
Recent Advances in Integrative Multi-Omics Research in Breast and Ovarian Cancer



{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Clinical Characterization of Breast and Ovarian Cancer
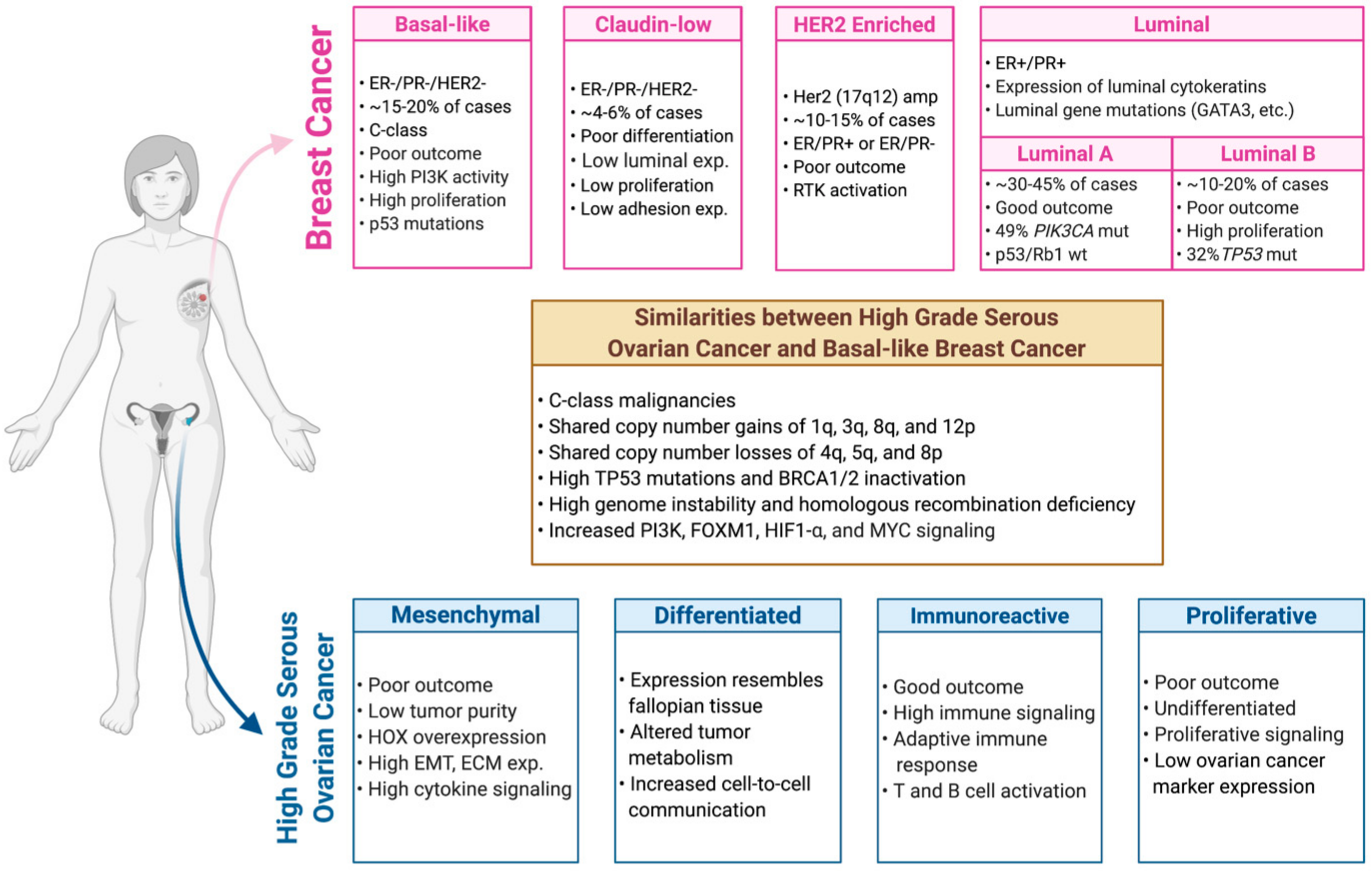
3. Molecular Classification and Characterization of Breast Cancer
4. Molecular Classification and Characterization of Ovarian Cancer
5. Genetic and Genomic Relationship between Breast and Ovarian Tumors
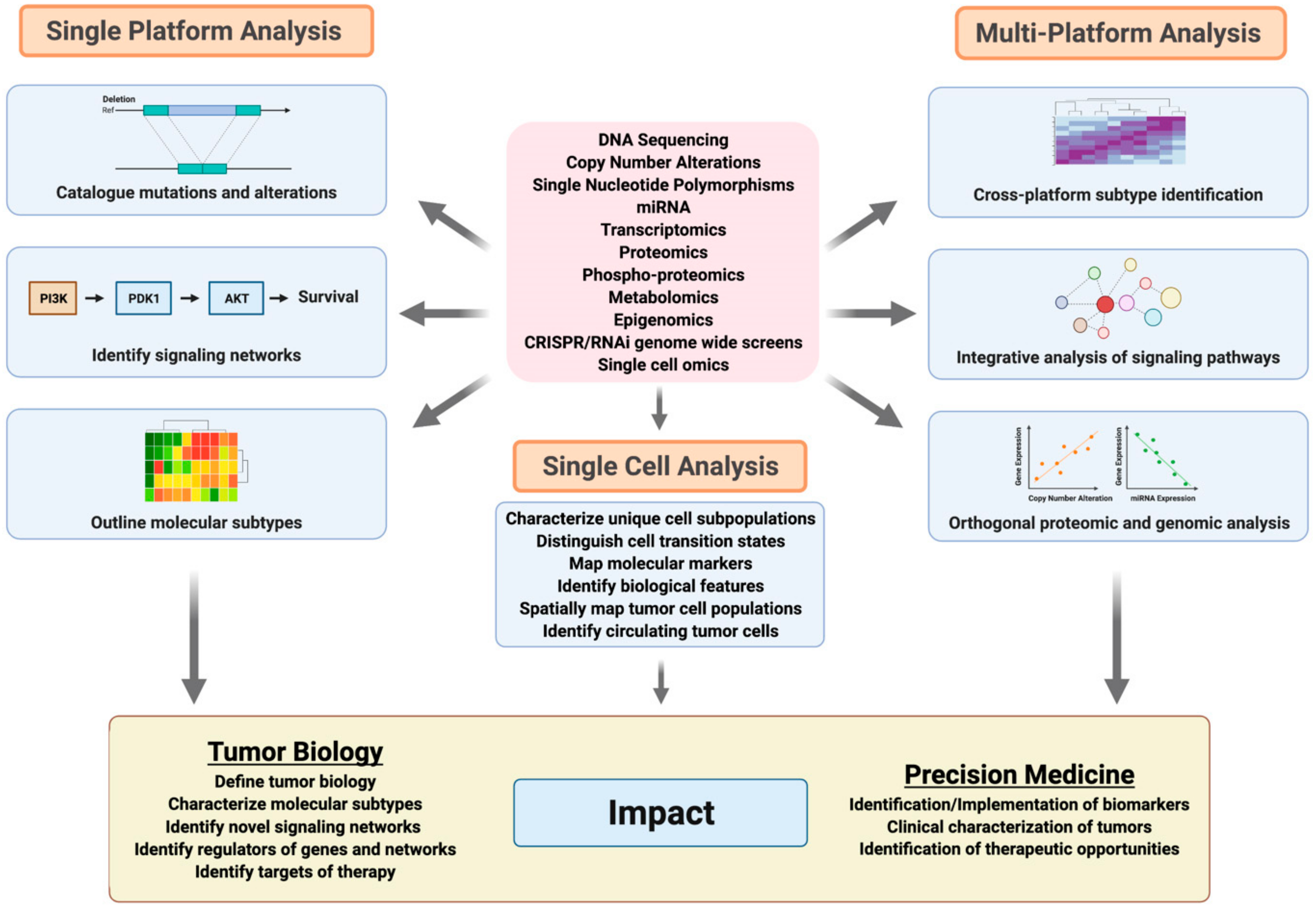
6. Advances in Genomic Analyses of Breast and Ovarian Cancer
7. Summary
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular portraits of human breast tumours. Nature 2012, 490, 61–70. [Google Scholar] [CrossRef] [Green Version]
- Cancer Genome Atlas Research Network. Integrated genomic analyses of ovarian carcinoma. Nature 2011, 474, 609–615. [Google Scholar] [CrossRef]
- Zhang, H.; Liu, T.; Zhang, Z.; Payne, S.H.; Zhang, B.; McDermott, J.E.; Zhou, J.-Y.; Petyuk, V.A.; Chen, L.; Ray, D.; et al. Integrated Proteogenomic Characterization of Human High-Grade Serous Ovarian Cancer. Cell 2016, 166, 755–765. [Google Scholar] [CrossRef] [Green Version]
- Mertins, P.; Mani, D.R.; Ruggles, K.V.; Gillette, M.A.; Clauser, K.R.; Wang, P.; Wang, X.; Qiao, J.W.; Cao, S.; Petralia, F.; et al. Proteogenomics connects somatic mutations to signalling in breast cancer. Nature 2016, 534, 55–62. [Google Scholar] [CrossRef] [Green Version]
- The ICGC/TCGA Pan-Cancer Analysis of Whole Genomes Consortium. Pan-cancer analysis of whole genomes. Nature 2020, 578, 82–93. [Google Scholar] [CrossRef] [Green Version]
- Alexandrov, L.B.; Kim, J.; Haradhvala, N.J.; Huang, M.N.; Tian Ng, A.W.; Wu, Y.; Boot, A.; Covington, K.R.; Gordenin, D.A.; Bergstrom, E.N.; et al. The repertoire of mutational signatures in human cancer. Nature 2020, 578, 94–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paczkowska, M.; Barenboim, J.; Sintupisut, N.; Fox, N.S.; Zhu, H.; Abd-Rabbo, D.; Mee, M.W.; Boutros, P.C.; Drivers, P.; Functional Interpretation Working Group; et al. Integrative pathway enrichment analysis of multivariate omics data. Nat. Commun. 2020, 11, 735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Curtis, C.; Shah, S.P.; Chin, S.F.; Turashvili, G.; Rueda, O.M.; Dunning, M.J.; Speed, D.; Lynch, A.G.; Samarajiwa, S.; Yuan, Y.; et al. The genomic and transcriptomic architecture of 2,000 breast tumours reveals novel subgroups. Nature 2012, 486, 346–352. [Google Scholar] [CrossRef] [PubMed]
- Berger, A.C.; Korkut, A.; Kanchi, R.S.; Hegde, A.M.; Lenoir, W.; Liu, W.; Liu, Y.; Fan, H.; Shen, H.; Ravikumar, V.; et al. A Comprehensive Pan-Cancer Molecular Study of Gynecologic and Breast Cancers. Cancer Cell 2018, 33, 690–705.e699. [Google Scholar] [CrossRef] [Green Version]
- Hoadley, K.A.; Yau, C.; Hinoue, T.; Wolf, D.M.; Lazar, A.J.; Drill, E.; Shen, R.; Taylor, A.M.; Cherniack, A.D.; Thorsson, V.; et al. Cell-of-Origin Patterns Dominate the Molecular Classification of 10,000 Tumors from 33 Types of Cancer. Cell 2018, 173, 291–304.e296. [Google Scholar] [CrossRef] [Green Version]
- Hutter, C.; Zenklusen, J.C. The Cancer Genome Atlas: Creating Lasting Value beyond Its Data. Cell 2018, 173, 283–285. [Google Scholar] [CrossRef]
- Lawrence, M.S.; Stojanov, P.; Polak, P.; Kryukov, G.V.; Cibulskis, K.; Sivachenko, A.; Carter, S.L.; Stewart, C.; Mermel, C.H.; Roberts, S.A.; et al. Mutational heterogeneity in cancer and the search for new cancer-associated genes. Nature 2013, 499, 214–218. [Google Scholar] [CrossRef]
- Vogelstein, B.; Papadopoulos, N.; Velculescu, V.E.; Zhou, S.; Diaz, L.A., Jr.; Kinzler, K.W. Cancer genome landscapes. Science 2013, 339, 1546–1558. [Google Scholar] [CrossRef]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef] [Green Version]
- Tucker, T.; Marra, M.; Friedman, J.M. Massively parallel sequencing: The next big thing in genetic medicine. Am. J. Hum. Genet. 2009, 85, 142–154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, F.; Paluch-Shimon, S.; Senkus, E.; Curigliano, G.; Aapro, M.S.; Andre, F.; Barrios, C.H.; Bergh, J.; Bhattacharyya, G.S.; Biganzoli, L.; et al. 5th ESO-ESMO international consensus guidelines for advanced breast cancer (ABC 5)(dagger). Ann. Oncol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Waks, A.G.; Winer, E.P. Breast Cancer Treatment: A Review. JAMA 2019, 321, 288–300. [Google Scholar] [CrossRef] [PubMed]
- Schmid, P.; Adams, S.; Rugo, H.S.; Schneeweiss, A.; Barrios, C.H.; Iwata, H.; Dieras, V.; Hegg, R.; Im, S.A.; Shaw Wright, G.; et al. Atezolizumab and Nab-Paclitaxel in Advanced Triple-Negative Breast Cancer. N. Engl. J. Med. 2018, 379, 2108–2121. [Google Scholar] [CrossRef] [PubMed]
- Burstein, H.J.; Lacchetti, C.; Anderson, H.; Buchholz, T.A.; Davidson, N.E.; Gelmon, K.A.; Giordano, S.H.; Hudis, C.A.; Solky, A.J.; Stearns, V.; et al. Adjuvant Endocrine Therapy for Women With Hormone Receptor-Positive Breast Cancer: ASCO Clinical Practice Guideline Focused Update. J. Clin. Oncol. 2019, 37, 423–438. [Google Scholar] [CrossRef] [PubMed]
- Breast Cancer Treatment (Adult) (PDQ(R)): Health Professional Version. In PDQ Cancer Information Summaries; National Cancer Institute: Bethesda, MD, USA, 2002.
