
Autism Spectrum Disorder from the Womb to Adulthood: Suggestions for a Paradigm Shift

 , ,
, , 

 ,
,  ,
,  ,
, 
 ,
, 
 ,
,  , , , , and
, , , , and 
Abstract
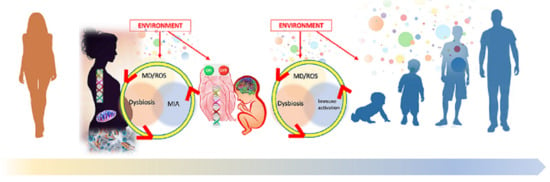
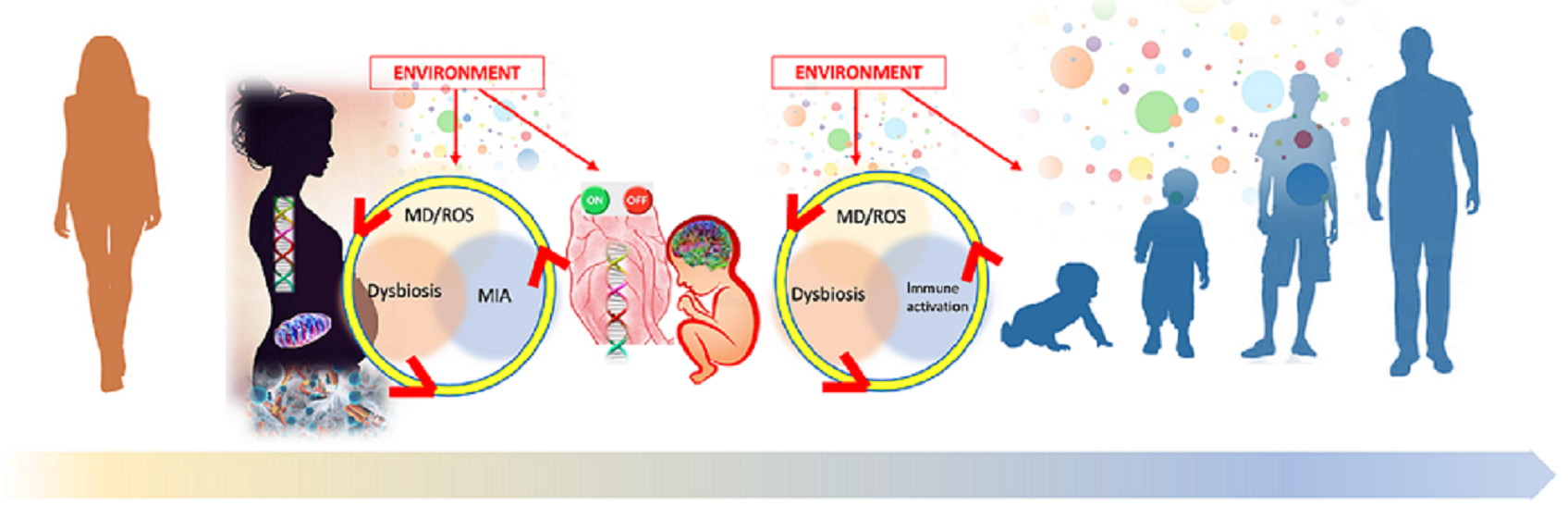
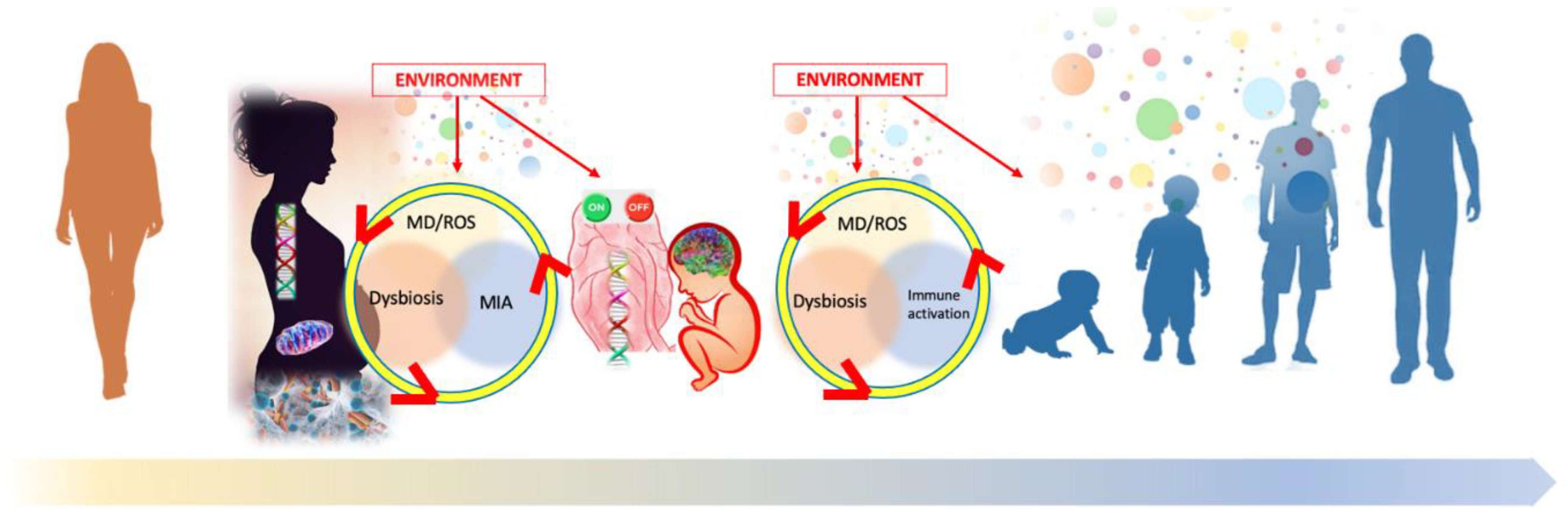
:
{kind=link}
{kind=link}
{kind=link}
1. Introduction
1.1. Clinical Features of People with ASD: Not Only a Behavioral Disorder
1.2. Genetics, Epigenetics, and Environment: Who’s to Blame?
1.3. Widening the Gaze: From ASD to the Epidemiological Transition of NCDs
1.4. Maternal Immune Response in Neurodevelopmental Disorders
1.5. Interplay between Epigenetics and Immune Response in Neurodevelopment
1.6. Take Home Message from the Interplay between MIA and Epigenetics in Pregnancy
1.7. Neuroinflammation and Gut-Brain Axis in People with ASD
1.8. Microbiome in the Crosstalk between Immune System, Gut, and Brain
1.9. Mitochondria/Oxidative Stress … and the ‘Bad Trio’
1.10. Metabolomics: A Promising ‘Meaningful Web’ Describing a Biochemical Fingerprint
2. New Methods for Renewed Diagnostic Tools: Machine Learning System in EEG
3. Discussion
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- American Psychiatric Association. Diagnostic and Statistical Manual of Mental Disorders (DSM-5), 5th ed.; American Psychiatric Association: Arlington, VA, USA, 2013. [Google Scholar]
- Aldinger, K.A.; Lane, C.J.; Veenstra-VanderWeele, J.; Levitt, P. Patterns of Risk for Multiple Co-Occurring Medical Conditions Replicate Across Distinct Cohorts of Children with Autism Spectrum Disorder. Autism. Res. 2015, 8, 771–781. [Google Scholar] [CrossRef]
- Maenner, M.J.; Shaw, K.A.; Baio, J.; Washington, A.; Patrick, M.; DiRienzo, M.; Christensen, D.L.; Wiggins, L.D.; Pettygrove, S.; Andrews, J.G.; et al. Prevalence of Autism Spectrum Disorder Among Children Aged 8 Years Autism and Developmental Disabilities Monitoring Network, 11 Sites, United States, 2016. MMWR Surveill. Summ. 2020, 69, 1–12. [Google Scholar] [CrossRef]
- Cusick, S.E.; Georgieff, M.K. The Role of Nutrition in Brain Development: The Golden Opportunity of the “First 1000 Days”. J. Pediatr. 2016, 175, 16–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, T.P.; Velazquez, M.A.; Eckert, J.J. Embryos, DOHaD and David Barker. J. Dev. Orig. Health Dis. 2015, 6, 377–383. [Google Scholar] [CrossRef] [PubMed]
- Barger, B.D.; Campbell, J.M.; McDonough, J.D. Prevalence and onset of regression within autism spectrum disorders: A meta-analytic review. J. Autism Dev. Disord. 2013, 43, 817–828. [Google Scholar] [CrossRef]
- Bölte, S.; Mahdi, S.; de Vries, P.J.; Granlund, M.; Robison, J.E.; Shulman, C.; Swedo, S.; Tonge, B.; Wong, V.; Zwaigenbaum, L.; et al. The Gestalt of functioning in autism spectrum disorder: Results of the international conference to develop final consensus International Classification of Functioning, Disability and Health core sets. Autism 2019, 23, 449–467. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jonsson, U.; Alaie, I.; Löfgren Wilteus, A.; Zander, E.; Marschik, P.B.; Coghill, D.; Bölte, S. Annual Research Review: Quality of life and childhood mental and behavioural disorders a critical review of the research. J. Child Psychol. Psychiatry 2017, 58, 439–469. [Google Scholar] [CrossRef] [PubMed]
- Holwerda, A.; van der Klink, J.J.; Groothoff, J.W.; Brouwer, S. Predictors for work participation in individuals with an Autism spectrum disorder: A systematic review. J. Occup. Rehabil. 2012, 22, 333–352. [Google Scholar] [CrossRef] [Green Version]
- Hertz-Picciotto, I.; Delwiche, L. The rise in autism and the role of age at diagnosis. Epidemiology 2009, 20, 84–90. [Google Scholar] [CrossRef] [Green Version]
- Mead, J.; Ashwood, P. Evidence supporting an altered immune response in ASD. Immunol. Lett. 2015, 163, 49–55. [Google Scholar] [CrossRef]
- McElhanon, B.O.; McCracken, C.; Karpen, S.; Sharp, W.G. Gastrointestinal symptoms in autism spectrum disorder: A meta-analysis. Pediatrics 2014, 133, 872–883. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandin, S.; Lichtenstein, P.; Kuja-Halkola, R.; Larsson, H.; Hultman, C.M.; Reichenberg, A. The familial risk of autism. JAMA 2014, 311, 1770–1777. [Google Scholar] [CrossRef] [PubMed]
- Tick, B.; Bolton, P.; Happé, F.; Rutter, M.; Rijsdijk, F. Heritability of autism spectrum disorders: A meta-analysis of twin studies. J. Child Psychol. Psychiatry 2016, 57, 585–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bai, D.; Yip, B.H.K.; Windham, G.C.; Sourander, A.; Francis, R.; Yoffe, R.; Glasson, E.; Mahjani, B.; Suominen, A.; Leonard, H.; et al. Association of Genetic and Environmental Factors With Autism in a 5-Country Cohort. JAMA Psychiatry 2019, 76, 1035–1043. [Google Scholar] [CrossRef] [PubMed]
- Grove, J.; Ripke, S.; Als, T.D.; Mattheisen, M.; Walters, R.K.; Won, H.; Pallesen, J.; Agerbo, E.; Andreassen, O.A.; Anney, R.; et al. Identification of common genetic risk variants for autism spectrum disorder. Nat. Genet. 2019, 51, 431–444. [Google Scholar] [CrossRef] [Green Version]
- Di Gregorio, E.; Riberi, E.; Belligni, E.F.; Biamino, E.; Spielmann, M.; Ala, U.; Calcia, A.; Bagnasco, I.; Carli, D.; Gai, G.; et al. Copy number variants analysis in a cohort of isolated and syndromic developmental delay/intellectual disability reveals novel genomic disorders, position effects and candidate disease genes. Clin. Genet. 2017, 92, 415–422. [Google Scholar] [CrossRef]
- Velinov, M. Genomic Copy Number Variations in the Autism Clinic-Work in Progress. Front. Cell. Neurosci. 2019, 13, 57. [Google Scholar] [CrossRef]
- Zhou, J.; Park, C.Y.; Theesfeld, C.L.; Wong, A.K.; Yuan, Y.; Scheckel, C.; Fak, J.J.; Funk, J.; Yao, K.; Tajima, Y.; et al. Whole-genome deep-learning analysis identifies contribution of noncoding mutations to autism risk. Nat. Genet. 2019, 51, 973–980. [Google Scholar] [CrossRef] [Green Version]
- Hu, V.W. From genes to environment: Using integrative genomics to build a “systems-level” understanding of autism spectrum disorders. Child Dev. 2013, 84, 89–103. [Google Scholar] [CrossRef] [Green Version]
- Biamino, E.; Di Gregorio, E.; Belligni, E.F.; Keller, R.; Riberi, E.; Gandione, M.; Calcia, A.; Mancini, C.; Giorgio, E.; Cavalieri, S.; et al. A novel 3q29 deletion associated with autism, intellectual disability, psychiatric disorders, and obesity. Am. J. Med. Genet B Neuropsychiatr. Genet. 2016, 171B, 290–299. [Google Scholar] [CrossRef]
- Cauda, F.; Nani, A.; Costa, T.; Palermo, S.; Tatu, K.; Manuello, J.; Duca, S.; Fox, P.T.; Keller, R. The morphometric co-atrophy networking of schizophrenia, autistic and obsessive spectrum disorders. Hum Brain Mapp. 2018, 39, 1898–1928. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mannion, A.; Leader, G. An investigation of comorbid psychological disorders, sleep problems, gastrointestinal symptoms and epilepsy in children and adolescents with autism spectrum disorder: A two year follow-up. Res. Autism Spectr. Disord. 2016, 22, 20–33. [Google Scholar] [CrossRef] [Green Version]
- Hallmayer, J.; Cleveland, S.; Torres, A.; Phillips, J.; Cohen, B.; Torigoe, T.; Miller, J.; Fedele, A.; Collins, J.; Smith, K.; et al. Genetic heritability and shared environmental factors among twin pairs with autism. Arch. Gen. Psychiatry 2011, 68, 1095–1102. [Google Scholar] [CrossRef]
- Vogel Ciernia, A.; LaSalle, J. The landscape of DNA methylation amid a perfect storm of autism aetiologies. Nat. Rev. Neurosci. 2016, 17, 411–423. [Google Scholar] [CrossRef] [Green Version]
- Hannon, E.; Schendel, D.; Ladd-Acosta, C.; Grove, J.; iPSYCH-Broad ASD Group; Hansen, C.S.; Andrews, S.V.; Hougaard, D.M.; Bresnahan, M.; Mors, O.; et al. Elevated polygenic burden for autism is associated with differential DNA methylation at birth. Genome Med. 2018, 10, 19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dupont, C.; Armant, D.R.; Brenner, C.A. Epigenetics: Definition, mechanisms and clinical perspective. Semin. Reprod. Med. 2009, 27, 351–357. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strohman, R.C. Linear genetics, non-linear epigenetics: Complementary approaches to understanding complex diseases. Integr. Physiol. Behav. Sci. 1995, 30, 273–282. [Google Scholar] [CrossRef]
- Goldberg, A.D.; Allis, C.D.; Bernstein, E. Epigenetics: A landscape takes shape. Cell 2007, 128, 635–638. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kiefer, J.C. Epigenetics in development. Dev. Dyn. 2007, 236, 1144–1156. [Google Scholar] [CrossRef]
