
Integrating Patient-Specific Information into Logic Models of Complex Diseases: Application to Acute Myeloid Leukemia

 , , and
, , and
Abstract
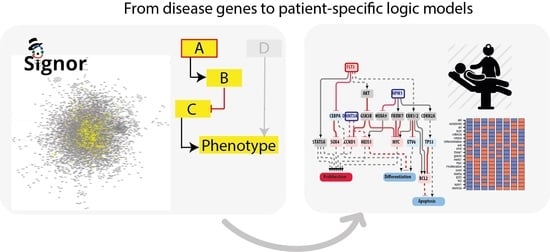
:
1. Introduction
2. Materials and Methods
2.1. Curation of AML-Relevant Causal Information
2.2. Assembly and Curation of a Network Linking Driver Genes and Cancer Hallmarks by Causal Relationships
2.3. Developing Boolean Models from Logic Networks
3. Results
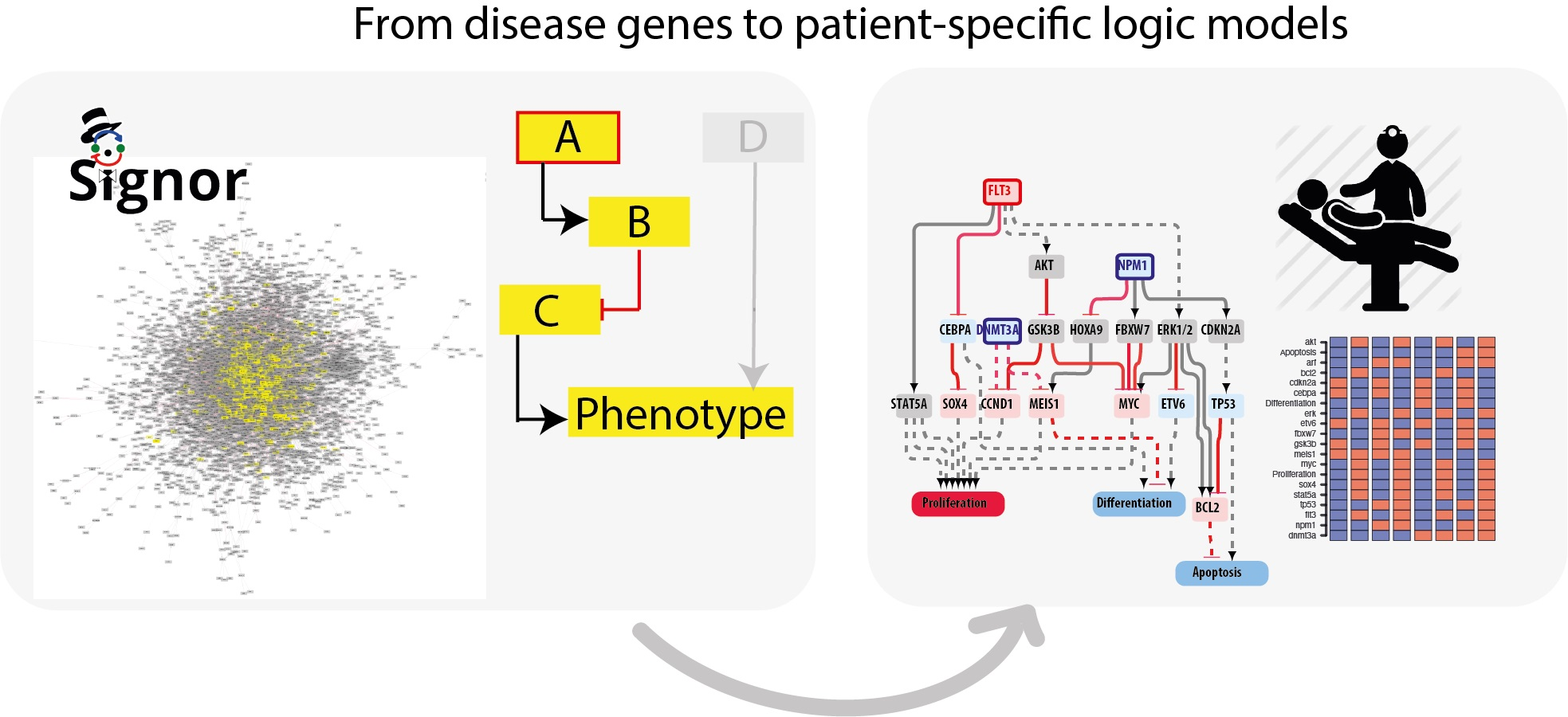
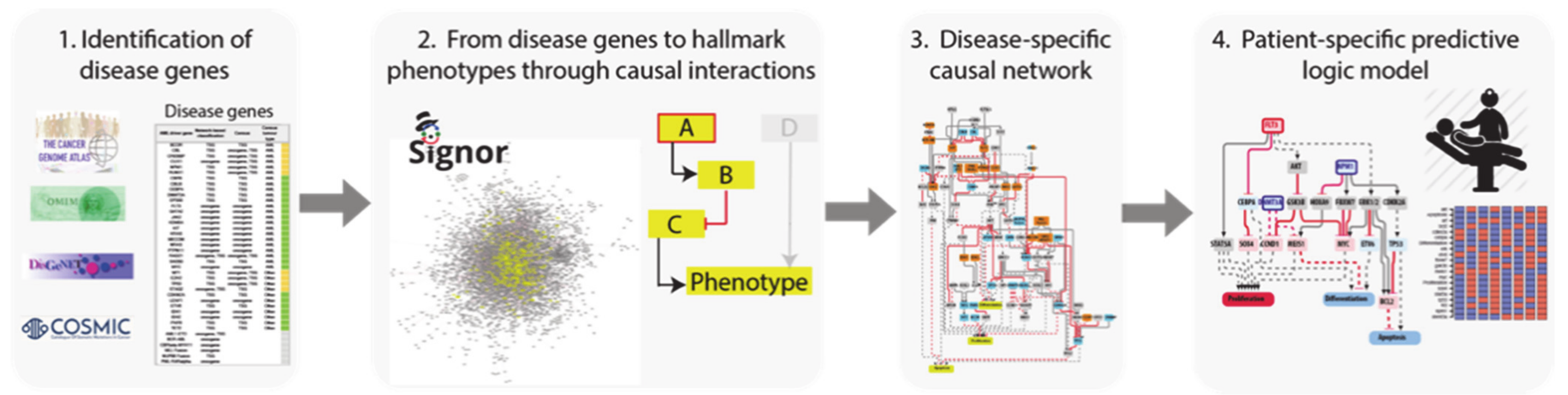
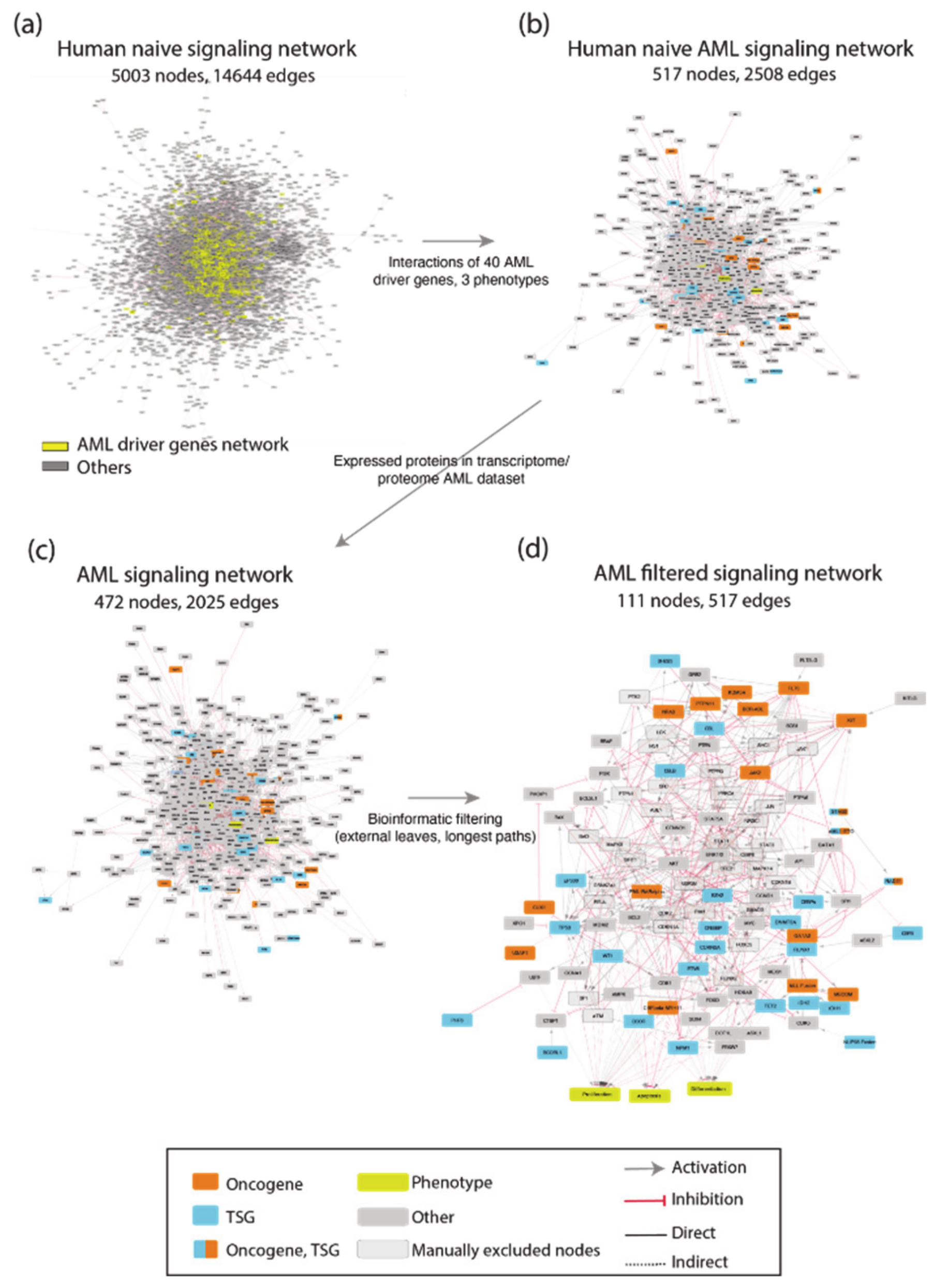
3.1. A Network-Based Strategy
- Identification of driver genes. First, we took advantage of different disease mutation databases to annotate as disease genes those genes that are found frequently mutated in patients diagnosed with the specific pathology.