- Ciriello, G.; Gatza, M.L.; Beck, A.H.; Wilkerson, M.D.; Rhie, S.K.; Pastore, A.; Zhang, H.; McLellan, M.; Yau, C.; Kandoth, C.; et al. Comprehensive Molecular Portraits of Invasive Lobular Breast Cancer. Cell 2015, 163, 506–519. [Google Scholar] [CrossRef] [Green Version]
- Keenan, T.; Moy, B.; Mroz, E.A.; Ross, K.; Niemierko, A.; Rocco, J.W.; Isakoff, S.; Ellisen, L.W.; Bardia, A. Comparison of the Genomic Landscape Between Primary Breast Cancer in African American Versus White Women and the Association of Racial Differences With Tumor Recurrence. J. Clin. Oncol. 2015, 33, 3621–3627. [Google Scholar] [CrossRef]
- Huo, D.; Hu, H.; Rhie, S.K.; Gamazon, E.R.; Cherniack, A.D.; Liu, J.; Yoshimatsu, T.F.; Pitt, J.J.; Hoadley, K.A.; Troester, M.; et al. Comparison of Breast Cancer Molecular Features and Survival by African and European Ancestry in The Cancer Genome Atlas. JAMA Oncol. 2017, 3, 1654–1662. [Google Scholar] [CrossRef]
- Ross, J.S.; Gay, L.M.; Wang, K.; Ali, S.M.; Chumsri, S.; Elvin, J.A.; Bose, R.; Vergilio, J.A.; Suh, J.; Yelensky, R.; et al. Nonamplification ERBB2 genomic alterations in 5605 cases of recurrent and metastatic breast cancer: An emerging opportunity for anti-HER2 targeted therapies. Cancer 2016, 122, 2654–2662. [Google Scholar] [CrossRef]
- Chalakur-Ramireddy, N.K.R.; Pakala, S.B. Combined drug therapeutic strategies for the effective treatment of Triple Negative Breast Cancer. Biosci. Rep. 2018, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marra, A.; Trapani, D.; Viale, G.; Criscitiello, C.; Curigliano, G. Practical classification of triple-negative breast cancer: Intratumoral heterogeneity, mechanisms of drug resistance, and novel therapies. NPJ Breast Cancer 2020, 6, 54. [Google Scholar] [CrossRef]
- Rozeboom, B.; Dey, N.; De, P. ER+ metastatic breast cancer: Past, present, and a prescription for an apoptosis-targeted future. Am. J.Cancer Res. 2019, 9, 2821–2831. [Google Scholar] [PubMed]
- Haque, M.M.; Desai, K.V. Pathways to Endocrine Therapy Resistance in Breast Cancer. Front. Endocrinol. 2019, 10, 573. [Google Scholar] [CrossRef] [PubMed]
- Razavi, P.; Chang, M.T.; Xu, G.; Bandlamudi, C.; Ross, D.S.; Vasan, N.; Cai, Y.; Bielski, C.M.; Donoghue, M.T.A.; Jonsson, P.; et al. The Genomic Landscape of Endocrine-Resistant Advanced Breast Cancers. Cancer Cell 2018, 34, 427–438.e426. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yardley, D.A.; Noguchi, S.; Pritchard, K.I.; Burris, H.A., 3rd; Baselga, J.; Gnant, M.; Hortobagyi, G.N.; Campone, M.; Pistilli, B.; Piccart, M.; et al. Everolimus plus exemestane in postmenopausal patients with HR(+) breast cancer: BOLERO-2 final progression-free survival analysis. Adv. Ther. 2013, 30, 870–884. [Google Scholar] [CrossRef] [Green Version]
- Narayan, P.; Prowell, T.M.; Gao, J.J.; Fernandes, L.L.; Li, E.; Jiang, X.; Qiu, J.; Fan, J.; Song, P.; Yu, J.; et al. FDA Approval Summary: Alpelisib plus fulvestrant for patients with HR-positive, HER2-negative, PIK3CA-mutated, advanced or metastatic breast cancer. Clin. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Cardoso, F.; Senkus, E.; Costa, A.; Papadopoulos, E.; Aapro, M.; Andre, F.; Harbeck, N.; Aguilar Lopez, B.; Barrios, C.H.; Bergh, J.; et al. 4th ESO-ESMO International Consensus Guidelines for Advanced Breast Cancer (ABC 4)dagger. Ann. Oncol. 2018, 29, 1634–1657. [Google Scholar] [CrossRef]
- Chopra, N.; Tovey, H.; Pearson, A.; Cutts, R.; Toms, C.; Proszek, P.; Hubank, M.; Dowsett, M.; Dodson, A.; Daley, F.; et al. Homologous recombination DNA repair deficiency and PARP inhibition activity in primary triple negative breast cancer. Nat. Commun. 2020, 11, 2662. [Google Scholar] [CrossRef] [PubMed]
- Lin, P.H.; Chen, M.; Tsai, L.W.; Lo, C.; Yen, T.C.; Huang, T.Y.; Chen, C.K.; Fan, S.C.; Kuo, S.H.; Huang, C.S. Using next-generation sequencing to redefine BRCAness in triple-negative breast cancer. Cancer Sci. 2020, 111, 1375–1384. [Google Scholar] [CrossRef] [Green Version]
- Kobel, M.; Kalloger, S.E.; Boyd, N.; McKinney, S.; Mehl, E.; Palmer, C.; Leung, S.; Bowen, N.J.; Ionescu, D.N.; Rajput, A.; et al. Ovarian carcinoma subtypes are different diseases: Implications for biomarker studies. PLoS Med. 2008, 5, e232. [Google Scholar] [CrossRef] [PubMed]
- Zorn, K.K.; Bonome, T.; Gangi, L.; Chandramouli, G.V.; Awtrey, C.S.; Gardner, G.J.; Barrett, J.C.; Boyd, J.; Birrer, M.J. Gene expression profiles of serous, endometrioid, and clear cell subtypes of ovarian and endometrial cancer. Clin. Cancer Res. 2005, 11, 6422–6430. [Google Scholar] [CrossRef] [Green Version]
- Bashashati, A.; Ha, G.; Tone, A.; Ding, J.; Prentice, L.M.; Roth, A.; Rosner, J.; Shumansky, K.; Kalloger, S.; Senz, J.; et al. Distinct evolutionary trajectories of primary high-grade serous ovarian cancers revealed through spatial mutational profiling. J. Pathol. 2013, 231, 21–34. [Google Scholar] [CrossRef] [PubMed]
- Beckmeyer-Borowko, A.B.; Peterson, C.E.; Brewer, K.C.; Otoo, M.A.; Davis, F.G.; Hoskins, K.F.; Joslin, C.E. The effect of time on racial differences in epithelial ovarian cancer (OVCA) diagnosis stage, overall and by histologic subtypes: A study of the National Cancer Database. Cancer Causes Control 2016, 27, 1261–1271. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shih Ie, M.; Kurman, R.J. Ovarian tumorigenesis: A proposed model based on morphological and molecular genetic analysis. Am. J. Pathol. 2004, 164, 1511–1518. [Google Scholar] [CrossRef]
- Jones, S.; Wang, T.L.; Shih Ie, M.; Mao, T.L.; Nakayama, K.; Roden, R.; Glas, R.; Slamon, D.; Diaz, L.A., Jr.; Vogelstein, B.; et al. Frequent mutations of chromatin remodeling gene ARID1A in ovarian clear cell carcinoma. Science 2010, 330, 228–231. [Google Scholar] [CrossRef] [Green Version]
- Wiegand, K.C.; Shah, S.P.; Al-Agha, O.M.; Zhao, Y.; Tse, K.; Zeng, T.; Senz, J.; McConechy, M.K.; Anglesio, M.S.; Kalloger, S.E.; et al. ARID1A mutations in endometriosis-associated ovarian carcinomas. N. Engl. J. Med. 2010, 363, 1532–1543. [Google Scholar] [CrossRef] [Green Version]
- Ahmed, A.A.; Etemadmoghadam, D.; Temple, J.; Lynch, A.G.; Riad, M.; Sharma, R.; Stewart, C.; Fereday, S.; Caldas, C.; Defazio, A.; et al. Driver mutations in TP53 are ubiquitous in high grade serous carcinoma of the ovary. J. Pathol. 2010, 221, 49–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kurman, R.J.; Shih Ie, M. Molecular pathogenesis and extraovarian origin of epithelial ovarian cancer—Shifting the paradigm. Hum. Pathol. 2011, 42, 918–931. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bowtell, D.D.; Bohm, S.; Ahmed, A.A.; Aspuria, P.-J.; Bast, R.C., Jr.; Beral, V.; Berek, J.S.; Birrer, M.J.; Blagden, S.; Bookman, M.A.; et al. Rethinking ovarian cancer II: Reducing mortality from high-grade serous ovarian cancer. Nat. Rev. Cancer 2015, 15, 668–679. [Google Scholar] [CrossRef] [PubMed]
- Vaughan, S.; Coward, J.I.; Bast, R.C.; Berchuck, A.; Berek, J.S.; Brenton, J.D.; Coukos, G.; Crum, C.C.; Drapkin, R.; Etemadmoghadam, D.; et al. Rethinking ovarian cancer: Recommendations for improving outcomes. Nat. Rev. Cancer 2011, 11, 719–725. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martignetti, J.A.; Camacho-Vanegas, O.; Priedigkeit, N.; Camacho, C.; Pereira, E.; Lin, L.; Garnar-Wortzel, L.; Miller, D.; Losic, B.; Shah, H.; et al. Personalized ovarian cancer disease surveillance and detection of candidate therapeutic drug target in circulating tumor DNA. Neoplasia 2014, 16, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Matulonis, U.A.; Sood, A.K.; Fallowfield, L.; Howitt, B.E.; Sehouli, J.; Karlan, B.Y. Ovarian cancer. Nat. Rev. Dis. Primers 2016, 2, 16061. [Google Scholar] [CrossRef] [PubMed]
- Bast, R.C., Jr.; Hennessy, B.; Mills, G.B. The biology of ovarian cancer: New opportunities for translation. Nat. Rev. Cancer 2009, 9, 415–428. [Google Scholar] [CrossRef]
- Schwarz, R.F.; Ng, C.K.; Cooke, S.L.; Newman, S.; Temple, J.; Piskorz, A.M.; Gale, D.; Sayal, K.; Murtaza, M.; Baldwin, P.J.; et al. Spatial and temporal heterogeneity in high-grade serous ovarian cancer: A phylogenetic analysis. PLoS Med. 2015, 12, e1001789. [Google Scholar] [CrossRef] [Green Version]
- Pokhriyal, R.; Hariprasad, R.; Kumar, L.; Hariprasad, G. Chemotherapy Resistance in Advanced Ovarian Cancer Patients. Biomark. Cancer 2019, 11, 1179299X19860815. [Google Scholar] [CrossRef] [PubMed]
- Konstantinopoulos, P.A.; Ceccaldi, R.; Shapiro, G.I.; D’Andrea, A.D. Homologous Recombination Deficiency: Exploiting the Fundamental Vulnerability of Ovarian Cancer. Cancer Discov. 2015, 5, 1137–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez-Martin, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Coleman, R.L.; Fleming, G.F.; Brady, M.F.; Swisher, E.M.; Steffensen, K.D.; Friedlander, M.; Okamoto, A.; Moore, K.N.; Efrat Ben-Baruch, N.; Werner, T.L.; et al. Veliparib with First-Line Chemotherapy and as Maintenance Therapy in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2403–2415. [Google Scholar] [CrossRef]
- Moore, K.; Colombo, N.; Scambia, G.; Kim, B.G.; Oaknin, A.; Friedlander, M.; Lisyanskaya, A.; Floquet, A.; Leary, A.; Sonke, G.S.; et al. Maintenance Olaparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2018, 379, 2495–2505. [Google Scholar] [CrossRef] [PubMed]
- Kaufman, B.; Shapira-Frommer, R.; Schmutzler, R.K.; Audeh, M.W.; Friedlander, M.; Balmana, J.; Mitchell, G.; Fried, G.; Stemmer, S.M.; Hubert, A.; et al. Olaparib monotherapy in patients with advanced cancer and a germline BRCA1/2 mutation. J. Clin. Oncol. 2015, 33, 244–250. [Google Scholar] [CrossRef]