- Spiers, H.; Hannon, E.; Schalkwyk, L.C.; Smith, R.; Wong, C.C.; O’Donovan, M.C.; Bray, N.J.; Mill, J. Methylomic trajectories across human fetal brain development. Genome Res. 2015, 25, 338–352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Linnér, A.; Almgren, M. Epigenetic programming-The important first 1000 days. Acta Paediatr. 2020, 109, 443–452. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Rubeis, S.; He, X.; Goldberg, A.P.; Poultney, C.S.; Samocha, K.; Cicek, A.E.; Kou, Y.; Liu, L.; Fromer, M.; Walker, S.; et al. Synaptic, transcriptional and chromatin genes disrupted in autism. Nature 2014, 515, 209–215. [Google Scholar] [CrossRef]
- Lasalle, J.M. Autism genes keep turning up chromatin. OA Autism 2013, 1, 14. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amir, R.E.; Van den Veyver, I.B.; Wan, M.; Tran, C.Q.; Francke, U.; Zoghbi, H.Y. Rett syndrome is caused by mutations in X-linked MECP2, encoding methyl-CpG-binding protein 2. Nat. Genet. 1999, 23, 185–188. [Google Scholar] [CrossRef]
- Chahrour, M.; Jung, S.Y.; Shaw, C.; Zhou, X.; Wong, S.T.; Qin, J.; Zoghbi, H.Y. MeCP2, a key contributor to neurological disease, activates and represses transcription. Science 2008, 320, 1224–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, Y.H.; Bressler, J.; Beaudet, A.L. Epigenetics and human disease. Annu. Rev. Genom. Hum. Genet. 2004, 5, 479–510. [Google Scholar] [CrossRef] [Green Version]
- Zhao, X.; Pak, C.; Smrt, R.D.; Jin, P. Epigenetics and Neural developmental disorders: Washington DC, September 18 and 19, 2006. Epigenetics 2007, 2, 126–134. [Google Scholar] [CrossRef] [Green Version]
- Dall’Aglio, L.; Muka, T.; Cecil, C.A.M.; Bramer, W.M.; Verbiest, M.M.P.J.; Nano, J.; Hidalgo, A.C.; Franco, O.H.; Tiemeier, H. The role of epigenetic modifications in neurodevelopmental disorders: A systematic review. Neurosci. Biobehav. Rev. 2018, 94, 17–30. [Google Scholar] [CrossRef] [Green Version]
- Stamova, B.; Ander, B.P.; Barger, N.; Sharp, F.R.; Schumann, C.M. Specific Regional and Age-Related Small Noncoding RNA Expression Patterns Within Superior Temporal Gyrus of Typical Human Brains Are Less Distinct in Autism Brains. J. Child Neurol. 2015, 30, 1930–1946. [Google Scholar] [CrossRef] [Green Version]
- Mundalil Vasu, M.; Anitha, A.; Thanseem, I.; Suzuki, K.; Yamada, K.; Takahashi, T.; Wakuda, T.; Iwata, K.; Tsujii, M.; Sugiyama, T.; et al. Serum microRNA profiles in children with autism. Mol. Autism 2014, 5, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hicks, S.D.; Rajan, A.T.; Wagner, K.E.; Barns, S.; Carpenter, R.L.; Middleton, F.A. Validation of a Salivary RNA Test for Childhood Autism Spectrum Disorder. Front. Genet. 2018, 9, 534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gluckman, P.D.; Hanson, M.A. Living with the past: Evolution, development, and patterns of disease. Science 2004, 305, 1733–1736. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gluckman, P.D.; Hanson, M.A.; Low, F.M. The role of developmental plasticity and epigenetics in human health. Birth Defects Res. C Embryo Today 2011, 93, 12–18. [Google Scholar] [CrossRef] [PubMed]
- Burgio, E. Environment and Fetal Programming: The origins of some current “pandemics”. J. Pediatr. Neonat. Individual. Med. 2015, 4, 2. [Google Scholar]
- Vineis, P.; Stringhini, S.; Porta, M. The environmental roots of non-communicable diseases (NCDs) and the epigenetic impacts of globalization. Environ. Res. 2014, 133, 424–430. [Google Scholar] [CrossRef]
- Godfrey, K.M.; Costello, P.M.; Lillycrop, K.A. The developmental environment, epigenetic biomarkers and long-term health. J. Dev. Orig. Health Dis. 2015, 6, 399–406. [Google Scholar] [CrossRef]
- Barker, D.J.; Eriksson, J.G.; Forsén, T.; Osmond, C. Fetal origins of adult disease: Strength of effects and biological basis. Int. J. Epidemiol. 2002, 31, 1235–1239. [Google Scholar] [CrossRef] [Green Version]
- Shapiro, J.A. A 21st century view of evolution: Genome system architecture, repetitive DNA, and natural genetic engineering. Gene 2005, 345, 91–100. [Google Scholar] [CrossRef]
- Nugent, B.M.; Bale, T.L. The omniscient placenta: Metabolic and epigenetic regulation of fetal programming. Front. Neuroendocrinol. 2015, 39, 28–37. [Google Scholar] [CrossRef] [Green Version]
- Coan, P.M.; Burton, G.J.; Ferguson-Smith, A.C. Imprinted genes in the placenta–A review. Placenta 2005, 26, S10–S20. [Google Scholar] [CrossRef]
- Hochberg, Z.; Feil, R.; Constancia, M.; Fraga, M.; Junien, C.; Carel, J.C.; Boileau, P.; Le Bouc, Y.; Deal, C.L.; Lillycrop, K.; et al. Child health, developmental plasticity, and epigenetic programming. Endocr. Rev. 2011, 32, 159–224. [Google Scholar] [CrossRef] [PubMed]
- LaSalle, J.M. A genomic point-of-view on environmental factors influencing the human brain methylome. Epigenetics 2011, 6, 862–869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Day, J.J.; Childs, D.; Guzman-Karlsson, M.C.; Kibe, M.; Moulden, J.; Song, E.; Tahir, A.; Sweatt, J.D. DNA methylation regulates associative reward learning. Nat. Neurosci. 2013, 16, 1445–1452. [Google Scholar] [CrossRef] [PubMed]
- Podobinska, M.; Szablowska-Gadomska, I.; Augustyniak, J.; Sandvig, I.; Sandvig, A.; Buzanska, L. Epigenetic Modulation of Stem Cells in Neurodevelopment: The Role of Methylation and Acetylation. Front. Cell. Neurosci. 2017, 11, 23. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gesundheit, B.; Rosenzweig, J.P.; Naor, D.; Lerer, B.; Zachor, D.A.; Procházka, V.; Melamed, M.; Kristt, D.A.; Steinberg, A.; Shulman, C.; et al. Immunological and autoimmune considerations of Autism Spectrum Disorders. J. Autoimmun. 2013, 44, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Filiano, A.J.; Gadani, S.P.; Kipnis, J. Interactions of innate and adaptive immunity in brain development and function. Brain Res. 2015, 1617, 18–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deverman, B.E.; Patterson, P.H. Cytokines and CNS development. Neuron 2009, 64, 61–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bilbo, S.D.; Schwarz, J.M. The immune system and developmental programming of brain and behavior. Front. Neuroendocrinol. 2012, 33, 267–286. [Google Scholar] [CrossRef] [Green Version]
- Schwarz, J.M.; Bilbo, S.D. Sex, glia, and development: Interactions in health and disease. Horm. Behav. 2012, 62, 243–253. [Google Scholar] [CrossRef] [Green Version]
- Kipnis, J. Immune system: The “seventh sense”. J. Exp. Med. 2018, 215, 397–398. [Google Scholar] [CrossRef] [Green Version]
- Chess, S. Autism in children with congenital rubella. J. Autism Child Schizophr. 1971, 1, 33–47. [Google Scholar] [CrossRef] [PubMed]
- Chess, S. Follow-up report on autism in congenital rubella. J. Autism Child Schizophr. 1977, 7, 69–81. [Google Scholar] [CrossRef] [PubMed]
- Patterson, P.H. Immune involvement in schizophrenia and autism: Etiology, pathology and animal models. Behav. Brain Res. 2009, 204, 313–321. [Google Scholar] [CrossRef]
- Reisinger, S.; Khan, D.; Kong, E.; Berger, A.; Pollak, A.; Pollak, D.D. The poly(I:C)-induced maternal immune activation model in preclinical neuropsychiatric drug discovery. Pharmacol. Ther. 2015, 149, 213–226. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Knuesel, I.; Chicha, L.; Britschgi, M.; Schobel, S.A.; Bodmer, M.; Hellings, J.A.; Toovey, S.; Prinssen, E.P. Maternal immune activation and abnormal brain development across CNS disorders. Nat. Rev. Neurol. 2014, 10, 643–660. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.K.; Magnusson, C.; Gardner, R.M.; Blomström, Å.; Newschaffer, C.J.; Burstyn, I.; Karlsson, H.; Dalman, C. Maternal hospitalization with infection during pregnancy and risk of autism spectrum disorders. Brain Behav. Immun. 2015, 44, 100–105. [Google Scholar] [CrossRef] [Green Version]
- Vianna, P.; Gomes, J.D.A.; Boquett, J.A.; Fraga, L.R.; Schuch, J.B.; Vianna, F.S.L.; Schuler-Faccini, L. Zika Virus as a Possible Risk Factor for Autism Spectrum Disorder: Neuroimmunological Aspects. Neuroimmunomodulation 2018, 25, 320–327. [Google Scholar] [CrossRef]
- Forestieri, S.; Marcialis, M.A.; Migliore, L.; Panisi, C.; Fanos, V. Relationship between pregnancy and coronavirus: What we know. J. Matern. Fetal Neonatal Med. 2020, 1–12. [Google Scholar] [CrossRef]