- Connecting driver genes to hallmark processes. Next, we used CancerGeneNet, a tool implemented in the resource SIGNOR, to connect the disease genes to hallmark disease phenotypes by causal relationships, obtaining a naïve network of cause–effect interactions.
- Generation of pathway modules. This large network is broken down into smaller modules representing functional path detailing how the most common co-occurring mutations may functionally interact to regulate hallmark phenotypes.
- Development of disease-specific Boolean network. The logic information underlying the module network-topology is translated into actionable Boolean models to be used to infer the combined effect of the different mutations on patient prognosis.
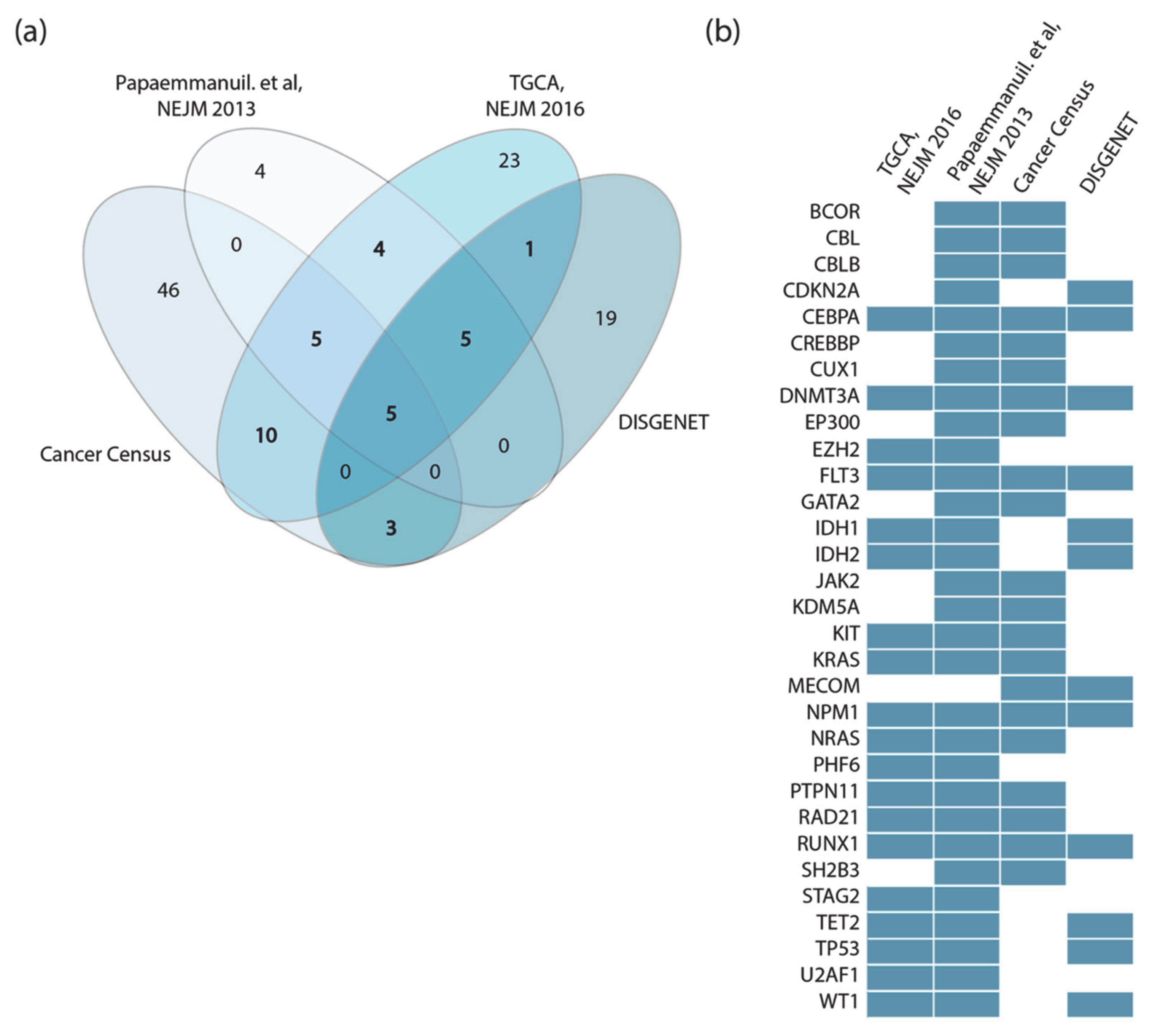
3.2. Identification of AML Driver Genes
- The TCGA AML dataset, consisting of genomic data of 200 clinically annotated adult cases of de novo AML patients. Fifty cases were characterized by whole-genome sequencing while for the remaining 150 only the exomes were sequenced. This study, which is one of the sequencing projects of the landmark cancer genomics program TCGA, enabled the identification of 23 significantly and recurrently mutated genes [9].
- Papaemmanuil et al., NEJM 2016, reported the mutational profile of 111 cancer driver genes in 1540 patients enrolled in three clinical trials. For each patient cytogenetic and clinical data are also available [8].
- Cancer Gene Census, a continuously updated resource storing a manually expert-annotated catalogue of genes containing mutations that have been causally implicated in cancer onset or progression [16].