- Del Campo, J.M.; Matulonis, U.A.; Malander, S.; Provencher, D.; Mahner, S.; Follana, P.; Waters, J.; Berek, J.S.; Woie, K.; Oza, A.M.; et al. Niraparib Maintenance Therapy in Patients With Recurrent Ovarian Cancer After a Partial Response to the Last Platinum-Based Chemotherapy in the ENGOT-OV16/NOVA Trial. J. Clin. Oncol. 2019, 37, 2968–2973. [Google Scholar] [CrossRef]
- Kristeleit, R.; Shapiro, G.I.; Burris, H.A.; Oza, A.M.; LoRusso, P.; Patel, M.R.; Domchek, S.M.; Balmana, J.; Drew, Y.; Chen, L.M.; et al. A Phase I-II Study of the Oral PARP Inhibitor Rucaparib in Patients with Germline BRCA1/2-Mutated Ovarian Carcinoma or Other Solid Tumors. Clin. Cancer Res. 2017, 23, 4095–4106. [Google Scholar] [CrossRef] [Green Version]
- Swisher, E.M.; Lin, K.K.; Oza, A.M.; Scott, C.L.; Giordano, H.; Sun, J.; Konecny, G.E.; Coleman, R.L.; Tinker, A.V.; O’Malley, D.M.; et al. Rucaparib in relapsed, platinum-sensitive high-grade ovarian carcinoma (ARIEL2 Part 1): An international, multicentre, open-label, phase 2 trial. Lancet Oncol. 2017, 18, 75–87. [Google Scholar] [CrossRef] [Green Version]
- Moore, K.N.; Secord, A.A.; Geller, M.A.; Miller, D.S.; Cloven, N.; Fleming, G.F.; Wahner Hendrickson, A.E.; Azodi, M.; DiSilvestro, P.; Oza, A.M.; et al. Niraparib monotherapy for late-line treatment of ovarian cancer (QUADRA): A multicentre, open-label, single-arm, phase 2 trial. Lancet Oncol. 2019, 20, 636–648. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Perol, D.; Gonzalez-Martin, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Maenpaa, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Wang, Q.; Peng, H.; Qi, X.; Wu, M.; Zhao, X. Targeted therapies in gynecological cancers: A comprehensive review of clinical evidence. Signal Transduct. Target. Ther. 2020, 5, 137. [Google Scholar] [CrossRef]
- Peters, M.L.; Garber, J.E.; Tung, N. Managing hereditary breast cancer risk in women with and without ovarian cancer. Gynecol. Oncol. 2017, 146, 205–214. [Google Scholar] [CrossRef]
- Yoshida, R. Hereditary breast and ovarian cancer (HBOC): Review of its molecular characteristics, screening, treatment, and prognosis. Breast Cancer 2020. [Google Scholar] [CrossRef]
- King, M.C.; Marks, J.H.; Mandell, J.B.; New York Breast Cancer Study Group. Breast and ovarian cancer risks due to inherited mutations in BRCA1 and BRCA2. Science 2003, 302, 643–646. [Google Scholar] [CrossRef] [PubMed]
- Momozawa, Y.; Iwasaki, Y.; Parsons, M.T.; Kamatani, Y.; Takahashi, A.; Tamura, C.; Katagiri, T.; Yoshida, T.; Nakamura, S.; Sugano, K.; et al. Germline pathogenic variants of 11 breast cancer genes in 7051 Japanese patients and 11,241 controls. Nat. Commun. 2018, 9, 4083. [Google Scholar] [CrossRef] [PubMed]
- Sun, J.; Meng, H.; Yao, L.; Lv, M.; Bai, J.; Zhang, J.; Wang, L.; Ouyang, T.; Li, J.; Wang, T.; et al. Germline Mutations in Cancer Susceptibility Genes in a Large Series of Unselected Breast Cancer Patients. Clin. Cancer Res. 2017, 23, 6113–6119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, W.X.; Allen, J.; Lai, K.N.; Mariapun, S.; Hasan, S.N.; Ng, P.S.; Lee, D.S.; Lee, S.Y.; Yoon, S.Y.; Lim, J.; et al. Inherited mutations in BRCA1 and BRCA2 in an unselected multiethnic cohort of Asian patients with breast cancer and healthy controls from Malaysia. J. Med. Genet. 2018, 55, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Tung, N.; Lin, N.U.; Kidd, J.; Allen, B.A.; Singh, N.; Wenstrup, R.J.; Hartman, A.R.; Winer, E.P.; Garber, J.E. Frequency of Germline Mutations in 25 Cancer Susceptibility Genes in a Sequential Series of Patients With Breast Cancer. J. Clin. Oncol. 2016, 34, 1460–1468. [Google Scholar] [CrossRef] [Green Version]
- Song, H.; Cicek, M.S.; Dicks, E.; Harrington, P.; Ramus, S.J.; Cunningham, J.M.; Fridley, B.L.; Tyrer, J.P.; Alsop, J.; Jimenez-Linan, M.; et al. The contribution of deleterious germline mutations in BRCA1, BRCA2 and the mismatch repair genes to ovarian cancer in the population. Hum. Mol. Genet. 2014, 23, 4703–4709. [Google Scholar] [CrossRef] [Green Version]
- Alsop, K.; Fereday, S.; Meldrum, C.; de Fazio, A.; Emmanuel, C.; George, J.; Dobrovic, A.; Birrer, M.J.; Webb, P.M.; Stewart, C.; et al. BRCA mutation frequency and patterns of treatment response in BRCA mutation-positive women with ovarian cancer: A report from the Australian Ovarian Cancer Study Group. J. Clin. Oncol. 2012, 30, 2654–2663. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Royer, R.; Li, S.; McLaughlin, J.R.; Rosen, B.; Risch, H.A.; Fan, I.; Bradley, L.; Shaw, P.A.; Narod, S.A. Frequencies of BRCA1 and BRCA2 mutations among 1,342 unselected patients with invasive ovarian cancer. Gynecol. Oncol. 2011, 121, 353–357. [Google Scholar] [CrossRef]
- Kang, E.; Kim, S.W. The Korean hereditary breast cancer study: Review and future perspectives. J. Breast Cancer 2013, 16, 245–253. [Google Scholar] [CrossRef] [Green Version]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.; Parmigiani, G. Meta-analysis of BRCA1 and BRCA2 penetrance. J. Clin. Oncol. 2007, 25, 1329–1333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antoniou, A.; Pharoah, P.D.; Narod, S.; Risch, H.A.; Eyfjord, J.E.; Hopper, J.L.; Loman, N.; Olsson, H.; Johannsson, O.; Borg, A.; et al. Average risks of breast and ovarian cancer associated with BRCA1 or BRCA2 mutations detected in case Series unselected for family history: A combined analysis of 22 studies. Am. J. Hum. Genet. 2003, 72, 1117–1130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Y.C.; McGuffog, L.; Healey, S.; Friedman, E.; Laitman, Y.; Paluch-Shimon, S.; Kaufman, B.; Swe, B.; Liljegren, A.; Lindblom, A.; et al. A nonsynonymous polymorphism in IRS1 modifies risk of developing breast and ovarian cancers in BRCA1 and ovarian cancer in BRCA2 mutation carriers. Cancer Epidemiol. Biomark. Prev. 2012, 21, 1362–1370. [Google Scholar] [CrossRef] [Green Version]
- Kuchenbaecker, K.B.; Neuhausen, S.L.; Robson, M.; Barrowdale, D.; McGuffog, L.; Mulligan, A.M.; Andrulis, I.L.; Spurdle, A.B.; Schmidt, M.K.; Schmutzler, R.K.; et al. Associations of common breast cancer susceptibility alleles with risk of breast cancer subtypes in BRCA1 and BRCA2 mutation carriers. Breast Cancer Res. BCR 2014, 16, 3416. [Google Scholar] [CrossRef] [Green Version]
- McLaughlin, J.R.; Rosen, B.; Moody, J.; Pal, T.; Fan, I.; Shaw, P.A.; Risch, H.A.; Sellers, T.A.; Sun, P.; Narod, S.A. Long-term ovarian cancer survival associated with mutation in BRCA1 or BRCA2. J. Natl. Cancer Inst. 2013, 105, 141–148. [Google Scholar] [CrossRef]
- Templeton, A.J.; Gonzalez, L.D.; Vera-Badillo, F.E.; Tibau, A.; Goldstein, R.; Seruga, B.; Srikanthan, A.; Pandiella, A.; Amir, E.; Ocana, A. Interaction between Hormonal Receptor Status, Age and Survival in Patients with BRCA1/2 Germline Mutations: A Systematic Review and Meta-Regression. PLoS ONE 2016, 11, e0154789. [Google Scholar] [CrossRef]
- Copson, E.R.; Maishman, T.C.; Tapper, W.J.; Cutress, R.I.; Greville-Heygate, S.; Altman, D.G.; Eccles, B.; Gerty, S.; Durcan, L.T.; Jones, L.; et al. Germline BRCA mutation and outcome in young-onset breast cancer (POSH): A prospective cohort study. Lancet Oncol. 2018, 19, 169–180. [Google Scholar] [CrossRef] [Green Version]
- Pernas, S.; Tolaney, S.M.; Winer, E.P.; Goel, S. CDK4/6 inhibition in breast cancer: Current practice and future directions. Ther. Adv. Med. Oncol. 2018, 10, 1758835918786451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Perou, C.M.; Sorlie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; Rees, C.A.; Pollack, J.R.; Ross, D.T.; Johnsen, H.; Akslen, L.A.; et al. Molecular portraits of human breast tumours. Nature 2000, 406, 747–752. [Google Scholar] [CrossRef] [PubMed]
- Sorlie, T.; Perou, C.M.; Tibshirani, R.; Aas, T.; Geisler, S.; Johnsen, H.; Hastie, T.; Eisen, M.B.; van de Rijn, M.; Jeffrey, S.S.; et al. Gene expression patterns of breast carcinomas distinguish tumor subclasses with clinical implications. Proc. Natl. Acad. Sci. USA 2001, 98, 10869–10874. [Google Scholar] [CrossRef] [Green Version]
- Sorlie, T.; Tibshirani, R.; Parker, J.; Hastie, T.; Marron, J.S.; Nobel, A.; Deng, S.; Johnsen, H.; Pesich, R.; Geisler, S.; et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc. Natl. Acad. Sci. USA 2003, 100, 8418–8423. [Google Scholar] [CrossRef] [Green Version]
- Hu, Z.; Fan, C.; Oh, D.S.; Marron, J.S.; He, X.; Qaqish, B.F.; Livasy, C.; Carey, L.A.; Reynolds, E.; Dressler, L.; et al. The molecular portraits of breast tumors are conserved across microarray platforms. BMC Genom. 2006, 7, 96. [Google Scholar] [CrossRef] [Green Version]
- Parker, J.S.; Mullins, M.; Cheang, M.C.; Leung, S.; Voduc, D.; Vickery, T.; Davies, S.; Fauron, C.; He, X.; Hu, Z.; et al. Supervised risk predictor of breast cancer based on intrinsic subtypes. J. Clin. Oncol. 2009, 27, 1160–1167. [Google Scholar] [CrossRef]
- Wallden, B.; Storhoff, J.; Nielsen, T.; Dowidar, N.; Schaper, C.; Ferree, S.; Liu, S.; Leung, S.; Geiss, G.; Snider, J.; et al. Development and verification of the PAM50-based Prosigna breast cancer gene signature assay. BMC Med. Genom. 2015, 8, 54. [Google Scholar] [CrossRef] [Green Version]
- Cheang, M.C.; Voduc, K.D.; Tu, D.; Jiang, S.; Leung, S.; Chia, S.K.; Shepherd, L.E.; Levine, M.N.; Pritchard, K.I.; Davies, S.; et al. Responsiveness of intrinsic subtypes to adjuvant anthracycline substitution in the NCIC.CTG MA.5 randomized trial. Clin. Cancer Res. 2012, 18, 2402–2412. [Google Scholar] [CrossRef] [Green Version]
- Martin, M.; Prat, A.; Rodriguez-Lescure, A.; Caballero, R.; Ebbert, M.T.; Munarriz, B.; Ruiz-Borrego, M.; Bastien, R.R.; Crespo, C.; Davis, C.; et al. PAM50 proliferation score as a predictor of weekly paclitaxel benefit in breast cancer. Breast Cancer Res. Treat. 2013, 138, 457–466. [Google Scholar] [CrossRef] [Green Version]