- Keil, A.; Daniels, J.L.; Forssen, U.; Hultman, C.; Cnattingius, S.; Söderberg, K.C.; Feychting, M.; Sparen, P. Parental autoimmune diseases associated with autism spectrum disorders in offspring. Epidemiology 2010, 21, 805–808. [Google Scholar] [CrossRef] [Green Version]
- Lyall, K.; Pauls, D.L.; Spiegelman, D.; Ascherio, A.; Santangelo, S.L. Pregnancy complications and obstetric suboptimality in association with autism spectrum disorders in children of the Nurses’ Health Study II. Autism Res. 2012, 5, 21–30. [Google Scholar] [CrossRef]
- Brown, A.S.; Surcel, H.M.; Hinkka-Yli-Salomäki, S.; Cheslack-Postava, K.; Bao, Y.; Sourander, A. Maternal thyroid autoantibody and elevated risk of autism in a national birth cohort. Prog. Neuropsychopharmacol. Biol. Psychiatry 2015, 57, 86–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Singer, H.S.; Morris, C.; Gause, C.; Pollard, M.; Zimmerman, A.W.; Pletnikov, M. Prenatal exposure to antibodies from mothers of children with autism produces neurobehavioral alterations: A pregnant dam mouse model. J. Neuroimmunol. 2009, 211, 39–48. [Google Scholar] [CrossRef]
- Braunschweig, D.; Van de Water, J. Maternal autoantibodies in autism. Arch. Neurol. 2012, 69, 693–699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, S.W.; Zhong, X.S.; Jiang, L.N.; Zheng, X.Y.; Xiong, Y.Q.; Ma, S.J.; Qiu, M.; Huo, S.T.; Ge, J.; Chen, Q. Maternal autoimmune diseases and the risk of autism spectrum disorders in offspring: A systematic review and meta-analysis. Behav. Brain Res. 2016, 296, 61–69. [Google Scholar] [CrossRef]
- Krakowiak, P.; Walker, C.K.; Tancredi, D.; Hertz-Picciotto, I.; Van de Water, J. Autism-specific maternal anti-fetal brain autoantibodies are associated with metabolic conditions. J. Autism Res. 2017, 10, 89–98. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Croen, L.A.; Braunschweig, D.; Haapanen, L.; Yoshida, C.K.; Fireman, B.; Grether, J.K.; Kharhfr, M.; Hansen, R.L.; Ashwood, P.; Van de Water, J. Maternal mid-pregnancy autoantibodies to fetal brain protein: The early markers for autism study. Biol. Psychiatry 2008, 64, 583–588. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ramaekers, V.T.; Sequeira, J.M.; DiDuca, M.; Vrancken, G.; Thomas, A.; Philippe, C.; Peters, M.; Jadot, A.; Quadros, E.V. Improving Outcome in Infantile Autism with Folate Receptor Autoimmunity and Nutritional Derangements: A Self-Controlled Trial. Autism. Res. Treat. 2019, 2019, 7486431. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sequeira, J.M.; Desai, A.; Berrocal-Zaragoza, M.I.; Murphy, M.M.; Fernandez-Ballart, J.D.; Quadros, E.V. Exposure to Folate Receptor Alpha Antibodies during Gestation and Weaning Leads to Severe Behavioral Deficits in Rats: A Pilot Study. PLoS ONE 2016, 11, e0152249. [Google Scholar] [CrossRef]
- Frye, R.E.; Rossignol, D.A.; Scahill, L.; McDougle, C.J.; Huberman, H.; Quadros, E.V. Treatment of folate metabolism abnormalities in Autism Spectrum Disorder. Semin. Pediatr. Neurol. 2020, 35, 100835. [Google Scholar] [CrossRef]
- Frye, R.E.; Sequeira, J.M.; Quadros, E.V.; James, S.J.; Rossignol, D.A. Cerebral folate receptor autoantibodiesin autism spectrum disorder. Mol. Psychiatry 2013, 18, 369–381. [Google Scholar] [CrossRef]
- Nardone, S.; Elliott, E. The Interaction between the Immune System and Epigenetics in the Etiology of Autism Spectrum Disorders. Front. Neurosci. 2016, 10, 329. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombardo, M.V.; Moon, H.M.; Su, J.; Palmer, T.D.; Courchesne, E.; Pramparo, T. Maternal immune activation dysregulation of the fetal brain transcriptome and relevance to the pathophysiology of autism spectrum disorder. Mol. Psychiatry 2018, 23, 1001–1013. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conway, F.; Brown, A.S. Maternal Immune Activation and Related Factors in the Risk of Offspring Psychiatric Disorders. Front. Psychiatry 2019, 10, 430. [Google Scholar] [CrossRef] [PubMed]
- Phillips, N.L.H.; Roth, T.L. Animal Models and Their Contribution to Our Understanding of the Relationship Between Environments, Epigenetic Modifications, and Behavior. Genes 2019, 10, 47. [Google Scholar] [CrossRef] [Green Version]
- Parker-Athill, E.C.; Tan, J. Maternal immune activation and autism spectrum disorder: Interleukin-6 signaling as a key mechanistic pathway. Neurosignals 2010, 18, 113–128. [Google Scholar] [CrossRef] [Green Version]
- Estes, M.L.; McAllister, A.K. Immune mediators in the brain and peripheral tissues in autism spectrum disorder. Nat. Rev. Neurosci. 2015, 16, 469–486. [Google Scholar] [CrossRef] [Green Version]
- Sweeten, T.L.; Bowyer, S.L.; Posey, D.J.; Halberstadt, G.M.; McDougle, C.J. Increased prevalence of familial autoimmunity in probands with pervasive developmental disorders. Pediatrics 2003, 112, e420. [Google Scholar] [CrossRef] [Green Version]
- Patel, S.; Masi, A.; Dale, R.C.; Whitehouse, A.J.O.; Pokorski, I.; Alvares, G.A.; Hickie, I.B.; Breen, E.; Guastella, A.J. Social impairments in autism spectrum disorder are related to maternal immune history profile. Mol. Psychiatry 2018, 23, 1794–1797. [Google Scholar] [CrossRef]
- Hompes, T.; Izzi, B.; Gellens, E.; Morreels, M.; Fieuws, S.; Pexsters, A.; Schops, G.; Dom, M.; Van Bree, R.; Freson, K.; et al. Investigating the influence of maternal cortisol and emotional state during pregnancy on the DNA methylation status of the glucocorticoid receptor gene (NR3C1) promoter region in cord blood. J. Psychiatr. Res. 2013, 47, 880–891. [Google Scholar] [CrossRef]
- Liu, Y.; Murphy, S.K.; Murtha, A.P.; Fuemmeler, B.F.; Schildkraut, J.; Huang, Z.; Overcash, F.; Kurtzberg, J.; Jirtle, R.; Iversen, E.S.; et al. Depression in pregnancy, infant birth weight and DNA methylation of imprint regulatory elements. Epigenetics 2012, 7, 735–746. [Google Scholar] [CrossRef] [Green Version]
- Carter, C.J.; Blizard, R.A. Autism genes are selectively targeted by environmental pollutants including pesticides, heavy metals, bisphenol A, phthalates and many others in food, cosmetics or household products. Neurochem. Int. 2016, S0197-0186, 30197–30198. [Google Scholar] [CrossRef] [PubMed]
- Estes, M.L.; McAllister, A.K. Maternal immune activation: Implications for neuropsychiatric disorders. Science 2016, 353, 772–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tang, B.; Jia, H.; Kast, R.J.; Thomas, E.A. Epigenetic changes at gene promoters in response to immune activation in utero. Brain Behav. Immun. 2013, 30, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Reisinger, S.N.; Kong, E.; Khan, D.; Schulz, S.; Ronovsky, M.; Berger, S.; Horvath, O.; Cabatic, M.; Berger, A.; Pollak, D.D. Maternal immune activation epigenetically regulates hippocampal serotonin transporter levels. Neurobiol. Stress 2016, 4, 34–43. [Google Scholar] [CrossRef] [Green Version]
- Richetto, J.; Massart, R.; Weber-Stadlbauer, U.; Szyf, M.; Riva, M.A.; Meyer, U. Genome-wide DNA Methylation Changes in a Mouse Model of Infection-Mediated Neurodevelopmental Disorders. Biol. Psychiatry 2017, 81, 265–276. [Google Scholar] [CrossRef]
- Alexopoulou, L.; Holt, A.C.; Medzhitov, R.; Flavell, R.A. Recognition of double-stranded RNA and activation of NF-kappaB by Toll-like receptor 3. Nature 2001, 413, 732–738. [Google Scholar] [CrossRef] [PubMed]
- Malkova, N.V.; Yu, C.Z.; Hsiao, E.Y.; Moore, M.J.; Patterson, P.H. Maternal immune activation yields offspring displaying mouse versions of the three core symptoms of autism. Brain Behav. Immun. 2012, 26, 607–616. [Google Scholar] [CrossRef] [Green Version]
- Da SilvaVaccaro, T.; Sorrentino, J.M.; Salvador, S.; Veit, T.; Souza, D.O.; de Almeida, R.F. Alterations in the MicroRNA of the Blood of Autism Spectrum Disorder Patients: Effects on Epigenetic Regulation and Potential Biomarkers. Behav. Sci. 2018, 8, 75. [Google Scholar] [CrossRef] [Green Version]
- Gupta, S.; Ellis, S.E.; Ashar, F.N.; Moes, A.; Bader, J.S.; Zhan, J.; West, A.B.; Arking, D.E. Transcriptome analysis reveals dysregulation of innate immune response genes and neuronal activity-dependent genes in autism. Nat. Commun. 2014, 5, 5748. [Google Scholar] [CrossRef]
- Nardone, S.; Sams, D.S.; Reuveni, E.; Getselter, D.; Oron, O.; Karpuj, M.; Elliott, E. DNA methylation analysis of the autistic brain reveals multiple dysregulated biological pathways. Transl. Psychiatry 2014, 4, e433. [Google Scholar] [CrossRef] [Green Version]