- DisGeNET, a resource containing one of the largest collections of gene lists associated with human diseases [17]. The list of genes that this resource links to AML is very large (102 genes). We only considered the 33 genes significantly associated to AML (Score_dga > 0.02).
3.3. Connecting AML Driver Genes to Hallmark Processes
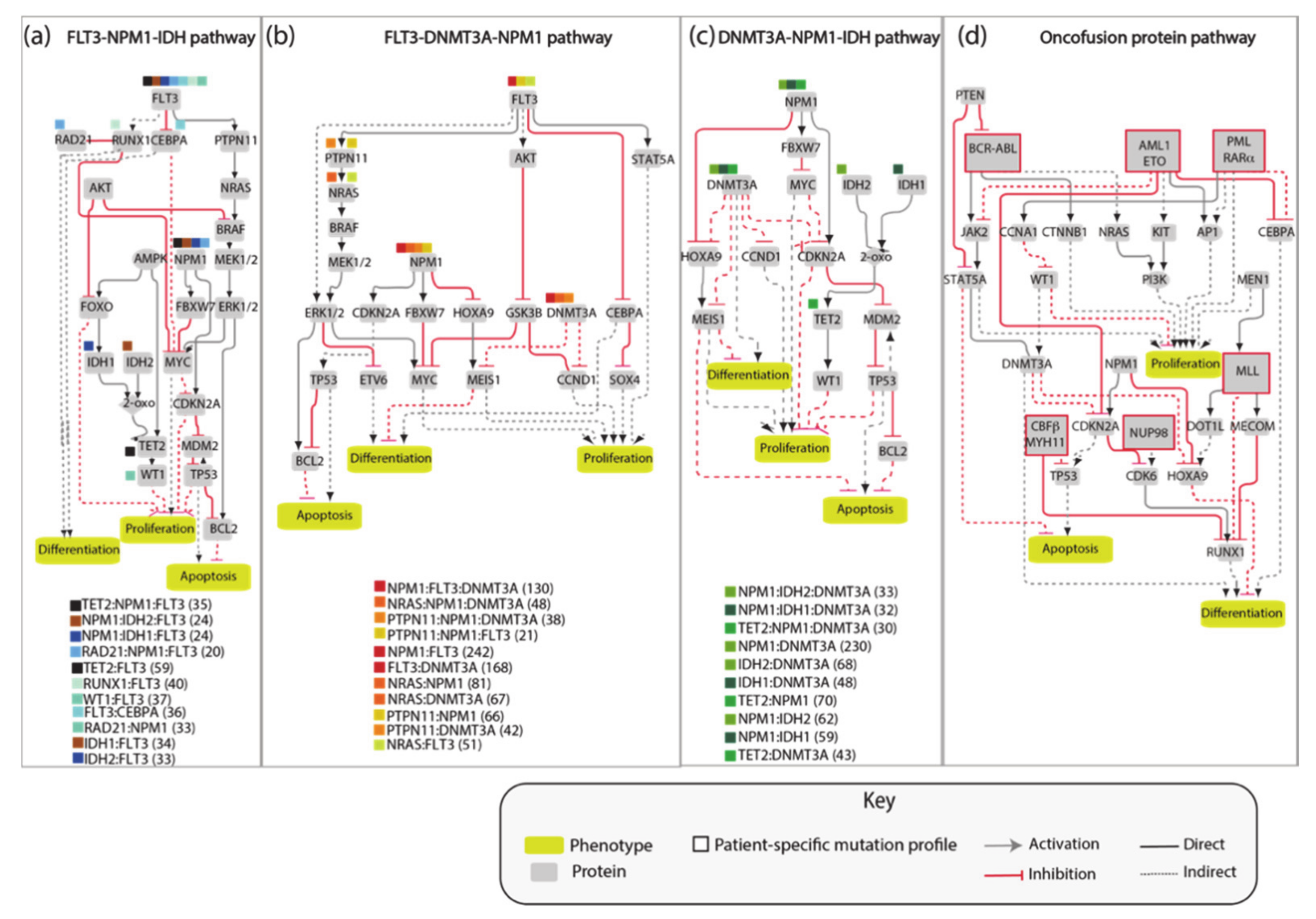
3.4. AML Modules
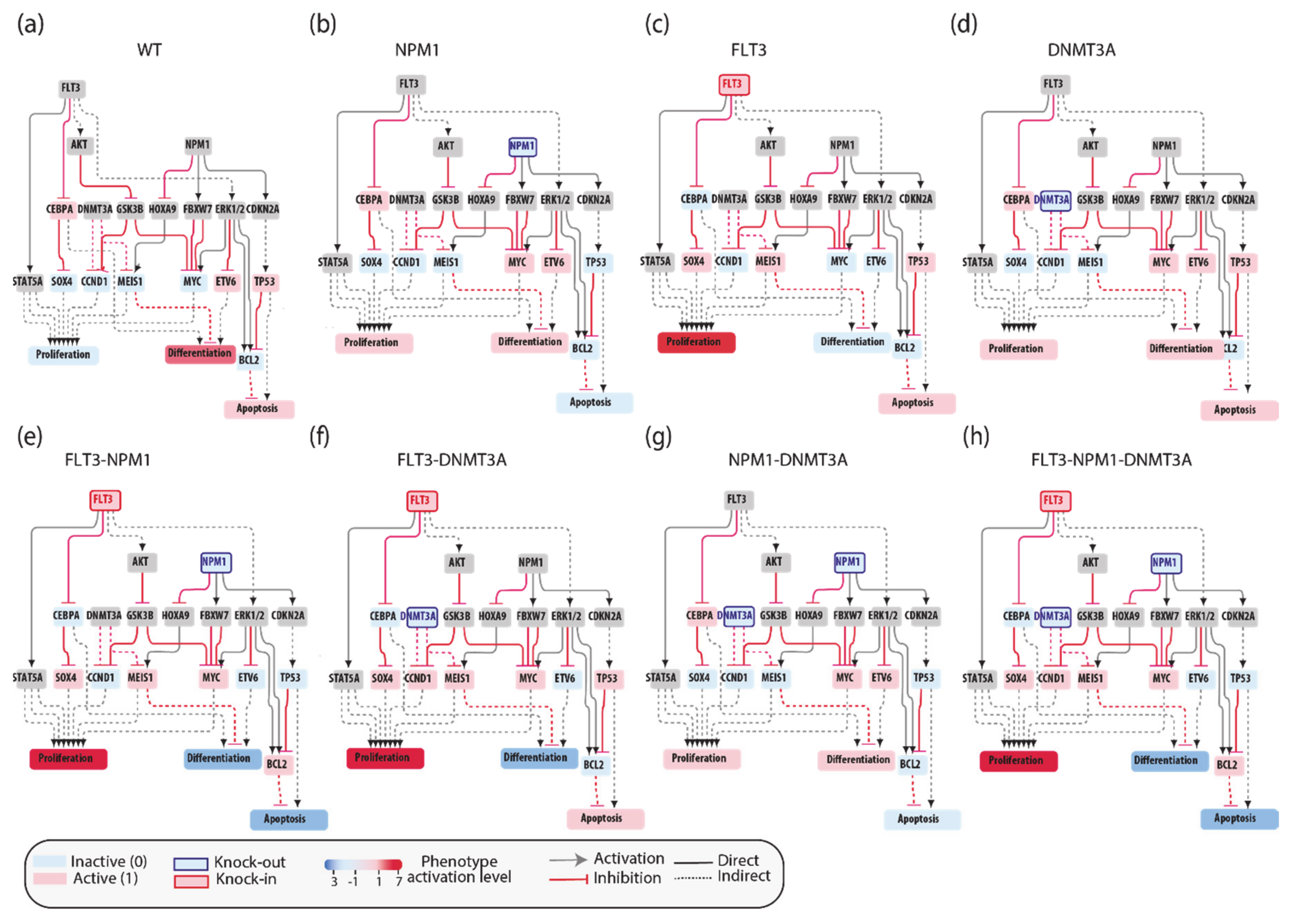
3.5. The FLT3-NPM1-DNMT3A Boolean Model
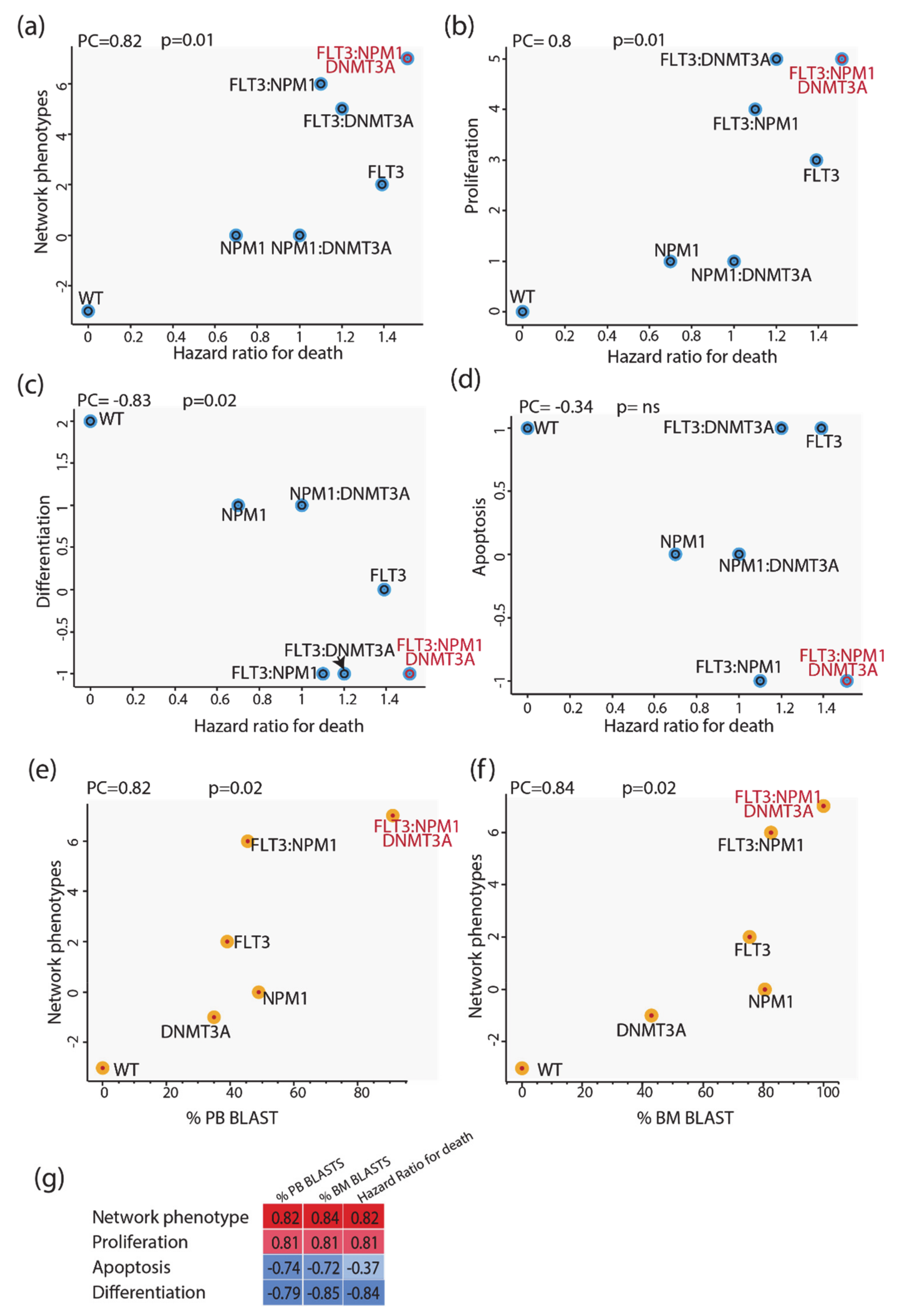
3.6. Predictive Power of Boolean Models
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Appendix A


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targets | Factors |
|---|---|
| flt3 | flt3 |
| npm1 | npm1 |
| akt | flt3 |
| Apoptosis | tp53 & !bcl2 |
| arf | npm1 |
| bcl2 | erk & !tp53 |
| ccnd1 | !(dnmt3a | gsk3b) |
| cebpa | !flt3 |
| Differentiation | (cebpa | etv6) & !meis1 |
| erk | flt3 |
| etv6 | !erk |
| fbxw7 | npm1 |
| gsk3b | !akt |
| hoxa9 | !npm1 |
| meis1 | !(dnmt3a & !hoxa9) |
| myc | erk & !(fbxw7 & gsk3b) |
| Proliferation | (myc | ccnd1 | sox4 | meis1 | stat5a) |
| sox4 | !cebpa |
| stat5a | flt3 |
| tp53 | arf |
References
- Brinkman, R.R.; Dubé, M.-P.; Rouleau, G.A.; Orr, A.C.; Samuels, M.E. Human monogenic disorders—A source of novel drug targets. Nat. Rev. Genet. 2006, 7, 249–260. [Google Scholar] [CrossRef] [PubMed]
- Glaser, R.L.; Goldbach-Mansky, R. The spectrum of monogenic autoinflammatory syndromes: Understanding disease mechanisms and use of targeted therapies. Curr. Allergy Asthma Rep. 2008, 8, 288–298. [Google Scholar] [CrossRef]
- McCarthy, M.I.; Abecasis, G.R.; Cardon, L.R.; Goldstein, D.B.; Little, J.; Ioannidis, J.P.A.; Hirschhorn, J.N. Genome-wide association studies for complex traits: Consensus, uncertainty and challenges. Nat. Rev. Genet. 2008, 9, 356–369. [Google Scholar] [CrossRef] [PubMed]
- Welch, J.S.; Ley, T.J.; Link, D.C.; Miller, C.A.; Larson, D.E.; Koboldt, D.C.; Wartman, L.D.; Lamprecht, T.L.; Liu, F.; Xia, J.; et al. The Origin and Evolution of Mutations in Acute Myeloid Leukemia. Cell 2012, 150, 264–278. [Google Scholar] [CrossRef] [Green Version]