- Herschkowitz, J.I.; Simin, K.; Weigman, V.J.; Mikaelian, I.; Usary, J.; Hu, Z.; Rasmussen, K.E.; Jones, L.P.; Assefnia, S.; Chandrasekharan, S.; et al. Identification of conserved gene expression features between murine mammary carcinoma models and human breast tumors. Genome Biol. 2007, 8, R76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Prat, A.; Parker, J.S.; Karginova, O.; Fan, C.; Livasy, C.; Herschkowitz, J.I.; He, X.; Perou, C.M. Phenotypic and molecular characterization of the claudin-low intrinsic subtype of breast cancer. Breast Cancer Res. 2010, 12, R68. [Google Scholar] [CrossRef] [Green Version]
- Lehmann, B.D.; Bauer, J.A.; Chen, X.; Sanders, M.E.; Chakravarthy, A.B.; Shyr, Y.; Pietenpol, J.A. Identification of human triple-negative breast cancer subtypes and preclinical models for selection of targeted therapies. J. Clin. Investig. 2011, 121, 2750–2767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehmann, B.D.; Jovanovic, B.; Chen, X.; Estrada, M.V.; Johnson, K.N.; Shyr, Y.; Moses, H.L.; Sanders, M.E.; Pietenpol, J.A. Refinement of Triple-Negative Breast Cancer Molecular Subtypes: Implications for Neoadjuvant Chemotherapy Selection. PLoS ONE 2016, 11, e0157368. [Google Scholar] [CrossRef]
- Bild, A.H.; Yao, G.; Chang, J.T.; Wang, Q.; Potti, A.; Chasse, D.; Joshi, M.B.; Harpole, D.; Lancaster, J.M.; Berchuck, A.; et al. Oncogenic pathway signatures in human cancers as a guide to targeted therapies. Nature 2006, 439, 353–357. [Google Scholar] [CrossRef]
- Gatza, M.L.; Lucas, J.E.; Barry, W.T.; Kim, J.W.; Wang, Q.; Crawford, M.D.; Datto, M.B.; Kelley, M.; Mathey-Prevot, B.; Potti, A.; et al. A pathway-based classification of human breast cancer. Proc. Natl. Acad. Sci. USA 2010, 107, 6994–6999. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gatza, M.L.; Kung, H.N.; Blackwell, K.L.; Dewhirst, M.W.; Marks, J.R.; Chi, J.T. Analysis of tumor environmental response and oncogenic pathway activation identifies distinct basal and luminal features in HER2-related breast tumor subtypes. Breast Cancer Res. BCR 2011, 13, R62. [Google Scholar] [CrossRef] [Green Version]
- Gatza, M.L.; Silva, G.O.; Parker, J.S.; Fan, C.; Perou, C.M. An integrated genomics approach identifies drivers of proliferation in luminal-subtype human breast cancer. Nat. Genet. 2014, 46, 1051–1059. [Google Scholar] [CrossRef] [Green Version]
- Bild, A.H.; Parker, J.S.; Gustafson, A.M.; Acharya, C.R.; Hoadley, K.A.; Anders, C.; Marcom, P.K.; Carey, L.A.; Potti, A.; Nevins, J.R.; et al. An integration of complementary strategies for gene-expression analysis to reveal novel therapeutic opportunities for breast cancer. Breast Cancer Res. BCR 2009, 11, R55. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weigman, V.J.; Chao, H.H.; Shabalin, A.A.; He, X.; Parker, J.S.; Nordgard, S.H.; Grushko, T.; Huo, D.; Nwachukwu, C.; Nobel, A.; et al. Basal-like Breast cancer DNA copy number losses identify genes involved in genomic instability, response to therapy, and patient survival. Breast Cancer Res. Treat. 2012, 133, 865–880. [Google Scholar] [CrossRef] [Green Version]
- Ciriello, G.; Miller, M.L.; Aksoy, B.A.; Senbabaoglu, Y.; Schultz, N.; Sander, C. Emerging landscape of oncogenic signatures across human cancers. Nat. Genet. 2013, 45, 1127–1133. [Google Scholar] [CrossRef] [Green Version]
- Valla, M.; Mjones, P.G.; Engstrom, M.J.; Ytterhus, B.; Bordin, D.L.; van Loon, B.; Akslen, L.A.; Vatten, L.J.; Opdahl, S.; Bofin, A.M. Characterization of FGD5 Expression in Primary Breast Cancers and Lymph Node Metastases. J. Histochem. Cytochem. 2018, 66, 787–799. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klaestad, E.; Opdahl, S.; Engstrom, M.J.; Ytterhus, B.; Wik, E.; Bofin, A.M.; Valla, M. MRPS23 amplification and gene expression in breast cancer; association with proliferation and the non-basal subtypes. Breast Cancer Res. Treat. 2020, 180, 73–86. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valla, M.; Engstrom, M.J.; Ytterhus, B.; Hansen, A.K.; Akslen, L.A.; Vatten, L.J.; Opdahl, S.; Bofin, A.M. FGD5 amplification in breast cancer patients is associated with tumour proliferation and a poorer prognosis. Breast Cancer Res. Treat. 2017, 162, 243–253. [Google Scholar] [CrossRef]
- Desmedt, C.; Zoppoli, G.; Gundem, G.; Pruneri, G.; Larsimont, D.; Fornili, M.; Fumagalli, D.; Brown, D.; Rothe, F.; Vincent, D.; et al. Genomic Characterization of Primary Invasive Lobular Breast Cancer. J. Clin. Oncol. 2016, 34, 1872–1881. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guido, L.P.; Gomez-Fernandez, C. Advances in the Molecular Taxonomy of Breast Cancer. Arch. Med. Res. 2020, 51, 777–783. [Google Scholar] [CrossRef] [PubMed]
- Bandura, D.R.; Baranov, V.I.; Ornatsky, O.I.; Antonov, A.; Kinach, R.; Lou, X.; Pavlov, S.; Vorobiev, S.; Dick, J.E.; Tanner, S.D. Mass cytometry: Technique for real time single cell multitarget immunoassay based on inductively coupled plasma time-of-flight mass spectrometry. Anal. Chem. 2009, 81, 6813–6822. [Google Scholar] [CrossRef]
- Ali, H.R.; Jackson, H.W.; Zanotelli, V.R.T.; Danenberg, E.; Fischer, J.R.; Bardwell, H.; Provenzano, E.; Ali, H.R.; Al Sa’d, M.; Alon, S.; et al. Imaging mass cytometry and multiplatform genomics define the phenogenomic landscape of breast cancer. Nat. Cancer 2020, 1, 163–175. [Google Scholar] [CrossRef]
- Giesen, C.; Wang, H.A.; Schapiro, D.; Zivanovic, N.; Jacobs, A.; Hattendorf, B.; Schuffler, P.J.; Grolimund, D.; Buhmann, J.M.; Brandt, S.; et al. Highly multiplexed imaging of tumor tissues with subcellular resolution by mass cytometry. Nat. Methods 2014, 11, 417–422. [Google Scholar] [CrossRef]
- Tothill, R.W.; Tinker, A.V.; George, J.; Brown, R.; Fox, S.B.; Lade, S.; Johnson, D.S.; Trivett, M.K.; Etemadmoghadam, D.; Locandro, B.; et al. Novel molecular subtypes of serous and endometrioid ovarian cancer linked to clinical outcome. Clin. Cancer Res. 2008, 14, 5198–5208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Helland, A.; Anglesio, M.S.; George, J.; Cowin, P.A.; Johnstone, C.N.; House, C.M.; Sheppard, K.E.; Etemadmoghadam, D.; Melnyk, N.; Rustgi, A.K.; et al. Deregulation of MYCN, LIN28B and LET7 in a molecular subtype of aggressive high-grade serous ovarian cancers. PLoS ONE 2011, 6, e18064. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Konecny, G.E.; Wang, C.; Hamidi, H.; Winterhoff, B.; Kalli, K.R.; Dering, J.; Ginther, C.; Chen, H.W.; Dowdy, S.; Cliby, W.; et al. Prognostic and therapeutic relevance of molecular subtypes in high-grade serous ovarian cancer. J. Natl. Cancer Inst. 2014, 106. [Google Scholar] [CrossRef] [PubMed]
- Chen, G.M.; Kannan, L.; Geistlinger, L.; Kofia, V.; Safikhani, Z.; Gendoo, D.M.A.; Parmigiani, G.; Birrer, M.; Haibe-Kains, B.; Waldron, L. Consensus on Molecular Subtypes of High-Grade Serous Ovarian Carcinoma. Clin. Cancer Res. 2018, 24, 5037–5047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verhaak, R.G.; Tamayo, P.; Yang, J.Y.; Hubbard, D.; Zhang, H.; Creighton, C.J.; Fereday, S.; Lawrence, M.; Carter, S.L.; Mermel, C.H.; et al. Prognostically relevant gene signatures of high-grade serous ovarian carcinoma. J. Clin. Investig. 2013, 123, 517–525. [Google Scholar] [CrossRef]
- Karagoz, K.; Mehta, G.A.; Khella, C.A.; Khanna, P.; Gatza, M.L. Integrative proteogenomic analyses of human tumours identifies ADNP as a novel oncogenic mediator of cell cycle progression in high-grade serous ovarian cancer with poor prognosis. EBioMedicine 2019, 50, 191–202. [Google Scholar] [CrossRef]
- Izar, B.; Tirosh, I.; Stover, E.H.; Wakiro, I.; Cuoco, M.S.; Alter, I.; Rodman, C.; Leeson, R.; Su, M.J.; Shah, P.; et al. A single-cell landscape of high-grade serous ovarian cancer. Nat. Med. 2020, 26, 1271–1279. [Google Scholar] [CrossRef]
- Way, G.P.; Rudd, J.; Wang, C.; Hamidi, H.; Fridley, B.L.; Konecny, G.E.; Goode, E.L.; Greene, C.S.; Doherty, J.A. Comprehensive Cross-Population Analysis of High-Grade Serous Ovarian Cancer Supports No More Than Three Subtypes. G3 (Bethesda) 2016, 6, 4097–4103. [Google Scholar] [CrossRef] [Green Version]
- Hu, Y.; Pan, J.; Shah, P.; Ao, M.; Thomas, S.N.; Liu, Y.; Chen, L.; Schnaubelt, M.; Clark, D.J.; Rodriguez, H.; et al. Integrated Proteomic and Glycoproteomic Characterization of Human High-Grade Serous Ovarian Carcinoma. Cell Rep. 2020, 33, 108276. [Google Scholar] [CrossRef]
- Tong, M.; Yu, C.; Zhan, D.; Zhang, M.; Zhen, B.; Zhu, W.; Wang, Y.; Wu, C.; He, F.; Qin, J.; et al. Molecular subtyping of cancer and nomination of kinase candidates for inhibition with phosphoproteomics: Reanalysis of CPTAC ovarian cancer. EBioMedicine 2019, 40, 305–317. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, K.; Tsunoda, T.; Shigemizu, D.; Fujiwara, H.; Hatae, M.; Fujiwara, H.; Masuzaki, H.; Katabuchi, H.; Kawakami, Y.; Okamoto, A.; et al. High-risk ovarian cancer based on 126-gene expression signature is uniquely characterized by downregulation of antigen presentation pathway. Clin. Cancer Res. 2012, 18, 1374–1385. [Google Scholar] [CrossRef] [Green Version]
- Perets, R.; Drapkin, R. It’s Totally Tubular....Riding The New Wave of Ovarian Cancer Research. Cancer Res. 2016, 76, 10–17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karst, A.M.; Drapkin, R. Ovarian cancer pathogenesis: A model in evolution. J. Oncol. 2010, 2010, 932371. [Google Scholar] [CrossRef] [PubMed]
- Jarboe, E.; Folkins, A.; Nucci, M.R.; Kindelberger, D.; Drapkin, R.; Miron, A.; Lee, Y.; Crum, C.P. Serous carcinogenesis in the fallopian tube: A descriptive classification. Int. J. Gynecol. Pathol. 2008, 27, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Engqvist, H.; Parris, T.Z.; Biermann, J.; Ronnerman, E.W.; Larsson, P.; Sundfeldt, K.; Kovacs, A.; Karlsson, P.; Helou, K. Integrative genomics approach identifies molecular features associated with early-stage ovarian carcinoma histotypes. Sci. Rep. 2020, 10, 7946. [Google Scholar] [CrossRef]