- Ashwood, P.; Krakowiak, P.; Hertz-Picciotto, I.; Hansen, R.; Pessah, I.; Van de Water, J. Elevated plasma cytokines in autism spectrum disorders provide evidence of immune dysfunction and are associated with impaired behavioral outcome. Brain Behav. Immun. 2011, 25, 40–45. [Google Scholar] [CrossRef] [Green Version]
- Guerini, F.R.; Bolognesi, E.; Chiappedi, M.; Manca, S.; Ghezzo, A.; Agliardi, C.; Zanette, M.; Littera, R.; Carcassi, C.; Sotgiu, S.; et al. Activating KIR molecules and their cognate ligands prevail in children with a diagnosis of ASD and in their mothers. Brain Behav. Immun. 2014, 36, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Boulanger, L.M.; Shatz, C.J. Immune signalling in neural development, synaptic plasticity and disease. Nat. Rev. Neurosci. 2004, 5, 521–531. [Google Scholar] [CrossRef]
- Comi, A.M.; Zimmerman, A.W.; Frye, V.H.; Law, P.A.; Peeden, J.N. Familial clustering of autoimmune disorders and evaluation of medical risk factors in autism. J. Child Neurol. 1999, 14, 388–394. [Google Scholar] [CrossRef]
- Croen, L.A.; Grether, J.K.; Yoshida, C.K.; Odouli, R.; Van de Water, J. Maternal autoimmune diseases, asthma and allergies, and childhood autism spectrum disorders: A case-control study. Arch. Pediatr. Adolesc. Med. 2005, 159, 151–157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Atladóttir, H.O.; Thorsen, P.; Østergaard, L.; Schendel, D.E.; Lemcke, S.; Abdallah, M.; Partner, E.T. Maternal infection requiring hospitalization during pregnancy and autism spectrum disorders. J. Autism Dev. Disord. 2010, 40, 1423–1430. [Google Scholar] [CrossRef]
- Altevogt, B.M.; Hanson, S.L.; Leshner, A.I. Autism and the environment: Challenges and opportunities for research. Pediatrics 2008, 121, 1225–1229. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Colucci, F. The role of KIR and HLA interactions in pregnancy complications. Immunogenetics 2017, 69, 557–565. [Google Scholar] [CrossRef]
- Torres, A.R.; Westover, J.B.; Gibbons, C.; Johnson, R.C.; Ward, D.C. Activating killer-cell immunoglobulin-like receptors (KIR) and their cognate HLA ligands are significantly increased in autism. Brain Behav. Immun. 2012, 26, 1122–1127. [Google Scholar] [CrossRef] [Green Version]
- Guerini, F.R.; Bolognesi, E.; Chiappedi, M.; Ghezzo, A.; Canevini, M.P.; Mensi, M.M.; Vignoli, A.; Agliardi, C.; Zanette, M.; Clerici, M. An HLA-G(∗)14bp insertion/deletion polymorphism associates with the development of autistic spectrum disorders. Brain Behav. Immun. 2015, 44, 207–212. [Google Scholar] [CrossRef]
- Christiansen, O.B.; Kolte, A.M.; Dahl, M.; Larsen, E.C.; Steffensen, R.; Nielsen, H.S.; Hviid, T.V. Maternal homozygocity for a 14 base pair insertion in exon 8 of the HLA-G gene and carriage of HLA class II alleles restricting HY immunity predispose to unexplained secondary recurrent miscarriage and low birth weight in children born to these patients. Hum. Immunol. 2012, 73, 699–705. [Google Scholar] [CrossRef] [PubMed]
- Hylenius, S.; Andersen, A.M.; Melbye, M.; Hviid, T.V. Association between HLA-G genotype and risk of pre-eclampsia: A case-control study using family triads. Mol. Hum. Reprod. 2004, 10, 237–246. [Google Scholar] [CrossRef] [Green Version]
- Guerini, F.R.; Bolognesi, E.; Chiappedi, M.; Ghezzo, A.; Manca, S.; Zanette, M.; Sotgiu, S.; Mensi, M.M.; Zanzottera, M.; Agliardi, C.; et al. HLA-G∗14bp Insertion and the KIR2DS1-HLAC2 Complex Impact on Behavioral Impairment in Children with Autism Spectrum Disorders. Neuroscience 2018, 370, 163–169. [Google Scholar] [CrossRef] [PubMed]
- Uhrberg, M. Shaping the human NK cell repertoire: An epigenetic glance at KIR gene regulation. Mol. Immunol. 2005, 42, 471–475. [Google Scholar] [CrossRef] [PubMed]
- Blomström, Å.; Karlsson, H.; Gardner, R.; Jörgensen, L.; Magnusson, C.; Dalman, C. Associations Between Maternal Infection During Pregnancy, Childhood Infections, and the Risk of Subsequent Psychotic Disorder--A Swedish Cohort Study of Nearly 2 Million Individuals. Schizophr. Bull. 2016, 42, 125–133. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kipnis, J. Multifaceted interactions between adaptive immunity and the central nervous system. Science 2016, 353, 766–771. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jones, K.L.; Croen, L.A.; Yoshida, C.K.; Heuer, L.; Hansen, R.; Zerbo, O.; DeLorenze, G.N.; Kharrazi, M.; Yolken, R.; Ashwood, P.; et al. Autism with intellectual disability is associated with increased levels of maternal cytokines and chemokines during gestation. Mol. Psychiatry 2017, 22, 273–279. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, G.B.; Yim, Y.S.; Wong, H.; Kim, S.; Kim, H.; Kim, S.V.; Hoeffer, C.A.; Littman, D.R.; Huh, J.R. The maternal interleukin-17a pathway in mice promotes autism-like phenotypes in offspring. Science 2016, 351, 933–939. [Google Scholar] [CrossRef] [Green Version]
- Zaretsky, M.V.; Alexander, J.M.; Byrd, W.; Bawdon, R.E. Transfer of inflammatory cytokines across the placenta. Obstet. Gynecol. 2004, 103, 546–550. [Google Scholar] [CrossRef]
- Smith, S.E.; Li, J.; Garbett, K.; Mirnics, K.; Patterson, P.H. Maternal immune activation alters fetal brain development through interleukin-6. J. Neurosci. 2007, 27, 10695–10702. [Google Scholar] [CrossRef] [Green Version]
- Boulanger, L.M. Immune proteins in brain development and synaptic plasticity. Neuron 2009, 64, 93–109. [Google Scholar] [CrossRef] [Green Version]
- Sotgiu, S.; Manca, S.; Gagliano, A.; Minutolo, A.; Melis, M.A.; Pisuttu, G.; Scoppola, C.; Bolognesi, E.; Clerici, M.; Guerini, F.R.; et al. Immune regulation of neurodevelopment at the mother-foetus interface: The case of autism. Clin. Transl. Immunol. 2020, e1211. [Google Scholar] [CrossRef]
- Aagaard, K.; Ma, J.; Antony, K.M.; Ganu, R.; Petrosino, J.; Versalovic, J. The placenta harbors a unique microbiome. Sci. Transl. Med. 2014, 6, 237ra65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gomez de Agüero, M.; Ganal-Vonarburg, S.C.; Fuhrer, T.; Rupp, S.; Uchimura, Y.; Li, H.; Steinert, A.; Heikenwalder, M.; Hapfelmeier, S.; Sauer, U.; et al. The maternal microbiota drives early postnatal innate immune development. Science 2016, 351, 1296–1302. [Google Scholar] [CrossRef] [PubMed]
- Romano-Keeler, J.; Weitkamp, J.H. Maternal influences on fetal microbial colonization and immune development. Pediatr. Res. 2015, 77, 189–195. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoffman, D.J.; Reynolds, R.M.; Hardy, D.B. Developmental origins of health and disease: Current knowledge and potential mechanisms. Nutr. Rev. 2017, 75, 951–970. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weaver, I.C.; Korgan, A.C.; Lee, K.; Wheeler, R.V.; Hundert, A.S.; Goguen, D. Stress and the Emerging Roles of Chromatin Remodeling in Signal Integration and Stable Transmission of Reversible Phenotypes. Front. Behav. Neurosci. 2017, 11, 41. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koren, O.; Goodrich, J.K.; Cullender, T.C.; Spor, A.; Laitinen, K.; Bäckhed, H.K.; Gonzalez, A.; Werner, J.J.; Angenent, L.T.; Knight, R.; et al. Host remodeling of the gut microbiome and metabolic changes during pregnancy. Cell 2012, 150, 470–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aagaard, K.; Riehle, K.; Ma, J.; Segata, N.; Mistretta, T.A.; Coarfa, C.; Raza, S.; Rosenbaum, S.; Van den Veyver, I.; Milosavljevic, A.; et al. A metagenomic approach to characterization of the vaginal microbiome signature in pregnancy. PLoS ONE 2012, 7, e36466. [Google Scholar] [CrossRef] [PubMed]
- Blaser, M.J.; Dominguez-Bello, M.G. The Human Microbiome before Birth. Cell Host. Microbe. 2016, 20, 558–560. [Google Scholar] [CrossRef] [Green Version]
- Hackman, D.A.; Farah, M.J.; Meaney, M.J. Socioeconomic status and the brain: Mechanistic insights from human and animal research. Nat. Rev. Neurosci. 2010, 11, 651–659. [Google Scholar] [CrossRef] [PubMed]
- Emberti Gialloreti, L.; Mazzone, L.; Benvenuto, A.; Fasano, A.; Alcon, A.G.; Kraneveld, A.; Moavero, R.; Raz, R.; Riccio, M.P.; Siracusano, M.; et al. Risk and Protective Environmental Factors Associated with Autism Spectrum Disorder: Evidence-Based Principles and Recommendations. J. Clin. Med. 2019, 8, 217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Raghavan, R.; Riley, A.W.; Volk, H.; Caruso, D.; Hironaka, L.; Sices, L.; Hong, X.; Wang, G.; Ji, Y.; Brucato, M.; et al. Maternal Multivitamin Intake, Plasma Folate and Vitamin B12 Levels and Autism Spectrum Disorder Risk in Offspring. Paediatr. Perinat. Epidemiol. 2018, 32, 100–111. [Google Scholar] [CrossRef] [PubMed]