- Gilliland, D.G.; Griffin, J.D. The roles of FLT3 in hematopoiesis and leukemia. Blood 2002, 100, 1532–1542. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, S. Current findings for recurring mutations in acute myeloid leukemia. J. Hematol. Oncol. 2011, 4, 36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dinardo, C.D.; Cortes, J.E. Mutations in AML: Prognostic and therapeutic implications. Hematology 2016, 2016, 348–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Papaemmanuil, E.; Gerstung, M.; Bullinger, L.; Gaidzik, V.I.; Paschka, P.; Roberts, N.D.; Potter, N.E.; Heuser, M.; Thol, F.; Bolli, N.; et al. Genomic Classification and Prognosis in Acute Myeloid Leukemia. N. Engl. J. Med. 2016, 374, 2209–2221. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research, N.; Ley, T.J.; Miller, C.; Ding, L.; Raphael, B.J.; Mungall, A.J.; Robertson, A.; Hoadley, K.; Triche, T.J., Jr.; Laird, P.W.; et al. Genomic and epigenomic landscapes of adult de novo acute myeloid leukemia. N. Engl. J. Med. 2013, 368, 2059–2074. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dutkowski, J.; Ideker, T. Protein Networks as Logic Functions in Development and Cancer. PLoS Comput. Biol. 2011, 7, e1002180. [Google Scholar] [CrossRef] [Green Version]
- Licata, L.; Surdo, P.L.; Iannuccelli, M.; Palma, A.; Micarelli, E.; Perfetto, L.; Peluso, D.; Calderone, A.; Castagnoli, L.; Cesareni, G. SIGNOR 2.0, the SIGnaling Network Open Resource 2.0: 2019 update. Nucleic Acids Res. 2019, 48, D504–D510. [Google Scholar] [CrossRef]
- Iannuccelli, M.; Micarelli, E.; Surdo, P.L.; Palma, A.; Perfetto, L.; Rozzo, I.; Castagnoli, L.; Licata, L.; Cesareni, G. CancerGeneNet: Linking driver genes to cancer hallmarks. Nucleic Acids Res. 2019, 48, D416–D421. [Google Scholar] [CrossRef]
- Nusinow, D.P.; Szpyt, J.; Ghandi, M.; Rose, C.M.; McDonald, E.R., 3rd; Kalocsay, M.; Jane-Valbuena, J.; Gelfand, E.; Schweppe, D.K.; Jedrychowski, M.; et al. Quantitative Proteomics of the Cancer Cell Line Encyclopedia. Cell 2020, 180, 387–402.e16. [Google Scholar] [CrossRef] [PubMed]
- Müssel, C.; Hopfensitz, M.; Kestler, H.A. BoolNet—An R package for generation, reconstruction and analysis of Boolean networks. Bioinformatics 2010, 26, 1378–1380. [Google Scholar] [CrossRef] [Green Version]
- Dorier, J.; Crespo, I.; Niknejad, A.; Liechti, R.; Ebeling, M.; Xenarios, I. Boolean regulatory network reconstruction using literature based knowledge with a genetic algorithm optimization method. BMC Bioinform. 2016, 17, 410. [Google Scholar] [CrossRef] [Green Version]
- Krumsiek, J.; Marr, C.; Schroeder, T.; Theis, F.J. Hierarchical Differentiation of Myeloid Progenitors Is Encoded in the Transcription Factor Network. PLoS ONE 2011, 6, e22649. [Google Scholar] [CrossRef] [Green Version]