- Tsang, T.; Wei, W.; Itamochi, H.; Tambouret, R.; Birrer, M. Integrated genomic analysis of clear cell ovarian cancers identified PRKCI as a potential therapeutic target. Oncotarget 2017, 8, 96482–96495. [Google Scholar] [CrossRef]
- Rehmani, H.; Li, Y.; Li, T.; Padia, R.; Calbay, O.; Jin, L.; Chen, H.; Huang, S. Addiction to protein kinase Ci due to PRKCI gene amplification can be exploited for an aptamer-based targeted therapy in ovarian cancer. Signal Transduct. Target. Ther. 2020, 5, 140. [Google Scholar] [CrossRef]
- Kurimchak, A.M.; Herrera-Montavez, C.; Brown, J.; Johnson, K.J.; Sodi, V.; Srivastava, N.; Kumar, V.; Deihimi, S.; O’Brien, S.; Peri, S.; et al. Functional proteomics interrogation of the kinome identifies MRCKA as a therapeutic target in high-grade serous ovarian carcinoma. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef]
- Coscia, F.; Lengyel, E.; Duraiswamy, J.; Ashcroft, B.; Bassani-Sternberg, M.; Wierer, M.; Johnson, A.; Wroblewski, K.; Montag, A.; Yamada, S.D.; et al. Multi-level Proteomics Identifies CT45 as a Chemosensitivity Mediator and Immunotherapy Target in Ovarian Cancer. Cell 2018, 175, 159–170.e116. [Google Scholar] [CrossRef] [Green Version]
- Hoadley, K.A.; Yau, C.; Wolf, D.M.; Cherniack, A.D.; Tamborero, D.; Ng, S.; Leiserson, M.D.M.; Niu, B.; McLellan, M.D.; Uzunangelov, V.; et al. Multiplatform analysis of 12 cancer types reveals molecular classification within and across tissues of origin. Cell 2014, 158, 929–944. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Bailey, M.H.; Porta-Pardo, E.; Thorsson, V.; Colaprico, A.; Bertrand, D.; Gibbs, D.L.; Weerasinghe, A.; Huang, K.L.; Tokheim, C.; et al. Perspective on Oncogenic Processes at the End of the Beginning of Cancer Genomics. Cell 2018, 173, 305–320.e310. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.C.; Birkbak, N.J.; Culhane, A.C.; Drapkin, R.; Fatima, A.; Tian, R.; Schwede, M.; Alsop, K.; Daniels, K.E.; Piao, H.; et al. Profiles of genomic instability in high-grade serous ovarian cancer predict treatment outcome. Clin. Cancer Res. 2012, 18, 5806–5815. [Google Scholar] [CrossRef] [Green Version]
- Wang, Z.C.; Lin, M.; Wei, L.J.; Li, C.; Miron, A.; Lodeiro, G.; Harris, L.; Ramaswamy, S.; Tanenbaum, D.M.; Meyerson, M.; et al. Loss of heterozygosity and its correlation with expression profiles in subclasses of invasive breast cancers. Cancer Res. 2004, 64, 64–71. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gorringe, K.L.; Jacobs, S.; Thompson, E.R.; Sridhar, A.; Qiu, W.; Choong, D.Y.; Campbell, I.G. High-resolution single nucleotide polymorphism array analysis of epithelial ovarian cancer reveals numerous microdeletions and amplifications. Clin. Cancer Res. 2007, 13, 4731–4739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takaya, H.; Nakai, H.; Takamatsu, S.; Mandai, M.; Matsumura, N. Homologous recombination deficiency status-based classification of high-grade serous ovarian carcinoma. Sci. Rep. 2020, 10, 2757. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mehta, G.A.; Parker, J.S.; Silva, G.O.; Hoadley, K.A.; Perou, C.M.; Gatza, M.L. Amplification of SOX4 promotes PI3K/Akt signaling in human breast cancer. Breast Cancer Res. Treat. 2017, 162, 439–450. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kwok-Shing Ng, P.; Kucherlapati, M.; Chen, F.; Liu, Y.; Tsang, Y.H.; de Velasco, G.; Jeong, K.J.; Akbani, R.; Hadjipanayis, A.; et al. A Pan-Cancer Proteogenomic Atlas of PI3K/AKT/mTOR Pathway Alterations. Cancer Cell 2017, 31, 820–832.e823. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zervantonakis, I.K.; Iavarone, C.; Chen, H.Y.; Selfors, L.M.; Palakurthi, S.; Liu, J.F.; Drapkin, R.; Matulonis, U.; Leverson, J.D.; Sampath, D.; et al. Systems analysis of apoptotic priming in ovarian cancer identifies vulnerabilities and predictors of drug response. Nat. Commun. 2017, 8, 365. [Google Scholar] [CrossRef] [PubMed]
- Hanrahan, A.J.; Schultz, N.; Westfal, M.L.; Sakr, R.A.; Giri, D.D.; Scarperi, S.; Janakiraman, M.; Olvera, N.; Stevens, E.V.; She, Q.B.; et al. Genomic complexity and AKT dependence in serous ovarian cancer. Cancer Discov. 2012, 2, 56–67. [Google Scholar] [CrossRef] [Green Version]
- Cheaib, B.; Auguste, A.; Leary, A. The PI3K/Akt/mTOR pathway in ovarian cancer: Therapeutic opportunities and challenges. Chin. J. Cancer 2015, 34, 4–16. [Google Scholar] [CrossRef]
- Pascual, J.; Turner, N.C. Targeting the PI3-kinase pathway in triple-negative breast cancer. Ann. Oncol. 2019, 30, 1051–1060. [Google Scholar] [CrossRef] [Green Version]
- Ellis, M.J.; Perou, C.M. The genomic landscape of breast cancer as a therapeutic roadmap. Cancer Discov. 2013, 3, 27–34. [Google Scholar] [CrossRef] [Green Version]
- Marcotte, R.; Brown, K.R.; Suarez, F.; Sayad, A.; Karamboulas, K.; Krzyzanowski, P.M.; Sircoulomb, F.; Medrano, M.; Fedyshyn, Y.; Koh, J.L.Y.; et al. Essential gene profiles in breast, pancreatic, and ovarian cancer cells. Cancer Discov. 2012, 2, 172–189. [Google Scholar] [CrossRef] [Green Version]
- Bailey, M.H.; Tokheim, C.; Porta-Pardo, E.; Sengupta, S.; Bertrand, D.; Weerasinghe, A.; Colaprico, A.; Wendl, M.C.; Kim, J.; Reardon, B.; et al. Comprehensive Characterization of Cancer Driver Genes and Mutations. Cell 2018, 173, 371–385. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kandoth, C.; Schultz, N.; Cherniack, A.D.; Akbani, R.; Liu, Y.; Shen, H.; Robertson, A.G.; Pashtan, I.; Shen, R.; et al.; Cancer Genome Atlas Research Network Integrated genomic characterization of endometrial carcinoma. Nature 2013, 497, 67–73. [Google Scholar] [CrossRef] [Green Version]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M.; Cancer Genome Atlas Research Network. The Cancer Genome Atlas Pan-Cancer analysis project. Nat. Genet. 2013, 45, 1113–1120. [Google Scholar] [CrossRef] [PubMed]
- Jacobsen, A.; Silber, J.; Harinath, G.; Huse, J.T.; Schultz, N.; Sander, C. Analysis of microRNA-target interactions across diverse cancer types. Nat. Struct. Mol. Biol. 2013, 20, 1325–1332. [Google Scholar] [CrossRef] [PubMed]
- Kandoth, C.; McLellan, M.D.; Vandin, F.; Ye, K.; Niu, B.; Lu, C.; Xie, M.; Zhang, Q.; McMichael, J.F.; Wyczalkowski, M.A.; et al. Mutational landscape and significance across 12 major cancer types. Nature 2013, 502, 333–339. [Google Scholar] [CrossRef] [Green Version]
- Reimand, J.; Wagih, O.; Bader, G.D. The mutational landscape of phosphorylation signaling in cancer. Sci. Rep. 2013, 3, 2651. [Google Scholar] [CrossRef] [Green Version]
- Tomczak, K.; Czerwinska, P.; Wiznerowicz, M. The Cancer Genome Atlas (TCGA): An immeasurable source of knowledge. Contemp. Oncol. 2015, 19, A68–A77. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Research Network. Integrated Genomic Characterization of Pancreatic Ductal Adenocarcinoma. Cancer Cell 2017, 32, 185–203.e113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kreike, B.; van Kouwenhove, M.; Horlings, H.; Weigelt, B.; Peterse, H.; Bartelink, H.; van de Vijver, M.J. Gene expression profiling and histopathological characterization of triple-negative/basal-like breast carcinomas. Breast Cancer Res. BCR 2007, 9, R65. [Google Scholar] [CrossRef] [Green Version]
- Thomas, S.N.; Friedrich, B.; Schnaubelt, M.; Chan, D.W.; Zhang, H.; Aebersold, R. Orthogonal Proteomic Platforms and Their Implications for the Stable Classification of High-Grade Serous Ovarian Cancer Subtypes. iScience 2020, 23, 101079. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Z.; Huang, K.; Gu, C.; Zhao, L.; Wang, N.; Wang, X.; Zhao, D.; Zhang, C.; Lu, Y.; Meng, Y. Molecular Subtyping of Serous Ovarian Cancer Based on Multi-omics Data. Sci. Rep. 2016, 6, 26001. [Google Scholar] [CrossRef] [PubMed]
- Bansal, M.; He, J.; Peyton, M.; Kustagi, M.; Iyer, A.; Comb, M.; White, M.; Minna, J.D.; Califano, A. Elucidating synergistic dependencies in lung adenocarcinoma by proteome-wide signaling-network analysis. PLoS ONE 2019, 14, e0208646. [Google Scholar] [CrossRef]
- Barretina, J.; Caponigro, G.; Stransky, N.; Venkatesan, K.; Margolin, A.A.; Kim, S.; Wilson, C.J.; Lehar, J.; Kryukov, G.V.; Sonkin, D.; et al. The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 2012, 483, 603–607. [Google Scholar] [CrossRef]
- Gordon, V.; Banerji, S. Molecular pathways: PI3K pathway targets in triple-negative breast cancers. Clin. Cancer Res. 2013, 19, 3738–3744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jordan, V.C. Tamoxifen as the first targeted long-term adjuvant therapy for breast cancer. Endocr. Relat. Cancer 2014, 21, R235–R246. [Google Scholar] [CrossRef] [Green Version]
- Kedves, A.T.; Gleim, S.; Liang, X.; Bonal, D.M.; Sigoillot, F.; Harbinski, F.; Sanghavi, S.; Benander, C.; George, E.; Gokhale, P.C.; et al. Recurrent ubiquitin B silencing in gynecological cancers establishes dependence on ubiquitin C. J. Clin. Investig. 2017, 127, 4554–4568. [Google Scholar] [CrossRef] [Green Version]
- Natrajan, R.; Wilkerson, P. From integrative genomics to therapeutic targets. Cancer Res. 2013, 73, 3483–3488. [Google Scholar] [CrossRef] [Green Version]
- Oza, A.M.; Tinker, A.V.; Oaknin, A.; Shapira-Frommer, R.; McNeish, I.A.; Swisher, E.M.; Ray-Coquard, I.; Bell-McGuinn, K.; Coleman, R.L.; O’Malley, D.M.; et al. Antitumor activity and safety of the PARP inhibitor rucaparib in patients with high-grade ovarian carcinoma and a germline or somatic BRCA1 or BRCA2 mutation: Integrated analysis of data from Study 10 and ARIEL2. Gynecol. Oncol. 2017, 147, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Park, J.T.; Chen, X.; Trope, C.G.; Davidson, B.; Shih Ie, M.; Wang, T.L. Notch3 overexpression is related to the recurrence of ovarian cancer and confers resistance to carboplatin. Am. J. Pathol. 2010, 177, 1087–1094. [Google Scholar] [CrossRef]
