable Importance (XGBoost) |
|---|---|
| Global minor allele frequency | 100.00 |
| Candidate adaptive polymorphism (CAP) | 10.00 |
| Evolutionary time | 4.66 |
| Non-reference evolutionary probability | 1.81 |
| Uniprot Topo domain | 1.62 |
| Evolutionary rate | 1.21 |
| Uniprot chain | 1.16 |
| Reference evolutionary probability | 0.77 |
| Uniprot domain | 0.50 |
| Uniprot helix | 0.21 |
| Uniprot repeat | 0.18 |
| Uniprot proteome | 0.10 |
| Uniprot disulfide | 0.07 |
| Uniprot variants | 0.07 |
| Method | Sensitivity | Specificity | Accuracy | AUC |
|---|---|---|---|---|
| SIFT | 0.59 | 0.42 | 0.50 | 0.51 |
| PolyPhen2 | 0.60 | 0.44 | 0.52 | 0.53 |
| CADD | 0.73 | 0.78 | 0.75 | 0.56 |
| EVOD | 0.64 | 0.50 | 0.57 | 0.57 |
| XGB-PGX | 0.95 | 0.68 | 0.82 | 0.84 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Scheinfeldt, L.B.; Brangan, A.; Kusic, D.M.; Kumar, S.; Gharani, N. Common Treatment, Common Variant: Evolutionary Prediction of Functional Pharmacogenomic Variants. J. Pers. Med. 2021, 11, 131. https://doi.org/10.3390/jpm11020131
Scheinfeldt LB, Brangan A, Kusic DM, Kumar S, Gharani N. Common Treatment, Common Variant: Evolutionary Prediction of Functional Pharmacogenomic Variants. Journal of Personalized Medicine. 2021; 11(2):131. https://doi.org/10.3390/jpm11020131
Chicago/Turabian StyleScheinfeldt, Laura B., Andrew Brangan, Dara M. Kusic, Sudhir Kumar, and Neda Gharani. 2021. "Common Treatment, Common Variant: Evolutionary Prediction of Functional Pharmacogenomic Variants" Journal of Personalized Medicine 11, no. 2: 131. https://doi.org/10.3390/jpm11020131
APA StyleScheinfeldt, L. B., Brangan, A., Kusic, D. M., Kumar, S., & Gharani, N. (2021). Common Treatment, Common Variant: Evolutionary Prediction of Functional Pharmacogenomic Variants. Journal of Personalized Medicine, 11(2), 131. https://doi.org/10.3390/jpm11020131