- Méndez, A.; Mendoza, L. A Network Model to Describe the Terminal Differentiation of B Cells. PLoS Comput. Biol. 2016, 12, e1004696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Silverbush, D.; Grosskurth, S.; Wang, D.; Powell, F.; Gottgens, B.; Dry, J.; Fisher, J. Cell-Specific Computational Modeling of the PIM Pathway in Acute Myeloid Leukemia. Cancer Res. 2016, 77, 827–838. [Google Scholar] [CrossRef] [Green Version]
- Palma, A.; Jarrah, A.S.; Tieri, P.; Cesareni, G.; Castiglione, F. Gene Regulatory Network Modeling of Macrophage Differentiation Corroborates the Continuum Hypothesis of Polarization States. Front. Physiol. 2018, 9, 1659. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siegle, L.; Schwab, J.D.; Kühlwein, S.D.; Lausser, L.; Tümpel, S.; Pfister, A.S.; Kühl, M.; Kestler, H.A. A Boolean network of the crosstalk between IGF and Wnt signaling in aging satellite cells. PLoS ONE 2018, 13, e0195126. [Google Scholar] [CrossRef] [Green Version]
- Sizek, H.; Hamel, A.; Deritei, D.; Campbell, S.; Regan, E.R. Boolean model of growth signaling, cell cycle and apoptosis predicts the molecular mechanism of aberrant cell cycle progression driven by hyperactive PI3K. PLoS Comput. Biol. 2019, 15, e1006402. [Google Scholar] [CrossRef] [PubMed]
- Saez-Rodriguez, J.; Alexopoulos, L.G.; Zhang, M.; Morris, M.K.; Lauffenburger, D.A.; Sorger, P.K. Comparing Signaling Networks between Normal and Transformed Hepatocytes Using Discrete Logical Models. Cancer Res. 2011, 71, 5400–5411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Menche, J.; Sharma, A.; Kitsak, M.; Ghiassian, S.D.; Vidal, M.; Loscalzo, J.; Barabási, A.-L. Uncovering disease-disease relationships through the incomplete interactome. Science 2015, 347, 1257601. [Google Scholar] [CrossRef] [Green Version]
- Barabási, A.-L.; Gulbahce, N.; Loscalzo, J. Network medicine: A network-based approach to human disease. Nat. Rev. Genet. 2010, 12, 56–68. [Google Scholar] [CrossRef] [Green Version]
- Chuang, H.; Lee, E.; Liu, Y.; Lee, D.; Ideker, T. Network-based classification of breast cancer metastasis. Mol. Syst. Biol. 2007, 3, 140. [Google Scholar] [CrossRef] [PubMed]