- Patel, N.; Weekes, D.; Drosopoulos, K.; Gazinska, P.; Noel, E.; Rashid, M.; Mirza, H.; Quist, J.; Braso-Maristany, F.; Mathew, S.; et al. Integrated genomics and functional validation identifies malignant cell specific dependencies in triple negative breast cancer. Nat. Commun. 2018, 9, 1044. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.T.; Kim, D.W.; Nakagawa, K.; Seto, T.; Crino, L.; Ahn, M.J.; De Pas, T.; Besse, B.; Solomon, B.J.; Blackhall, F.; et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N. Engl. J. Med. 2013, 368, 2385–2394. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsherniak, A.; Vazquez, F.; Montgomery, P.G.; Weir, B.A.; Kryukov, G.; Cowley, G.S.; Gill, S.; Harrington, W.F.; Pantel, S.; Krill-Burger, J.M.; et al. Defining a Cancer Dependency Map. Cell 2017, 170, 564–576.e516. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Uzilov, A.V.; Ding, W.; Fink, M.Y.; Antipin, Y.; Brohl, A.S.; Davis, C.; Lau, C.Y.; Pandya, C.; Shah, H.; Kasai, Y.; et al. Development and clinical application of an integrative genomic approach to personalized cancer therapy. Genome Med. 2016, 8, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, J.; Mouradov, D.; Wang, X.; Jorissen, R.N.; Chambers, M.C.; Zimmerman, L.J.; Vasaikar, S.; Love, C.G.; Li, S.; Lowes, K.; et al. Colorectal Cancer Cell Line Proteomes Are Representative of Primary Tumors and Predict Drug Sensitivity. Gastroenterology 2017, 153, 1082–1095. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; McLeod, H.L.; Weinshilboum, R.M. Genomics and drug response. N. Engl. J. Med. 2011, 364, 1144–1153. [Google Scholar] [CrossRef] [Green Version]
- Xie, H.; Wang, W.; Sun, F.; Deng, K.; Lu, X.; Liu, H.; Zhao, W.; Zhang, Y.; Zhou, X.; Li, K.; et al. Proteomics analysis to reveal biological pathways and predictive proteins in the survival of high-grade serous ovarian cancer. Sci. Rep. 2017, 7, 9896. [Google Scholar] [CrossRef] [Green Version]
- Yang, J.Y.; Yoshihara, K.; Tanaka, K.; Hatae, M.; Masuzaki, H.; Itamochi, H.; Takano, M.; Ushijima, K.; Tanyi, J.L.; Cancer Genome Atlas Research Network; et al. Predicting time to ovarian carcinoma recurrence using protein markers. J. Clin. Investig. 2013, 123, 3740–3750. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yarden, Y.; Pines, G. The ERBB network: At last, cancer therapy meets systems biology. Nat. Rev. Cancer 2012, 12, 553–563. [Google Scholar] [CrossRef]
- Wang, W.; Baladandayuthapani, V.; Morris, J.S.; Broom, B.M.; Manyam, G.; Do, K.A. iBAG: Integrative Bayesian analysis of high-dimensional multiplatform genomics data. Bioinformatics 2013, 29, 149–159. [Google Scholar] [CrossRef]
- Alvarez, M.J.; Shen, Y.; Giorgi, F.M.; Lachmann, A.; Ding, B.B.; Ye, B.H.; Califano, A. Functional characterization of somatic mutations in cancer using network-based inference of protein activity. Nat. Genet. 2016, 48, 838–847. [Google Scholar] [CrossRef]
- Paull, E.O.; Carlin, D.E.; Niepel, M.; Sorger, P.K.; Haussler, D.; Stuart, J.M. Discovering causal pathways linking genomic events to transcriptional states using Tied Diffusion Through Interacting Events (TieDIE). Bioinformatics 2013, 29, 2757–2764. [Google Scholar] [CrossRef] [Green Version]
- Drake, J.M.; Paull, E.O.; Graham, N.A.; Lee, J.K.; Smith, B.A.; Titz, B.; Stoyanova, T.; Faltermeier, C.M.; Uzunangelov, V.; Carlin, D.E.; et al. Phosphoproteome Integration Reveals Patient-Specific Networks in Prostate Cancer. Cell 2016, 166, 1041–1054. [Google Scholar] [CrossRef] [Green Version]
- Lim, B.; Woodward, W.A.; Wang, X.; Reuben, J.M.; Ueno, N.T. Inflammatory breast cancer biology: The tumour microenvironment is key. Nat. Rev. Cancer 2018, 18, 485–499. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Yang, Y.; Yang, J.; Zhao, X.; Wei, X. Tumor Microenvironment in Ovarian Cancer: Function and Therapeutic Strategy. Front. Cell Dev. Biol. 2020, 8, 758. [Google Scholar] [CrossRef]
- Yoshihara, K.; Shahmoradgoli, M.; Martinez, E.; Vegesna, R.; Kim, H.; Torres-Garcia, W.; Trevino, V.; Shen, H.; Laird, P.W.; Levine, D.A.; et al. Inferring tumour purity and stromal and immune cell admixture from expression data. Nat. Commun. 2013, 4, 2612. [Google Scholar] [CrossRef] [PubMed]
- Dai, Z.; Gu, X.Y.; Xiang, S.Y.; Gong, D.D.; Man, C.F.; Fan, Y. Research and application of single-cell sequencing in tumor heterogeneity and drug resistance of circulating tumor cells. Biomark. Res. 2020, 8, 60. [Google Scholar] [CrossRef] [PubMed]
- Carter, B.; Zhao, K. The epigenetic basis of cellular heterogeneity. Nat. Rev. Genet. 2020. [Google Scholar] [CrossRef] [PubMed]
- Nam, A.S.; Chaligne, R.; Landau, D.A. Integrating genetic and non-genetic determinants of cancer evolution by single-cell multi-omics. Nat. Rev. Genet. 2021, 22, 3–18. [Google Scholar] [CrossRef]
- Wagner, D.E.; Klein, A.M. Lineage tracing meets single-cell omics: Opportunities and challenges. Nat. Rev. Genet. 2020, 21, 410–427. [Google Scholar] [CrossRef]
- Stuart, T.; Satija, R. Integrative single-cell analysis. Nat. Rev. Genet. 2019, 20, 257–272. [Google Scholar] [CrossRef] [PubMed]
- Kiselev, V.Y.; Andrews, T.S.; Hemberg, M. Challenges in unsupervised clustering of single-cell RNA-seq data. Nat. Rev. Genet. 2019, 20, 273–282. [Google Scholar] [CrossRef]
- Prakadan, S.M.; Shalek, A.K.; Weitz, D.A. Scaling by shrinking: Empowering single-cell ‘omics’ with microfluidic devices. Nat. Rev. Genet. 2017, 18, 345–361. [Google Scholar] [CrossRef] [PubMed]
- Woodworth, M.B.; Girskis, K.M.; Walsh, C.A. Building a lineage from single cells: Genetic techniques for cell lineage tracking. Nat. Rev. Genet. 2017, 18, 230–244. [Google Scholar] [CrossRef] [PubMed]
- Gawad, C.; Koh, W.; Quake, S.R. Single-cell genome sequencing: Current state of the science. Nat. Rev. Genet. 2016, 17, 175–188. [Google Scholar] [CrossRef]
- Schwartzman, O.; Tanay, A. Single-cell epigenomics: Techniques and emerging applications. Nat. Rev. Genet. 2015, 16, 716–726. [Google Scholar] [CrossRef]
- Stegle, O.; Teichmann, S.A.; Marioni, J.C. Computational and analytical challenges in single-cell transcriptomics. Nat. Rev. Genet. 2015, 16, 133–145. [Google Scholar] [CrossRef]
- Liberali, P.; Snijder, B.; Pelkmans, L. Single-cell and multivariate approaches in genetic perturbation screens. Nat. Rev. Genet. 2015, 16, 18–32. [Google Scholar] [CrossRef]
- Chuah, S.; Chew, V. High-dimensional immune-profiling in cancer: Implications for immunotherapy. J. Immunother. Cancer 2020, 8, e000363. [Google Scholar] [CrossRef]
- Chattopadhyay, P.K.; Gierahn, T.M.; Roederer, M.; Love, J.C. Single-cell technologies for monitoring immune systems. Nat. Immunol. 2014, 15, 128–135. [Google Scholar] [CrossRef] [PubMed]
- Gibellini, L.; De Biasi, S.; Porta, C.; Lo Tartaro, D.; Depenni, R.; Pellacani, G.; Sabbatini, R.; Cossarizza, A. Single-Cell Approaches to Profile the Response to Immune Checkpoint Inhibitors. Front. Immunol. 2020, 11, 490. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Landhuis, E. Single-cell approaches to immune profiling. Nature 2018, 557, 595–597. [Google Scholar] [CrossRef] [PubMed]
- Jaitin, D.A.; Kenigsberg, E.; Keren-Shaul, H.; Elefant, N.; Paul, F.; Zaretsky, I.; Mildner, A.; Cohen, N.; Jung, S.; Tanay, A.; et al. Massively parallel single-cell RNA-seq for marker-free decomposition of tissues into cell types. Science 2014, 343, 776–779. [Google Scholar] [CrossRef] [PubMed]
- Villani, A.C.; Satija, R.; Reynolds, G.; Sarkizova, S.; Shekhar, K.; Fletcher, J.; Griesbeck, M.; Butler, A.; Zheng, S.; Lazo, S.; et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science 2017, 356. [Google Scholar] [CrossRef] [Green Version]
- Azizi, E.; Carr, A.J.; Plitas, G.; Cornish, A.E.; Konopacki, C.; Prabhakaran, S.; Nainys, J.; Wu, K.; Kiseliovas, V.; Setty, M.; et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell 2018, 174, 1293–1308.e1236. [Google Scholar] [CrossRef] [Green Version]
- Savas, P.; Virassamy, B.; Ye, C.; Salim, A.; Mintoff, C.P.; Caramia, F.; Salgado, R.; Byrne, D.J.; Teo, Z.L.; Dushyanthen, S.; et al. Single-cell profiling of breast cancer T cells reveals a tissue-resident memory subset associated with improved prognosis. Nat. Med. 2018, 24, 986–993. [Google Scholar] [CrossRef]
- Lim, B.; Lin, Y.; Navin, N. Advancing Cancer Research and Medicine with Single-Cell Genomics. Cancer Cell 2020, 37, 456–470. [Google Scholar] [CrossRef]
- Wang, Q.; Guldner, I.H.; Golomb, S.M.; Sun, L.; Harris, J.A.; Lu, X.; Zhang, S. Single-cell profiling guided combinatorial immunotherapy for fast-evolving CDK4/6 inhibitor-resistant HER2-positive breast cancer. Nat. Commun. 2019, 10, 3817. [Google Scholar] [CrossRef] [Green Version]
- Alshetaiwi, H.; Pervolarakis, N.; McIntyre, L.L.; Ma, D.; Nguyen, Q.; Rath, J.A.; Nee, K.; Hernandez, G.; Evans, K.; Torosian, L.; et al. Defining the emergence of myeloid-derived suppressor cells in breast cancer using single-cell transcriptomics. Sci. Immunol. 2020, 5. [Google Scholar] [CrossRef]
- Wan, C.; Keany, M.P.; Dong, H.; Al-Alem, L.F.; Pandya, U.M.; Lazo, S.; Boehnke, K.; Lynch, K.N.; Xu, R.; Zarrella, D.T.; et al. Enhanced efficacy of simultaneous PD-1 and PD-L1 immune checkpoint blockade in high grade serous ovarian cancer. Cancer Res. 2020. [Google Scholar] [CrossRef]
- Bartoschek, M.; Oskolkov, N.; Bocci, M.; Lövrot, J.; Larsson, C.; Sommarin, M.; Madsen, C.D.; Lindgren, D.; Pekar, G.; Karlsson, G.; et al. Spatially and functionally distinct subclasses of breast cancer-associated fibroblasts revealed by single cell RNA sequencing. Nat. Commun. 2018, 9, 5150. [Google Scholar] [CrossRef] [Green Version]