- Bilbo, S.D.; Block, C.L.; Bolton, J.L.; Hanamsagar, R.; Tran, P.K. Beyond infection Maternal immune activation by environmental factors, microglial development, and relevance for autism spectrum disorders. Exp. Neurol. 2018, 299, 241–251. [Google Scholar] [CrossRef]
- Grossi, E.; Migliore, L.; Muratori, F. Pregnancy risk factors related to autism: An Italian case-control study in mothers of children with autism spectrum disorders (ASD), their siblings and of typically developing children. J. Dev. Orig. Health Dis. 2018, 9, 442–449. [Google Scholar] [CrossRef] [PubMed]
- Zhang, T.; Sidorchuk, A.; Sevilla-Cermeno, L.; Vilaplana-Perez, A.; Chang, Z.; Larsson, H.; Mataix-Cols, D.; Fernandez de la Cruz, L. Association of cesarean delivery with risk of neurodevelopmental and psychiatric disorders in the offspring: A systematic review and meta-analysis. JAMA Netw. Open 2019, 2, e1910236. [Google Scholar] [CrossRef] [PubMed]
- Vargas, D.L.; Nascimbene, C.; Krishnan, C.; Zimmerman, A.W.; Pardo, C.A. Neuroglial activation and neuroinflammation in the brain of patients with autism. Ann. Neurol. 2005, 57, 67–81. [Google Scholar] [CrossRef] [PubMed]
- Matta, S.M.; Hill-Yardin, E.L.; Crack, P.J. The influence of neuroinflammation in Autism Spectrum Disorder. Brain Behav. Immun. 2019, 79, 75–90. [Google Scholar] [CrossRef]
- Onore, C.; Careaga, M.; Ashwood, P. The role of immune dysfunction in the pathophysiology of autism. Brain Behav. Immun. 2012, 26, 383–392. [Google Scholar] [CrossRef] [Green Version]
- Saresella, M.; Marventano, I.; Guerini, F.R.; Mancuso, R.; Ceresa, L.; Zanzottera, M.; Rusconi, B.; Maggioni, E.; Tinelli, C.; Clerici, M. An autistic endophenotype results in complex immune dysfunction in healthy siblings of autistic children. Biol. Psychiatry 2009, 66, 978–984. [Google Scholar] [CrossRef]
- Li, Q.; Zhou, J.M. The microbiota-gut-brain axis and its potential therapeutic role in autism spectrum disorder. Neuroscience 2016, 324, 131–139. [Google Scholar] [CrossRef] [PubMed]
- El Aidy, S.; Dinan, T.G.; Cryan, J.F. Gut Microbiota: The Conductor in the Orchestra of Immune-Neuroendocrine Communication. Clin. Ther. 2015, 37, 954–967. [Google Scholar] [CrossRef] [PubMed]
- Mayer, E.A. Gut feelings: The emerging biology of gut-brain communication. Nat. Rev. Neurosci. 2011, 12, 453–466. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Frye, R.E. Evidence linking oxidative stress, mitochondrial dysfunction, and inflammation in the brain of individuals with autism. Front. Physiol. 2014, 5, 150. [Google Scholar] [CrossRef] [Green Version]
- Turnbaugh, P.J.; Ley, R.E.; Hamady, M.; Fraser-Liggett, C.M.; Knight, R.; Gordon, J.I. The human microbiome project. Nature 2007, 449, 804–810. [Google Scholar] [CrossRef]
- Qin, J.; Li, R.; Raes, J.; Arumugam, M.; Burgdorf, K.S.; Manichanh, C.; Nielsen, T.; Pons, N.; Levenez, F.; Yamada, T.; et al. A human gut microbial gene catalogue established by metagenomic sequencing. Nature 2010, 464, 59–65. [Google Scholar] [CrossRef] [Green Version]
- Littman, D.R.; Pamer, E.G. Role of the commensal microbiota in normal and pathogenic host immune responses. Cell Host Microbe 2011, 10, 311–323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blumberg, R.; Powrie, F. Microbiota, disease, and back to health: A metastable journey. Sci. Transl. Med. 2012, 4, 137rv7. [Google Scholar] [CrossRef] [Green Version]
- Hooper, L.V.; Littman, D.R.; Macpherson, A.J. Interactions between the microbiota and the immune system. Science 2012, 336, 1268–1273. [Google Scholar] [CrossRef] [Green Version]
- Ho, P.; Ross, D.A. More Than a Gut Feeling: The Implications of the Gut Microbiota in Psychiatry. Biol. Psychiatry 2017, 81, e35–e37. [Google Scholar] [CrossRef] [Green Version]
- Vuong, H.E.; Hsiao, E.Y. Emerging Roles for the Gut Microbiome in Autism Spectrum Disorder. Biol. Psychiatry 2017, 81, 411–423. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costello, E.K.; Stagaman, K.; Dethlefsen, L.; Bohannan, B.J.; Relman, D.A. The application of ecological theory toward an understanding of the human microbiome. Science 2012, 336, 1255–1262. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ferretti, P.; Pasolli, E.; Tett, A.; Asnicar, F.; Gorfer, V.; Fedi, S.; Armanini, F.; Truong, D.T.; Manara, S.; Zolfo, M.; et al. Mother-to-Infant Microbial Transmission from Different Body Sites Shapes the Developing Infant Gut Microbiome. Cell Host Microbe 2018, 24, 133–145.e5. [Google Scholar] [CrossRef] [PubMed]
- Francino, M.P. Early development of the gut microbiota and immune health. Pathogens 2014, 3, 769–790. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borre, Y.E.; O’Keeffe, G.W.; Clarke, G.; Stanton, C.; Dinan, T.G.; Cryan, J.F. Microbiota and neurodevelopmental windows: Implications for brain disorders. Trends Mol. Med. 2014, 20, 509–518. [Google Scholar] [CrossRef]
- Erny, D.; Hrabě Macfabe, D.F.; Short-chain, A.L.; Jaitin, D.; Wieghofer, P.; Staszewski, O.; David, E.; Keren-Shaul, H.; Mahlakoiv, T.; Jakobshagen, K.; et al. Host microbiota constantly control maturation and function of microglia in the CNS. Nat. Neurosci. 2015, 18, 965–977. [Google Scholar] [CrossRef]
- Saresella, M.; Piancone, F.; Marventano, I.; Zoppis, M.; Hernis, A.; Zanette, M.; Trabattoni, D.; Chiappedi, M.; Ghezzo, A.; Canevini, M.P.; et al. Multiple inflammasome complexes are activated in autistic spectrum disorders. Brain Behav. Immun. 2016, 57, 125–133. [Google Scholar] [CrossRef]
- Finegold, S.M.; Molitoris, D.; Song, Y.; Liu, C.; Vaisanen, M.L.; Bolte, E.; McTeague, M.; Sandler, R.; Wexler, H.; Marlowe, E.M.; et al. Gastrointestinal microflora studies in late-onset autism. Clin. Infect. Dis. 2002, 35, S6–S16. [Google Scholar] [CrossRef]
- Macfabe, D.F. Short-chain fatty acid fermentation products of the gut microbiome: Implications in autism spectrum disorders. Microb. Ecol. Health Dis. 2012, 23. [Google Scholar] [CrossRef]
- De Angelis, M.; Piccolo, M.; Vannini, L.; Siragusa, S.; De Giacomo, A.; Serrazzanetti, D.I.; Cristofori, F.; Guerzoni, M.E.; Gobbetti, M.; Francavilla, R. Fecal microbiota and metabolome of children with autism and pervasive developmental disorder not otherwise specified. PLoS ONE 2013, 8, e76993. [Google Scholar] [CrossRef] [Green Version]
- Fasano, A. Leaky gut and autoimmune diseases. Clin. Rev. Allergy Immunol. 2012, 42, 71–78. [Google Scholar] [CrossRef] [PubMed]
- Hsiao, E.Y.; McBride, S.W.; Hsien, S.; Sharon, G.; Hyde, E.R.; McCue, T.; Codelli, J.A.; Chow, J.; Reisman, S.E.; Petrosino, J.F.; et al. Microbiota modulate behavioral and physiological abnormalities associated with neurodevelopmental disorders. Cell 2013, 155, 1451–1463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carey, N.; La Thangue, N.B. Histone deacetylase inhibitors: Gathering pace. Curr. Opin. Pharmacol. 2006, 6, 369–375. [Google Scholar] [CrossRef] [PubMed]
- Lanza, M.; Campolo, M.; Casili, G.; Filippone, A.; Paterniti, I.; Cuzzocrea, S.; Esposito, E. Sodium butyrate exerts neuroprotective effects in spinal cord injury. Mol. Neurobiol. 2019, 56, 3937–3947. [Google Scholar] [CrossRef]
- Kratsman, N.; Getselter, D.; Elliott, E. Sodium butyrate attenuates social behavior deficits and modifies the transcription of inhibitory/excitatory genes in the frontal cortex of an autism model. Neuropharmacology 2016, 102, 136–145. [Google Scholar] [CrossRef]
- Hong, J.; Jia, Y.; Pan, S.; Jia, L.; Li, H.; Han, Z.; Cai, D.; Zhao, R. Butyrate alleviates high fat diet-induced obesity through activation of adiponectin-mediated pathway and stimulation of mitochon- drial function in the skeletal muscle of mice. Oncotarget 2016, 7, 56071–56082. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Li, E.; Sun, Z.; Fu, D.; Duan, G.; Jiang, M.; Yu, Y.; Mei, L.; Yang, P.; Tang, Y.; et al. Altered gut microbiota and short chain fatty acids in Chinese children with autism spectrum disorder. Sci. Rep. 2019, 9, 287. [Google Scholar] [CrossRef]
- Rose, S.; Bennuri, S.C.; Davis, J.E.; Wynne, R.; Slattery, J.C.; Tippett, M.; Delhey, L.; Melnyk, S.; Kahler, S.G.; MacFabe, D.F.; et al. Butyrate enhances mitochondrial function during oxidative stress in cell lines from boys with autism. Transl. Psychiatry 2018, 8, 42. [Google Scholar] [CrossRef]
- Kaur, H.; Singh, Y.; Singh, S.; Singh, R.B. Gut microbiome mediated epigenetic regulation of brain disorder and application of machine learning for multi-omics data analysis. Genome 2020. [Google Scholar] [CrossRef]