- Hofree, M.; Shen, J.P.; Carter, H.; Gross, A.; Ideker, T. Network-based stratification of tumor mutations. Nat. Methods 2013, 10, 1108–1115. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.-A.; Cho, N.-Y.; Przytycka, T.M. Understanding Genotype-Phenotype Effects in Cancer via Network Approaches. PLoS Comput. Biol. 2016, 12, e1004747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pe’Er, D.; Hacohen, N. Principles and Strategies for Developing Network Models in Cancer. Cell 2011, 144, 864–873. [Google Scholar] [CrossRef] [Green Version]
- Stolovitzky, G.; Monroe, D.; Califano, A. Dialogue on Reverse-Engineering Assessment and Methods: The DREAM of High-Throughput Pathway Inference. Ann. N. Y. Acad. Sci. 2007, 1115, 1–22. [Google Scholar] [CrossRef]
- Henriques, D.; Villaverde, A.F.; Rocha, M.; Saez-Rodriguez, J.; Banga, J.R. Data-driven reverse engineering of signaling pathways using ensembles of dynamic models. PLoS Comput. Biol. 2017, 13, e1005379. [Google Scholar] [CrossRef] [Green Version]
- Fazekas, D.; Koltai, M.; Türei, D.; Módos, D.; Pálfy, M.; Dúl, Z.; Zsákai, L.; Szalay-Bekő, M.; Lenti, K.; Farkas, I.J.; et al. SignaLink —A signaling pathway resource with multi-layered regulatory networks. BMC Syst. Biol. 2013, 7, 7. [Google Scholar] [CrossRef] [Green Version]
- De Kouchkovsky, I.; Abdul-Hay, M. Acute myeloid leukemia: A comprehensive review and 2016 update. Blood Cancer J. 2016, 6, e441. [Google Scholar] [CrossRef]
- Watson, A.P.; Shabaneh, A.; Wang, J.; Dehm, S.M.; Rao, A.; Ryan, C.J. Triple Aberrant Prostate Cancer (TAPC)—Aggregate role of aberrations in TP53, PTEN and RB1 on ETS gene fusions and prognosis in metastatic castrate resistant prostate cancer. Am. J. Clin. Exp. Urol. 2020, 8, 106–115. [Google Scholar] [PubMed]
- Ayers, M.; Lunceford, J.; Nebozhyn, M.; Murphy, E.; Loboda, A.; Kaufman, D.R.; Albright, A.; Cheng, J.D.; Kang, S.P.; Shankaran, V.; et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J. Clin. Invest. 2017, 127, 2930–2940. [Google Scholar] [CrossRef] [PubMed]
- Fennell, D.A.; Myrand, S.P.; Nguyen, T.S.; Ferry, D.; Kerr, K.M.; Maxwell, P.; Moore, S.D.; Visseren-Grul, C.; Das, M.; Nicolson, M.C. Association between Gene Expression Profiles and Clinical Outcome of Pemetrexed-Based Treatment in Patients with Advanced Non-Squamous Non-Small Cell Lung Cancer: Exploratory Results from a Phase II Study. PLoS ONE 2014, 9, e107455. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Béal, J.; Montagud, A.; Traynard, P.; Barillot, E.; Calzone, L. Personalization of Logical Models With Multi-Omics Data Allows Clinical Stratification of Patients. Front. Physiol. 2019, 9, 1965. [Google Scholar] [CrossRef]