- Jimenez-Sanchez, A.; Cybulska, P.; Mager, K.L.; Koplev, S.; Cast, O.; Couturier, D.L.; Memon, D.; Selenica, P.; Nikolovski, I.; Mazaheri, Y.; et al. Unraveling tumor-immune heterogeneity in advanced ovarian cancer uncovers immunogenic effect of chemotherapy. Nat. Genet. 2020, 52, 582–593. [Google Scholar] [CrossRef]
- Zhu, Y.; Zhang, Z.; Jiang, Z.; Liu, Y.; Zhou, J. CD38 Predicts Favorable Prognosis by Enhancing Immune Infiltration and Antitumor Immunity in the Epithelial Ovarian Cancer Microenvironment. Front. Genet. 2020, 11, 369. [Google Scholar] [CrossRef]
- Lyons, Y.A.; Wu, S.Y.; Overwijk, W.W.; Baggerly, K.A.; Sood, A.K. Immune cell profiling in cancer: Molecular approaches to cell-specific identification. NPJ Precis. Oncol. 2017, 1, 26. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gonzalez, H.; Hagerling, C.; Werb, Z. Roles of the immune system in cancer: From tumor initiation to metastatic progression. Genes Dev. 2018, 32, 1267–1284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhu, L.; Narloch, J.L.; Onkar, S.; Joy, M.; Broadwater, G.; Luedke, C.; Hall, A.; Kim, R.; Pogue-Geile, K.; Sammons, S.; et al. Metastatic breast cancers have reduced immune cell recruitment but harbor increased macrophages relative to their matched primary tumors. J. Immunother. Cancer 2019, 7, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Segovia-Mendoza, M.; Morales-Montor, J. Immune Tumor Microenvironment in Breast Cancer and the Participation of Estrogen and Its Receptors in Cancer Physiopathology. Front. Immunol. 2019, 10, 348. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tekpli, X.; Lien, T.; Rossevold, A.H.; Nebdal, D.; Borgen, E.; Ohnstad, H.O.; Kyte, J.A.; Vallon-Christersson, J.; Fongaard, M.; Due, E.U.; et al. An independent poor-prognosis subtype of breast cancer defined by a distinct tumor immune microenvironment. Nat. Commun. 2019, 10, 5499. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wagner, J.; Rapsomaniki, M.A.; Chevrier, S.; Anzeneder, T.; Langwieder, C.; Dykgers, A.; Rees, M.; Ramaswamy, A.; Muenst, S.; Soysal, S.D.; et al. A Single-Cell Atlas of the Tumor and Immune Ecosystem of Human Breast Cancer. Cell 2019, 177, 1330–1345.e1318. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suva, M.L.; Tirosh, I. Single-Cell RNA Sequencing in Cancer: Lessons Learned and Emerging Challenges. Mol. Cell 2019, 75, 7–12. [Google Scholar] [CrossRef] [PubMed]
- Ding, S.; Chen, X.; Shen, K. Single-cell RNA sequencing in breast cancer: Understanding tumor heterogeneity and paving roads to individualized therapy. Cancer Commun. 2020, 40, 329–344. [Google Scholar] [CrossRef]
- Fan, J.; Slowikowski, K.; Zhang, F. Single-cell transcriptomics in cancer: Computational challenges and opportunities. Exp. Mol. Med. 2020, 52, 1452–1465. [Google Scholar] [CrossRef]
- Hoffman, J.A.; Papas, B.N.; Trotter, K.W.; Archer, T.K. Single-cell RNA sequencing reveals a heterogeneous response to Glucocorticoids in breast cancer cells. Commun. Biol. 2020, 3, 126. [Google Scholar] [CrossRef]
- Fan, J.; Lee, H.O.; Lee, S.; Ryu, D.E.; Lee, S.; Xue, C.; Kim, S.J.; Kim, K.; Barkas, N.; Park, P.J.; et al. Linking transcriptional and genetic tumor heterogeneity through allele analysis of single-cell RNA-seq data. Genome Res. 2018, 28, 1217–1227. [Google Scholar] [CrossRef]
- Ren, X.; Kang, B.; Zhang, Z. Understanding tumor ecosystems by single-cell sequencing: Promises and limitations. Genome Biol. 2018, 19, 211. [Google Scholar] [CrossRef]
- Chung, W.; Eum, H.H.; Lee, H.O.; Lee, K.M.; Lee, H.B.; Kim, K.T.; Ryu, H.S.; Kim, S.; Lee, J.E.; Park, Y.H.; et al. Single-cell RNA-seq enables comprehensive tumour and immune cell profiling in primary breast cancer. Nat. Commun. 2017, 8, 15081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karaayvaz, M.; Cristea, S.; Gillespie, S.M.; Patel, A.P.; Mylvaganam, R.; Luo, C.C.; Specht, M.C.; Bernstein, B.E.; Michor, F.; Ellisen, L.W. Unravelling subclonal heterogeneity and aggressive disease states in TNBC through single-cell RNA-seq. Nat. Commun. 2018, 9, 3588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hong, S.P.; Chan, T.E.; Lombardo, Y.; Corleone, G.; Rotmensz, N.; Bravaccini, S.; Rocca, A.; Pruneri, G.; McEwen, K.R.; Coombes, R.C.; et al. Single-cell transcriptomics reveals multi-step adaptations to endocrine therapy. Nat. Commun. 2019, 10, 3840. [Google Scholar] [CrossRef] [Green Version]
- Geistlinger, L.; Oh, S.; Ramos, M.; Schiffer, L.; LaRue, R.S.; Henzler, C.M.; Munro, S.A.; Daughters, C.; Nelson, A.C.; Winterhoff, B.J.; et al. Multiomic Analysis of Subtype Evolution and Heterogeneity in High-Grade Serous Ovarian Carcinoma. Cancer Res. 2020, 80, 4335–4345. [Google Scholar] [CrossRef]
- Lourenco, A.R.; Ban, Y.; Crowley, M.J.; Lee, S.B.; Ramchandani, D.; Du, W.; Elemento, O.; George, J.T.; Jolly, M.K.; Levine, H.; et al. Differential Contributions of Pre- and Post-EMT Tumor Cells in Breast Cancer Metastasis. Cancer Res. 2020, 80, 163–169. [Google Scholar] [CrossRef] [Green Version]
- Baslan, T.; Kendall, J.; Volyanskyy, K.; McNamara, K.; Cox, H.; D’Italia, S.; Ambrosio, F.; Riggs, M.; Rodgers, L.; Leotta, A.; et al. Novel insights into breast cancer copy number genetic heterogeneity revealed by single-cell genome sequencing. Elife 2020, 9. [Google Scholar] [CrossRef]
- Almendro, V.; Cheng, Y.K.; Randles, A.; Itzkovitz, S.; Marusyk, A.; Ametller, E.; Gonzalez-Farre, X.; Munoz, M.; Russnes, H.G.; Helland, A.; et al. Inference of tumor evolution during chemotherapy by computational modeling and in situ analysis of genetic and phenotypic cellular diversity. Cell Rep. 2014, 6, 514–527. [Google Scholar] [CrossRef] [Green Version]
- Ding, L.; Ley, T.J.; Larson, D.E.; Miller, C.A.; Koboldt, D.C.; Welch, J.S.; Ritchey, J.K.; Young, M.A.; Lamprecht, T.; McLellan, M.D.; et al. Clonal evolution in relapsed acute myeloid leukaemia revealed by whole-genome sequencing. Nature 2012, 481, 506–510. [Google Scholar] [CrossRef]
- Kulkarni, A.; Al-Hraishawi, H.; Simhadri, S.; Hirshfield, K.M.; Chen, S.; Pine, S.; Jeyamohan, C.; Sokol, L.; Ali, S.; Teo, M.L.; et al. BRAF Fusion as a Novel Mechanism of Acquired Resistance to Vemurafenib in BRAF(V600E) Mutant Melanoma. Clin. Cancer Res. 2017, 23, 5631–5638. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, V.D.; Samusik, N.; Chen, T.J.; Savig, E.S.; Aghaeepour, N.; Quigley, D.A.; Huang, Y.W.; Giangarra, V.; Borowsky, A.D.; Hubbard, N.E.; et al. Commonly Occurring Cell Subsets in High-Grade Serous Ovarian Tumors Identified by Single-Cell Mass Cytometry. Cell Rep. 2018, 22, 1875–1888. [Google Scholar] [CrossRef] [Green Version]
- Kim, C.; Gao, R.; Sei, E.; Brandt, R.; Hartman, J.; Hatschek, T.; Crosetto, N.; Foukakis, T.; Navin, N.E. Chemoresistance Evolution in Triple-Negative Breast Cancer Delineated by Single-Cell Sequencing. Cell 2018, 173, 879–893.e813. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mucino-Olmos, E.A.; Vazquez-Jimenez, A.; Avila-Ponce de Leon, U.; Matadamas-Guzman, M.; Maldonado, V.; Lopez-Santaella, T.; Hernandez-Hernandez, A.; Resendis-Antonio, O. Unveiling functional heterogeneity in breast cancer multicellular tumor spheroids through single-cell RNA-seq. Sci. Rep. 2020, 10, 12728. [Google Scholar] [CrossRef] [PubMed]
- Yeo, S.K.; Zhu, X.; Okamoto, T.; Hao, M.; Wang, C.; Lu, P.; Lu, L.J.; Guan, J.L. Single-cell RNA-sequencing reveals distinct patterns of cell state heterogeneity in mouse models of breast cancer. Elife 2020, 9. [Google Scholar] [CrossRef] [PubMed]
- Zhang, A.W.; O’Flanagan, C.; Chavez, E.A.; Lim, J.L.P.; Ceglia, N.; McPherson, A.; Wiens, M.; Walters, P.; Chan, T.; Hewitson, B.; et al. Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling. Nat. Methods 2019, 16, 1007–1015. [Google Scholar] [CrossRef]
- Hu, Z.; Artibani, M.; Alsaadi, A.; Wietek, N.; Morotti, M.; Shi, T.; Zhong, Z.; Santana Gonzalez, L.; El-Sahhar, S.; KaramiNejadRanjbar, M.; et al. The Repertoire of Serous Ovarian Cancer Non-genetic Heterogeneity Revealed by Single-Cell Sequencing of Normal Fallopian Tube Epithelial Cells. Cancer Cell 2020, 37, 226–242.e227. [Google Scholar] [CrossRef]
- Marcotte, R.; Sayad, A.; Brown, K.R.; Sanchez-Garcia, F.; Reimand, J.; Haider, M.; Virtanen, C.; Bradner, J.E.; Bader, G.D.; Mills, G.B.; et al. Functional Genomic Landscape of Human Breast Cancer Drivers, Vulnerabilities, and Resistance. Cell 2016, 164, 293–309. [Google Scholar] [CrossRef] [Green Version]
- Cheung, H.W.; Cowley, G.S.; Weir, B.A.; Boehm, J.S.; Rusin, S.; Scott, J.A.; East, A.; Ali, L.D.; Lizotte, P.H.; Wong, T.C.; et al. Systematic investigation of genetic vulnerabilities across cancer cell lines reveals lineage-specific dependencies in ovarian cancer. Proc. Natl. Acad. Sci. USA 2011, 108, 12372–12377. [Google Scholar] [CrossRef] [Green Version]
- Chen, M.M.; Li, J.; Mills, G.B.; Liang, H. Predicting Cancer Cell Line Dependencies from the Protein Expression Data of Reverse-Phase Protein Arrays. JCO Clin. Cancer Inform. 2020, 4, 357–366. [Google Scholar] [CrossRef]
- Lord, C.J.; Quinn, N.; Ryan, C.J. Integrative analysis of large-scale loss-of-function screens identifies robust cancer-associated genetic interactions. Elife 2020, 9. [Google Scholar] [CrossRef]
- Price, C.; Gill, S.; Ho, Z.V.; Davidson, S.M.; Merkel, E.; McFarland, J.M.; Leung, L.; Tang, A.; Kost-Alimova, M.; Tsherniak, A.; et al. Genome-Wide Interrogation of Human Cancers Identifies EGLN1 Dependency in Clear Cell Ovarian Cancers. Cancer Res. 2019, 79, 2564–2579. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dempster, J.M.; Pacini, C.; Pantel, S.; Behan, F.M.; Green, T.; Krill-Burger, J.; Beaver, C.M.; Younger, S.T.; Zhivich, V.; Najgebauer, H.; et al. Agreement between two large pan-cancer CRISPR-Cas9 gene dependency data sets. Nat. Commun. 2019, 10, 5817. [Google Scholar] [CrossRef] [Green Version]