- Sadeghiyeh, T.; Dastgheib, S.A.; Mirzaee-Khoramabadi, K.; Morovati-Sharifabad, M.; Akbarian-Bafghi, M.J.; Poursharif, Z.; Mirjalili, S.R.; Neamatzadeh, H. Association of MTHFR 677C>T and 1298A>C polymorphisms with susceptibility to autism: A systematic review and meta-analysis. Asian J. Psychiatr. 2019, 46, 54–61. [Google Scholar] [CrossRef]
- Cryan, J.F.; Dinan, T.G. Mind-altering microorganisms: The impact of the gut microbiota on brain and behaviour. Nat. Rev. Neurosci. 2012, 13, 701–712. [Google Scholar] [CrossRef] [PubMed]
- MacFabe, D.F.; Cain, N.E.; Boon, F.; Ossenkopp, K.P.; Cain, D.P. Effects of the enteric bacterial metabolic product propionic acid on object-directed behavior, social behavior, cognition, and neuroinflammation in adolescent rats: Relevance to autism spectrum disorder. Behav. Brain Res. 2011, 217, 47–54. [Google Scholar] [CrossRef] [PubMed]
- Strati, F.; Cavalieri, D.; Albanese, D.; De Felice, C.; Donati, C.; Hayek, J.; Jousson, O.; Leoncini, S.; Renzi, D.; Calabrò, A.; et al. New evidences on the altered gut microbiota in autism spectrum disorders. Microbiome 2017, 5, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, H.T.; Taur, Y.; Walkup, J.T. Gut Microbiota and Autism: Key Concepts and Findings. J. Autism Dev. Disord. 2017, 47, 480–489. [Google Scholar] [CrossRef]
- de Magistris, L.; Familiari, V.; Pascotto, A.; Sapone, A.; Frolli, A.; Iardino, P.; Carteni, M.; De Rosa, M.; Francavilla, R.; Riegler, G.; et al. Alterations of the intestinal barrier in patients with autism spectrum disorders and in their first-degree relatives. J. Pediatr. Gastroenterol. Nutr. 2010, 51, 418–424. [Google Scholar] [CrossRef] [Green Version]
- Fasano, A. Zonulin and its regulation of intestinal barrier function: The biological door to inflammation, autoimmunity, and cancer. Physiol. Rev. 2011, 91, 151–175. [Google Scholar] [CrossRef] [Green Version]
- Fasano, A.; Hill, I. Serum Zonulin, Gut Permeability, and the Pathogenesis of Autism Spectrum Disorders: Cause, Effect, or an Epiphenomenon? J. Pediatr. 2017, 188, 15–17. [Google Scholar] [CrossRef] [Green Version]
- Kushak, R.I.; Buie, T.M.; Murray, K.F.; Newburg, D.S.; Chen, C.; Nestoridi, E.; Winter, H.S. Evaluation of Intestinal Function in Children with Autism and Gastrointestinal Symptoms. J. Pediatr. Gastroenterol. Nutr. 2016, 62, 687–691. [Google Scholar] [CrossRef]
- Babinská, K.; Tomova, A.; Celušáková, H.; Babková, J.; Repiská, G.; Kubranská, A.; Filčíková, D.; Siklenková, L.; Ostatníková, D. Fecal calprotectin levels correlate with main domains of the autism diagnostic interview-revised (ADI-R) in a sample of individuals with autism spectrum disorders from Slovakia. Physiol. Res. 2017, 66, S517–S522. [Google Scholar] [CrossRef]
- Nikolov, R.N.; Bearss, K.E.; Lettinga, J.; Erickson, C.; Rodowski, M.; Aman, M.G.; McCracken, J.T.; McDougle, C.J.; Tierney, E.; Vitiello, B.; et al. Gastrointestinal symptoms in a sample of children with pervasive developmental disorders. J. Autism Dev. Disord. 2009, 39, 405–413. [Google Scholar] [CrossRef]
- Buie, T.; Campbell, D.B.; Fuchs, G.J., 3rd; Furuta, G.T.; Levy, J.; Vandewater, J.; Whitaker, A.H.; Atkins, D.; Bauman, M.L.; Beaudet, A.L.; et al. Evaluation, diagnosis, and treatment of gastrointestinal disorders in individuals with ASDs: A consensus report. Pediatrics 2010, 125, S1–S18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, R.; Chieregato, S.; Bari, S.; Castaldo, R.; Rutto, F.; Chiocchetti, A.; Dianzani, U. Autism in Adulthood: Clinical and Demographic Characteristics of a Cohort of Five Hundred Persons with Autism Analyzed by a Novel Multistep Network Model. Brain Sci. 2020, 10, 416. [Google Scholar] [CrossRef]
- Adams, J.B.; Johansen, L.J.; Powell, L.D.; Quig, D.; Rubin, R.A. Gastrointestinal flora and gastrointestinal status in children with autism--comparisons to typical children and correlation with autism severity. BMC Gastroenterol. 2011, 11, 22. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whitney, D.G.; Shapiro, D.N. National Prevalence of Pain among Children and Adolescents with Autism Spectrum Disorders. JAMA Pediatr. 2019, 173, 1203–1205. [Google Scholar] [CrossRef] [PubMed]
- Iwata, B.A.; Deleon, I.G.; Roscoe, E.M. Reliability and validity of the functional analysis screening tool. J. Appl. Behav. Anal. 2013, 46, 271–284. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E.; Rossignol, D.A. Identification and Treatment of Pathophysiological Comorbidities of Autism Spectrum Disorder to Achieve Optimal Outcomes. Clin. Med. Insights Pediatr. 2016, 10, 43–56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
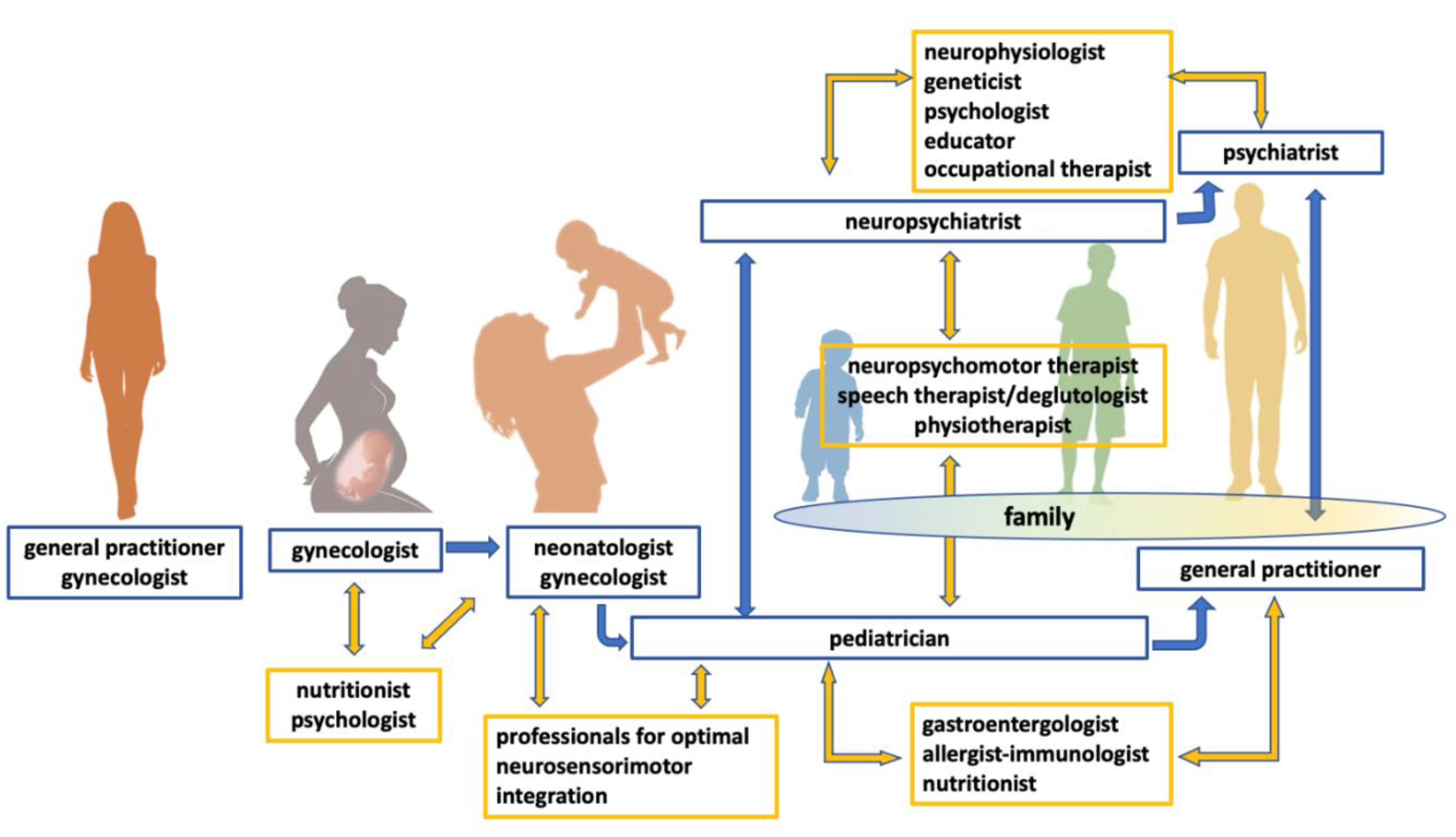
- Guinchat, V.; Cravero, C.; Lefèvre-Utile, J.; Cohen, D. Multidisciplinary treatment plan for challenging behaviors in neurodevelopmental disorders. Handb. Clin. Neurol. 2020, 174, 301–321. [Google Scholar] [CrossRef]
- Keller, R.; Basta, R.; Salerno, L.; Elia, M. Autism, epilepsy, and synaptopathies: A not rare association. Neurol. Sci. 2017, 38, 1353–1361. [Google Scholar] [CrossRef]
- Mouridsen, S.E.; Rich, B.; Isager, T. A longitudinal study of epilepsy and other central nervous system diseases in individuals with and without a history of infantile autism. Brain Dev. 2011, 33, 361–366. [Google Scholar] [CrossRef]
- Berg, A.T.; Plioplys, S. Epilepsy and autism: Is there a special relationship? Epilepsy Behav. 2012, 23, 193–198. [Google Scholar] [CrossRef] [Green Version]
- Theoharides, T.C.; Zhang, B. Neuro-inflammation, blood-brain barrier, seizures and autism. J. Neuroinflamm. 2011, 8, 168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valenti, D.; Lidia de Bari, L.; Bianca De Filippis, B.; Alexandra Henrion-Caude, A.; Vacca, R.A. Mitochondrial dysfunction as a central actor in intellectual disability-related diseases: An overview of Down syndrome, autism, Fragile X and Rett syndrome. Neurosci. Biobehav. Rev. 2014, 46, 202–217. [Google Scholar] [CrossRef] [PubMed]
- Chandel, N.S. Mitochondria as signaling organelles. BMC Biol. 2014, 12, 34. [Google Scholar] [CrossRef] [PubMed]
- Khacho, M.; Slack, R.S. Mitochondrial and Reactive Oxygen Species Signaling Coordinate Stem Cell Fate Decisions and Life Long Maintenance. Antioxid. Redox Signal. 2018, 28, 1090–1101. [Google Scholar] [CrossRef]
- Bahat, A.; Gross, A. Mitochondrial plasticity in cell fate regulation. J. Biol. Chem. 2019, 294, 13852–13863. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wright, R.H.; Fernandez-Fuentes, N.; Oliva, B.; Beato, M. Insight into the machinery that oils chromatin dynamics. Nucleus 2016, 7, 532–539. [Google Scholar] [CrossRef] [Green Version]
- Khacho, M.; Harris, R.; Slack, R.S. Mitochondria as central regulators of neural stem cell fate and cognitive function. Nat. Rev. Neurosci. 2019, 20, 34–48. [Google Scholar] [CrossRef]
- Mandò, C.; Anelli, G.M.; Novielli, C.; Panina-Bordignon, P.; Massari, M.; Mazzocco, M.I.; Cetin, I. Impact of Obesity and Hyperglycemia on Placental Mitochondria. Oxid. Med. Cell. Longev. 2018, 2018, 2378189. [Google Scholar] [CrossRef] [PubMed]
- Farrell, A.; Alahari, S.; Ermini, L.; Tagliaferro, A.; Litvack, M.; Post, M.; Caniggia, I. Faulty oxygen sensing disrupts angiomotin function in trophoblast cell migration and predisposes to preeclampsia. JCI Insight 2019, 4, e127009. [Google Scholar] [CrossRef] [PubMed]
- Figueiredo, A.S.; Schumacher, A. The T helper type 17/regulatory T cell paradigm in pregnancy. Immunology 2016, 148, 13–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Qian, J.; Sun, F.; Li, M.; Ye, J.; Li, M.; Du, M.; Li, D. Bidirectional regulation between 1st trimester HTR8/SVneo trophoblast cells and in vitro differentiated Th17/Treg cells suggest a fetal-maternal regulatory loop in human pregnancy. Am. J. Reprod. Immunol. 2019, 81, e13106. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rose, S.; Niyazov, D.M.; Rossignol, D.A.; Goldenthal, M.; Kahler, S.G.; Frye, R.E. Clinical and Molecular Characteristics of Mitochondrial Dysfunction in Autism Spectrum Disorder. Mol. Diagn. Ther. 2018, 22, 571–593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zorov, D.B.; Filburn, C.R.; Klotz, L.O.; Zweier, J.L.; Sollott, S.J. Reactive oxygen species (ROS)-induced ROS release: A new phenomenon accompanying induction of the mitochondrial permeability transition in cardiac myocytes. J. Exp. Med. 2000, 192, 1001–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tschopp, J.; Schroder, K. NLRP3 inflammasome activation: The convergence of multiple signalling pathways on ROS production? Nat. Rev. Immunol. 2010, 10, 210–215. [Google Scholar] [CrossRef]