- Fumiã, H.F.; Martins, M.L. Boolean Network Model for Cancer Pathways: Predicting Carcinogenesis and Targeted Therapy Outcomes. PLoS ONE 2013, 8, e69008. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Flobak, Åsmund; Baudot, A.; Remy, E.; Thommesen, L.; Thieffry, D.; Kuiper, M.; Lægreid, A. Discovery of Drug Synergies in Gastric Cancer Cells Predicted by Logical Modeling. PLoS Comput. Biol. 2015, 11, e1004426. [Google Scholar] [CrossRef] [Green Version]
- Saez-Rodriguez, J.; Alexopoulos, L.G.; Epperlein, J.; Samaga, R.; A Lauffenburger, D.; Klamt, S.; Sorger, P.K. Discrete logic modelling as a means to link protein signalling networks with functional analysis of mammalian signal transduction. Mol. Syst. Biol. 2009, 5, 331. [Google Scholar] [CrossRef]
- Rodriguez, A.; Crespo, I.; Androsova, G.; Del Sol, A. Discrete Logic Modelling Optimization to Contextualize Prior Knowledge Networks Using PRUNET. PLoS ONE 2015, 10, e0127216. [Google Scholar] [CrossRef] [Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Palma, A.; Iannuccelli, M.; Rozzo, I.; Licata, L.; Perfetto, L.; Massacci, G.; Castagnoli, L.; Cesareni, G.; Sacco, F. Integrating Patient-Specific Information into Logic Models of Complex Diseases: Application to Acute Myeloid Leukemia. J. Pers. Med. 2021, 11, 117. https://doi.org/10.3390/jpm11020117
Palma A, Iannuccelli M, Rozzo I, Licata L, Perfetto L, Massacci G, Castagnoli L, Cesareni G, Sacco F. Integrating Patient-Specific Information into Logic Models of Complex Diseases: Application to Acute Myeloid Leukemia. Journal of Personalized Medicine. 2021; 11(2):117. https://doi.org/10.3390/jpm11020117
Chicago/Turabian StylePalma, Alessandro, Marta Iannuccelli, Ilaria Rozzo, Luana Licata, Livia Perfetto, Giorgia Massacci, Luisa Castagnoli, Gianni Cesareni, and Francesca Sacco. 2021. "Integrating Patient-Specific Information into Logic Models of Complex Diseases: Application to Acute Myeloid Leukemia" Journal of Personalized Medicine 11, no. 2: 117. https://doi.org/10.3390/jpm11020117
APA StylePalma, A., Iannuccelli, M., Rozzo, I., Licata, L., Perfetto, L., Massacci, G., Castagnoli, L., Cesareni, G., & Sacco, F. (2021). Integrating Patient-Specific Information into Logic Models of Complex Diseases: Application to Acute Myeloid Leukemia. Journal of Personalized Medicine, 11(2), 117. https://doi.org/10.3390/jpm11020117