- Hofree, M.; Shen, J.P.; Carter, H.; Gross, A.; Ideker, T. Network-based stratification of tumor mutations. Nat. Methods 2013, 10, 1108–1115. [Google Scholar] [CrossRef]
- Tamborero, D.; Gonzalez-Perez, A.; Perez-Llamas, C.; Deu-Pons, J.; Kandoth, C.; Reimand, J.; Lawrence, M.S.; Getz, G.; Bader, G.D.; Ding, L.; et al. Comprehensive identification of mutational cancer driver genes across 12 tumor types. Sci. Rep. 2013, 3, 2650. [Google Scholar] [CrossRef]
- Alexandrov, L.B.; Nik-Zainal, S.; Wedge, D.C.; Aparicio, S.A.; Behjati, S.; Biankin, A.V.; Bignell, G.R.; Bolli, N.; Borg, A.; Borresen-Dale, A.L.; et al. Signatures of mutational processes in human cancer. Nature 2013, 500, 415–421. [Google Scholar] [CrossRef] [Green Version]
- Petljak, M.; Alexandrov, L.B.; Brammeld, J.S.; Price, S.; Wedge, D.C.; Grossmann, S.; Dawson, K.J.; Ju, Y.S.; Iorio, F.; Tubio, J.M.C.; et al. Characterizing Mutational Signatures in Human Cancer Cell Lines Reveals Episodic APOBEC Mutagenesis. Cell 2019, 176, 1282–1294.e1220. [Google Scholar] [CrossRef] [Green Version]
- Ha, M.J.; Banerjee, S.; Akbani, R.; Liang, H.; Mills, G.B.; Do, K.A.; Baladandayuthapani, V. Personalized Integrated Network Modeling of the Cancer Proteome Atlas. Sci. Rep. 2018, 8, 14924. [Google Scholar] [CrossRef]
- Chiu, A.M.; Mitra, M.; Boymoushakian, L.; Coller, H.A. Integrative analysis of the inter-tumoral heterogeneity of triple-negative breast cancer. Sci. Rep. 2018, 8, 11807. [Google Scholar] [CrossRef] [PubMed]
- Vaske, C.J.; Benz, S.C.; Sanborn, J.Z.; Earl, D.; Szeto, C.; Zhu, J.; Haussler, D.; Stuart, J.M. Inference of patient-specific pathway activities from multi-dimensional cancer genomics data using PARADIGM. Bioinformatics 2010, 26, i237–i245. [Google Scholar] [CrossRef] [PubMed]
- Sanchez-Vega, F.; Mina, M.; Armenia, J.; Chatila, W.K.; Luna, A.; La, K.C.; Dimitriadoy, S.; Liu, D.L.; Kantheti, H.S.; Saghafinia, S.; et al. Oncogenic Signaling Pathways in The Cancer Genome Atlas. Cell 2018, 173, 321–337.e310. [Google Scholar] [CrossRef] [Green Version]
- Sicklick, J.K.; Kato, S.; Okamura, R.; Schwaederle, M.; Hahn, M.E.; Williams, C.B.; De, P.; Krie, A.; Piccioni, D.E.; Miller, V.A.; et al. Molecular profiling of cancer patients enables personalized combination therapy: The I-PREDICT study. Nat. Med. 2019, 25, 744–750. [Google Scholar] [CrossRef] [PubMed]
- Woodhouse, R.; Li, M.; Hughes, J.; Delfosse, D.; Skoletsky, J.; Ma, P.; Meng, W.; Dewal, N.; Milbury, C.; Clark, T.; et al. Clinical and analytical validation of FoundationOne Liquid CDx, a novel 324-Gene cfDNA-based comprehensive genomic profiling assay for cancers of solid tumor origin. PLoS ONE 2020, 15, e0237802. [Google Scholar] [CrossRef]
- Whitwell, H.J.; Worthington, J.; Blyuss, O.; Gentry-Maharaj, A.; Ryan, A.; Gunu, R.; Kalsi, J.; Menon, U.; Jacobs, I.; Zaikin, A.; et al. Improved early detection of ovarian cancer using longitudinal multimarker models. Br. J. Cancer 2020, 122, 847–856. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Russell, M.R.; Graham, C.; D’Amato, A.; Gentry-Maharaj, A.; Ryan, A.; Kalsi, J.K.; Whetton, A.D.; Menon, U.; Jacobs, I.; Graham, R.L.J. Diagnosis of epithelial ovarian cancer using a combined protein biomarker panel. Br. J. Cancer 2019, 121, 483–489. [Google Scholar] [CrossRef] [Green Version]
- Russell, M.R.; Graham, C.; D’Amato, A.; Gentry-Maharaj, A.; Ryan, A.; Kalsi, J.K.; Ainley, C.; Whetton, A.D.; Menon, U.; Jacobs, I.; et al. A combined biomarker panel shows improved sensitivity for the early detection of ovarian cancer allowing the identification of the most aggressive type II tumours. Br. J. Cancer 2017, 117, 666–674. [Google Scholar] [CrossRef]
- Dochez, V.; Caillon, H.; Vaucel, E.; Dimet, J.; Winer, N.; Ducarme, G. Biomarkers and algorithms for diagnosis of ovarian cancer: CA125, HE4, RMI and ROMA, a review. J. Ovarian Res. 2019, 12, 28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fong, P.C.; Boss, D.S.; Yap, T.A.; Tutt, A.; Wu, P.; Mergui-Roelvink, M.; Mortimer, P.; Swaisland, H.; Lau, A.; O’Connor, M.J.; et al. Inhibition of poly(ADP-ribose) polymerase in tumors from BRCA mutation carriers. N. Engl. J. Med. 2009, 361, 123–134. [Google Scholar] [CrossRef] [Green Version]
- Moore, R.G.; Miller, M.C.; Disilvestro, P.; Landrum, L.M.; Gajewski, W.; Ball, J.J.; Skates, S.J. Evaluation of the diagnostic accuracy of the risk of ovarian malignancy algorithm in women with a pelvic mass. Obstet. Gynecol. 2011, 118, 280–288. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bristow, R.E.; Smith, A.; Zhang, Z.; Chan, D.W.; Crutcher, G.; Fung, E.T.; Munroe, D.G. Ovarian malignancy risk stratification of the adnexal mass using a multivariate index assay. Gynecol. Oncol. 2013, 128, 252–259. [Google Scholar] [CrossRef] [PubMed]
- Ueland, F.R.; Desimone, C.P.; Seamon, L.G.; Miller, R.A.; Goodrich, S.; Podzielinski, I.; Sokoll, L.; Smith, A.; van Nagell, J.R., Jr.; Zhang, Z. Effectiveness of a multivariate index assay in the preoperative assessment of ovarian tumors. Obstet. Gynecol. 2011, 117, 1289–1297. [Google Scholar] [CrossRef]
- Sparano, J.A.; Gray, R.J.; Makower, D.F.; Pritchard, K.I.; Albain, K.S.; Hayes, D.F.; Geyer, C.E., Jr.; Dees, E.C.; Goetz, M.P.; Olson, J.A., Jr.; et al. Adjuvant Chemotherapy Guided by a 21-Gene Expression Assay in Breast Cancer. N. Engl. J. Med. 2018, 379, 111–121. [Google Scholar] [CrossRef] [Green Version]
- Dubsky, P.; Brase, J.C.; Jakesz, R.; Rudas, M.; Singer, C.F.; Greil, R.; Dietze, O.; Luisser, I.; Klug, E.; Sedivy, R.; et al. The EndoPredict score provides prognostic information on late distant metastases in ER+/HER2- breast cancer patients. Br. J. Cancer 2013, 109, 2959–2964. [Google Scholar] [CrossRef] [Green Version]
- Cardoso, F.; van’t Veer, L.J.; Bogaerts, J.; Slaets, L.; Viale, G.; Delaloge, S.; Pierga, J.Y.; Brain, E.; Causeret, S.; DeLorenzi, M.; et al. 70-Gene Signature as an Aid to Treatment Decisions in Early-Stage Breast Cancer. N. Engl. J. Med. 2016, 375, 717–729. [Google Scholar] [CrossRef] [Green Version]
- Losk, K.; Freedman, R.A.; Laws, A.; Kantor, O.; Mittendorf, E.A.; Tan-Wasielewski, Z.; Trippa, L.; Lin, N.U.; Winer, E.P.; King, T.A. Oncotype DX testing in node-positive breast cancer strongly impacts chemotherapy use at a comprehensive cancer center. Breast Cancer Res. Treat. 2021, 185, 215–227. [Google Scholar] [CrossRef]
- Cheng, R.; Kong, X.; Wang, X.; Fang, Y.; Wang, J. Oncotype DX Breast Recurrence Score Distribution and Chemotherapy Benefit Among Women of Different Age Groups With HR-Positive, HER2-Negative, Node-Negative Breast Cancer in the SEER Database. Front. Oncol. 2020, 10, 1583. [Google Scholar] [CrossRef]
- Cardoso, F.; Veer, L.v.t.; Poncet, C.; Cardozo, J.L.; Delaloge, S.; Pierga, J.-Y.; Vuylsteke, P.; Brain, E.; Viale, G.; Kuemmel, S.; et al. MINDACT: Long-term results of the large prospective trial testing the 70-gene signature MammaPrint as guidance for adjuvant chemotherapy in breast cancer patients. J. Clin. Oncol. 2020, 38, 506. [Google Scholar] [CrossRef]
- Pereira, E.; Camacho-Vanegas, O.; Anand, S.; Sebra, R.; Catalina Camacho, S.; Garnar-Wortzel, L.; Nair, N.; Moshier, E.; Wooten, M.; Uzilov, A.; et al. Personalized Circulating Tumor DNA Biomarkers Dynamically Predict Treatment Response and Survival In Gynecologic Cancers. PLoS ONE 2015, 10, e0145754. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.H.; Long, G.V.; Menzies, A.M.; Lo, S.; Guminski, A.; Whitbourne, K.; Peranec, M.; Scolyer, R.; Kefford, R.F.; Rizos, H.; et al. Association Between Circulating Tumor DNA and Pseudoprogression in Patients With Metastatic Melanoma Treated with Anti-Programmed Cell Death 1 Antibodies. JAMA Oncol. 2018, 4, 717–721. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ogasawara, A.; Hihara, T.; Shintani, D.; Yabuno, A.; Ikeda, Y.; Tai, K.; Fujiwara, K.; Watanabe, K.; Hasegawa, K. Evaluation of Circulating Tumor DNA in Patients with Ovarian Cancer Harboring Somatic PIK3CA or KRAS Mutations. Cancer Res. Treat. 2020, 52, 1219–1228. [Google Scholar] [CrossRef] [PubMed]
- Noguchi, T.; Sakai, K.; Iwahashi, N.; Matsuda, K.; Matsukawa, H.; Yahata, T.; Toujima, S.; Nishio, K.; Ino, K. Changes in the gene mutation profiles of circulating tumor DNA detected using CAPP-Seq in neoadjuvant chemotherapy-treated advanced ovarian cancer. Oncol. Lett. 2020, 19, 2713–2720. [Google Scholar] [CrossRef]
- Liggett, T.E.; Melnikov, A.; Yi, Q.; Replogle, C.; Hu, W.; Rotmensch, J.; Kamat, A.; Sood, A.K.; Levenson, V. Distinctive DNA methylation patterns of cell-free plasma DNA in women with malignant ovarian tumors. Gynecol. Oncol. 2011, 120, 113–120. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Khella, C.A.; Mehta, G.A.; Mehta, R.N.; Gatza, M.L. Recent Advances in Integrative Multi-Omics Research in Breast and Ovarian Cancer. J. Pers. Med. 2021, 11, 149. https://doi.org/10.3390/jpm11020149
Khella CA, Mehta GA, Mehta RN, Gatza ML. Recent Advances in Integrative Multi-Omics Research in Breast and Ovarian Cancer. Journal of Personalized Medicine. 2021; 11(2):149. https://doi.org/10.3390/jpm11020149
Chicago/Turabian StyleKhella, Christen A., Gaurav A. Mehta, Rushabh N. Mehta, and Michael L. Gatza. 2021. "Recent Advances in Integrative Multi-Omics Research in Breast and Ovarian Cancer" Journal of Personalized Medicine 11, no. 2: 149. https://doi.org/10.3390/jpm11020149
APA StyleKhella, C. A., Mehta, G. A., Mehta, R. N., & Gatza, M. L. (2021). Recent Advances in Integrative Multi-Omics Research in Breast and Ovarian Cancer. Journal of Personalized Medicine, 11(2), 149. https://doi.org/10.3390/jpm11020149