- Engin, A. Endothelial Dysfunction in Obesity. Adv. Exp. Med. Biol. 2017, 960, 345–379. [Google Scholar] [CrossRef]
- Newsholme, P.; Cruzat, V.F.; Keane, K.N.; Carlessi, R.; Homem de Bittencourt, P.I., Jr. Molecular mechanisms of ROS production and oxidative stress in diabetes. Biochem. J. 2016, 473, 4527–4550. [Google Scholar] [CrossRef]
- Jomova, K.; Jenisova, Z.; Feszterova, M.; Baros, S.; Liska, J.; Hudecova, D.; Rhodes, C.J.; Valko, M. Arsenic: Toxicity, oxidative stress and human disease. J. Appl. Toxicol. 2011, 31, 95–107. [Google Scholar] [CrossRef]
- Meyer, U. Developmental neuroinflammation and schizophrenia. Prog. Neuropsychopharmacol. Biol. Psychiatry 2013, 42, 20–34. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Frye, R.E. A review of research trends in physiological abnormalities in autism spectrum disorders: Immune dysregulation, inflammation, oxidative stress, mitochondrial dysfunction and environmental toxicant exposures. Mol. Psychiatry 2012, 17, 389–401. [Google Scholar] [CrossRef] [Green Version]
- Rossignol, D.A.; Frye, R.E. Mitochondrial dysfunction in autism spectrum disorders: A systematic review and meta-analysis. Mol. Psychiatry 2012, 17, 290–314. [Google Scholar] [CrossRef] [Green Version]
- Walker, S.J.; Langefeld, C.D.; Zimmerman, K.; Schwartz, M.Z.; Krigsman, A. A molecular biomarker for prediction of clinical outcome in children with ASD, constipation, and intestinal inflammation. Sci. Rep. 2019, 9, 5987. [Google Scholar] [CrossRef]
- Rose, S.; Bennuri, S.C.; Murray, K.F.; Buie, T.; Winter, H.; Frye, R.E. Mitochondrial dysfunction in the gastrointestinal mucosa of children with autism: A blinded case-control study. PLoS ONE 2017, 12, e0186377. [Google Scholar] [CrossRef]
- Vacca, R.A.; Bawari, S.; Valenti, D.; Tewari, D.; Nabavi, S.F.; Shirooie, S.; Sah, A.N.; Volpicella, M.; Braidy, N.; Nabavi, S.M. Down syndrome: Neurobiological alterations and therapeutic targets. Neurosci. Biobehav. Rev. 2019, 98, 234–255. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E. Biomarker of abnormal energy metabolism in children with autism spectrum disorder. N. Am. J. Med. Sci. 2012, 5, 141–147. [Google Scholar] [CrossRef]
- El-Ansary, A.K.; Bacha, A.G.; Al-Ayahdi, L.Y. Plasma fatty acids as diagnostic markers in autistic patients from Saudi Arabia. Lipids Health Dis. 2011, 10, 62. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostafa, G.A.; El-Gamal, H.A.; El-Wakkad, A.S.E.; El-Shorbagy, O.E.; Hamza, M.M. Polyunsaturated fatty acids, carnitine and lactate as biological markers of brain energy in autistic children. Int. J. Child Neuropsychiatry 2005, 2, 179–188. [Google Scholar]
- Frye, R.E.; Melnyk, S.; Macfabe, D.F. Unique acyl-carnitine profiles are potential biomarkers for acquired mitochondrial disease in autism spectrum disorder. Transl. Psychiatry 2013, 3, e220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- MacFabe, D.F.; Cain, D.P.; Rodriguez-Capote, K.; Franklin, A.E.; Hoffman, J.E.; Boon, F.; Taylor, A.R.; Kavaliers, M.; Ossenkopp, K.P. Neurobiological effects of intraventricular propionic acid in rats: Possible role of short chain fatty acids on the pathogenesis and characteristics of autism spectrum disorders. Behav. Brain Res. 2007, 176, 149–169. [Google Scholar] [CrossRef]
- Foley, K.A.; MacFabe, D.F.; Kavaliers, M.; Ossenkopp, K.P. Sexually dimorphic effects of prenatal exposure to lipopolysaccharide, and prenatal and postnatal exposure to propionic acid, on acoustic startle response and prepulse inhibition in adolescent rats: Relevance to autism spectrum disorders. Behav. Brain Res. 2015, 278, 244–256. [Google Scholar] [CrossRef] [PubMed]
- Frye, R.E.; Delatorre, R.; Taylor, H.; Slattery, J.; Melnyk, S.; Chowdhury, N.; James, S.J. Redox metabolism abnormalities in autistic children associated with mitochondrial disease. Transl. Psychiatry 2013, 3, e273. [Google Scholar] [CrossRef]
- Frye, R.E.; Rose, S.; Slattery, J.; MacFabe, D.F. Gastrointestinal dysfunction in autism spectrum disorder: The role of the mitochondria and the enteric microbiome. Microb. Ecol. Health Dis. 2015, 26, 27458. [Google Scholar] [CrossRef] [PubMed]
- Amiot, A.; Tchikviladzé, M.; Joly, F.; Slama, A.; Hatem, D.C.; Jardel, C.; Messing, B.; Lombès, A. Frequency of mitochondrial defects in patients with chronic intestinal pseudo-obstruction. Gastroenterology 2009, 137, 101–109. [Google Scholar] [CrossRef] [PubMed]
- Rossignol, D.A.; Genuis, S.J.; Frye, R.E. Environmental toxicants and autism spectrum disorders: A systematic review. Transl. Psychiatry 2014, 4, e360. [Google Scholar] [CrossRef] [PubMed]
- Parker, W.; Hornik, C.D.; Bilbo, S.; Holzknecht, Z.E.; Gentry, L.; Rao, R.; Lin, S.S.; Herbert, M.R.; Nevison, C.D. The role of oxidative stress, inflammation and acetaminophen exposure from birth to early childhood in the induction of autism. J. Int. Med. Res. 2017, 45, 407–438. [Google Scholar] [CrossRef]
- Atladóttir, H.Ó.; Henriksen, T.B.; Schendel, D.E.; Partner, E.T. Autism after infection, febrile episodes, and antibiotic use during pregnancy: An exploratory study. Pediatrics 2012, 130, e1447–e1454. [Google Scholar] [CrossRef] [Green Version]
- Fallon, J. Could one of the most widely prescribed antibiotics amoxicillin/clavulanate “augmentin” be a risk factor for autism? Med. Hypotheses 2005, 64, 312–315. [Google Scholar] [CrossRef]
- Kalghatgi, S.; Spina, C.S.; Costello, J.C.; Liesa, M.; Morones-Ramirez, J.R.; Slomovic, S.; Molina, A.; Shirihai, O.S.; Collins, J.J. Bactericidal antibiotics induce mitochondrial dysfunction and oxidative damage in Mammalian cells. Sci. Transl. Med. 2013, 5, 192ra85. [Google Scholar] [CrossRef] [Green Version]
- Mezzelani, A.; Landini, M.; Facchiano, F.; Raggi, M.E.; Villa, L.; Molteni, M.; De Santis, B.; Brera, C.; Caroli, A.M.; Milanesi, L.; et al. Environment, dysbiosis, immunity and sex-specific susceptibility: A translational hypothesis for regressive autism pathogenesis. Nutr. Neurosci. 2015, 18, 145–161. [Google Scholar] [CrossRef] [Green Version]
- Good, P. Evidence the U.S. autism epidemic initiated by acetaminophen (Tylenol) is aggravated by oral antibiotic amoxicillin/clavulanate (Augmentin) and now exponentially by herbicide glyphosate (Roundup). Clin. Nutr. Espen 2018, 23, 171–183. [Google Scholar] [CrossRef]
- Bullón, P.; Román-Malo, L.; Marín-Aguilar, F.; Alvarez-Suarez, J.M.; Giampieri, F.; Battino, M.; Cordero, M.D. Lipophilic antioxidants prevent lipopolysaccharide-induced mitochondrial dysfunction through mitochondrial biogenesis improvement. Pharmacol. Res. 2015, 91, 1–8. [Google Scholar] [CrossRef]
- Hsiao, E.Y. Gastrointestinal issues in autism spectrum disorder. Harv. Rev. Psychiatry 2014, 22, 104–111. [Google Scholar] [CrossRef]
- Abdelli, L.S.; Samsam, A.; Naser, S.A. Propionic Acid Induces Gliosis and Neuro-inflammation through Modulation of PTEN/AKT Pathway in Autism Spectrum Disorder. Sci. Rep. 2019, 9, 8824. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harayama, T.; Riezman, H. Understanding the diversity of membrane lipid composition. Nat. Rev. Mol. Cell Biol. 2018, 19, 281–296. [Google Scholar] [CrossRef] [PubMed]
- Ghezzo, A.; Visconti, P.; Abruzzo, P.M.; Bolotta, A.; Ferreri, C.; Gobbi, G.; Malisardi, G.; Manfredini, S.; Marini, M.; Nanetti, L.; et al. Oxidative Stress and Erythrocyte Membrane Alterations in Children with Autism: Correlation with Clinical Features. PLoS ONE 2013, 8, e66418. [Google Scholar] [CrossRef] [Green Version]
- Ferreri, C.; Masi, A.; Sansone, A.; Giacometti, G.; Larocca, A.V.; Menounou, G.; Scanferlato, R.; Tortorella, S.; Rota, D.; Conti, M.; et al. Fatty Acids in Membranes as Homeostatic, Metabolic and Nutritional Biomarkers: Recent Advancements in Analytics and Diagnostics. Diagnostics 2017, 7, 1. [Google Scholar] [CrossRef] [PubMed]
- Giacometti, G.; Ferreri, C.; Sansone, A.; Chatgilialoglu, C.; Marzetti, C.; Spyratou, E.; Georgakilas, G.A.; Marini, M.; Abruzzo, P.M.; Bolotta, A.; et al. High predictive values of RBC membrane-based diagnostics by biophotonics in an integrated approach for Autism Spectrum Disorders. Sci. Rep. 2017, 7, 9854. [Google Scholar] [CrossRef] [PubMed]
- Bolotta, A.; Battistelli, M.; Falcieri, E.; Ghezzo, A.; Manara, M.C.; Manfredini, S.; Marini, M.; Posar, A.; Visconti, P.; Abruzzo, P.M. Oxidative Stress in Autistic Children Alters Erythrocyte Shape in the Absence of Quantitative Protein Alterations and of Loss of Membrane Phospholipid Asymmetry. Oxid. Med. Cell Longev. 2018, 2018, 6430601. [Google Scholar] [CrossRef]
- Anwar, A.; Abruzzo, P.M.; Pasha, S.; Rajpoot, K.; Bolotta, A.; Ghezzo, A.; Marini, M.; Posar, A.; Visconti, P.; Thornalley, P.J.; et al. Advanced glycation endproducts, dityrosine and arginine transporter dysfunction in autism a source of biomarkers for clinical diagnosis. Mol. Autism 2018, 9, 3. [Google Scholar] [CrossRef]
- Abruzzo, P.M.; Matté, A.; Bolotta, A.; Federti, E.; Ghezzo, A.; Guarnieri, T.; Marini, M.; Posar, A.; Siciliano, A.; De Franceschi, L.; et al. Plasma peroxiredoxin changes and inflammatory cytokines support the involvement of neuro-inflammation and oxidative stress in Autism Spectrum Disorder. J. Transl. Med. 2019, 17, 332. [Google Scholar] [CrossRef]
- Angajala, A.; Lim, S.; Phillips, J.B.; Kim, J.H.; Yates, C.; You, Z.; Tan, M. Diverse Roles of Mitochondria in Immune Responses: Novel Insights Into Immuno-Metabolism. Front. Immunol. 2018, 9, 1605. [Google Scholar] [CrossRef]
- Fanos, V.; Noto, A.; Mussap, M. The juniper bush of autism spectrum disorder (ASD): Metabolomics, microbiomics, acetaminophen. What else? J. Pediatr. Neonat. Individual. Med. 2018, 7, e070205. [Google Scholar] [CrossRef]
- Hazlett, H.C.; Gu, H.; Munsell, B.C.; Kim, S.H.; Styner, M.; Wolff, J.J.; Elison, J.T.; Swanson, M.R.; Zhu, H.; Botteron, K.N.; et al. Early brain development in infants at high risk for autism spectrum disorder. Nature 2017, 542, 348–351. [Google Scholar] [CrossRef] [PubMed]
- Zamboni, N.; Saghatelian, A.; Patti, G.J. Defining the metabolome: Size, flux, and regulation. Mol. Cell. 2015, 58, 699–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zampieri, M.; Sekar, K.; Zamboni, N.; Sauer, U. Frontiers of high-throughput metabolomics. Curr. Opin. Chem. Biol. 2017, 36, 15–23. [Google Scholar] [CrossRef]
- Rajula, H.S.R.; Mauri, M.; Fanos, V. Scale-free networks in metabolomics. Bioinformation 2018, 14, 140–144. [Google Scholar] [CrossRef]
- Elsabbagh, M.; Johnson, M.H. Getting answers from babies about autism. Trends Cogn. Sci. 2010, 14, 81–87. [Google Scholar] [CrossRef]
- Estes, M.L.; McAllister, A.K. IMMUNOLOGY. Maternal TH17 cells take a toll on baby’s brain. Science 2016, 351, 919–920. [Google Scholar] [CrossRef] [Green Version]
- Manchia, M.; Fanos, V. Targeting aggression in severe mental illness: The predictive role of genetic, epigenetic, and metabolomic markers. Prog. Neuropsychopharmacol. Biol. Psychiatry 2017, 77, 32–41. [Google Scholar] [CrossRef]
- Faa, G.; Manchia, M.; Pintus, R.; Gerosa, C.; Marcialis, M.A.; Fanos, V. Fetal programming of neuropsychiatric disorders. Birth Defects Res. C Embryo Today. 2016, 108, 207–223. [Google Scholar] [CrossRef]
- Fanni, D.; Gerosa, C.; Rais, M.; Ravarino, A.; Van Eyken, P.; Fanos, V.; Faa, G. The role of neuropathological markers in the interpretation of neuropsychiatric disorders: Focus on fetal and perinatal programming. Neurosci. Lett. 2018, 669, 75–82. [Google Scholar] [CrossRef]
- Hagenbeek, F.A.; Kluft, C.; Hankemeier, T.; Bartels, M.; Draisma, H.H.; Middeldorp, C.M.; Berger, R.; Noto, A.; Lussu, M.; Pool, R.; et al. Discovery of biochemical biomarkers for aggression: A role for metabolomics in psychiatry. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2016, 171, 719–732. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mussap, M.; Noto, A.; Fanos, V. Metabolomics of autism spectrum disorders: Early insights regarding mammalian-microbial cometabolites. Expert Rev. Mol. Diagn. 2016, 16, 869–881. [Google Scholar] [CrossRef] [PubMed]
- Bitar, T.; Mavel, S.; Emond, P.; Nadal-Desbarats, L.; Lefèvre, A.; Mattar, H.; Soufia, M.; Blasco, H.; Vourc’h, P.; Hleihel, W.; et al. Identification of metabolic pathway disturbances using multimodal metabolomics in autistic disorders in a Middle Eastern population. J. Pharm. Biomed. Anal. 2018, 152, 57–65. [Google Scholar] [CrossRef]
- Noto, A.; Fanos, V.; Barberini, L.; Grapov, D.; Fattuoni, C.; Zaffanello, M.; Casanova, A.; Fenu, G.; De Giacomo, A.; De Angelis, M.; et al. The urinary metabolomics profile of an Italian autistic children population and their unaffected siblings. J. Matern. Fetal Neonatal Med. 2014, 27, 46–52. [Google Scholar] [CrossRef] [PubMed]
- Lussu, M.; Noto, A.; Masili, A.; Rinaldi, A.C.; Dessì, A.; De Angelis, M.; De Giacomo, A.; Fanos, V.; Atzori, L.; Francavilla, R. The urinary 1 H-NMR metabolomics profile of an italian autistic children population and their unaffected siblings. Autism Res. 2017, 10, 1058–1066. [Google Scholar] [CrossRef]
- Wang, J.; Barstein, J.; Ethridge, L.E.; Mosconi, M.W.; Takarae, Y.; Sweeney, J.A. Resting state EEG abnormalities in autism spectrum disorders. J. Neurodev. Disord. 2013, 5, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Belmonte, M.K.; Allen, G.; Beckel-Mitchener, A.; Boulanger, L.M.; Carper, R.A.; Webb, S.J. Autism and abnormal development of brain connectivity. J. Neurosci. 2004, 24, 9228–9231. [Google Scholar] [CrossRef]
- Assaf, M.; Jagannathan, K.; Calhoun, V.D.; Miller, L.; Stevens, M.C.; Sahl, R.; O’Boyle, J.G.; Schultz, R.T.; Pearlson, G.D. Abnormal functional connectivity of default mode sub-networks in autism spectrum disorder patients. Neuroimage 2010, 53, 247–256. [Google Scholar] [CrossRef] [Green Version]
- Minshew, N.J.; Keller, T.A. The nature of brain dysfunction in autism: Functional brain imaging studies. Curr. Opin. Neurol. 2010, 23, 124–130. [Google Scholar] [CrossRef] [Green Version]
- Sato, J.R.; Calebe Vidal, M.; de Siqueira Santos, S.; Brauer Massirer, K.; Fujita, A. Complex Network Measures in Autism Spectrum Disorders. IEEE/ACM Trans. Comput. Biol. Bioinform. 2018, 15, 581–587. [Google Scholar] [CrossRef]
- Nelson, C.A., 3rd. Introduction to special issue on The Role of Connectivity in Developmental Disorders: Genetic and Neural Network Approaches. Dev. Sci. 2016, 19, 523. [Google Scholar] [CrossRef] [PubMed]
- Bosl, W.; Tierney, A.; Tager-Flusberg, H.; Nelson, C. EEG complexity as a biomarker for autism spectrum disorder risk. BMC Med. 2011, 9, 18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hutsler, J.J.; Casanova, M.F. Review: Cortical construction in autism spectrum disorder: Columns; connectivity and the subplate. Neuropathol. Appl. Neurobiol. 2016, 42, 115–134. [Google Scholar] [CrossRef] [PubMed]
- Buscema, M.; Vernieri, F.; Massini, G.; Scrascia, F.; Breda, M.; Rossini, P.M.; Grossi, E. An improved I-FAST system for the diagnosis of Alzheimer’s disease from unprocessed electroencephalograms by using robust invariant features. Artif. Intell. Med. 2015, 64, 59–74. [Google Scholar] [CrossRef]
- Grossi, E.; Olivieri, C.; Buscema, M. Diagnosis of autism through EEG processed by advanced computational algorithms: A pilot study. Comput. Methods Programs Biomed. 2017, 142, 73–79. [Google Scholar] [CrossRef]
- Grossi, E.; Buscema, M.; Della Torre, F.; Swatzyna, R.J. The “MS-ROM/IFAST” Model, a Novel Parallel Nonlinear EEG Analysis Technique, Distinguishes ASD Subjects from Children Affected With Other Neuropsychiatric Disorders with High Degree of Accuracy. Clin. EEG Neurosci. 2019, 50, 319–331. [Google Scholar] [CrossRef]
- Wahlqvist, M.L. Ecosystem Dependence of Healthy Localities, Food and People. Ann. Nutr. Metab. 2016, 69, 75–78. [Google Scholar] [CrossRef]
- Godoy-Vitorino, F. Human microbial ecology and the rising new medicine. Ann. Transl. Med. 2019, 7, 342. [Google Scholar] [CrossRef]
- Shedlock, K.; Susi, A.; Gorman, G.H.; Hisle-Gorman, E.; Erdie-Lalena, C.R.; Nylund, C.M. Autism Spectrum Disorders and Metabolic Complications of Obesity. J. Pediatr. 2016, 178, 183–187.e1. [Google Scholar] [CrossRef]
- Penninx, B.W.J.H.; Lange, S.M.M. Metabolic syndrome in psychiatric patients: Overview, mechanisms, and implications. Dialogues Clin. Neurosci. 2018, 20, 63–73. [Google Scholar] [CrossRef]
- Huke, V.; Turk, J.; Saeidi, S.; Kent, A.; Morgan, J.F. Autism spectrum disorders in eating disorder populations: A systematic review. Eur. Eat. Disord. Rev. 2013, 21, 345–351. [Google Scholar] [CrossRef] [PubMed]
- Fahmie, T.A.; Iwata, B.A.; Jann, K.E. Comparison of edible and leisure reinforcers. J. Appl. Behav. Anal. 2015, 48, 331–343. [Google Scholar] [CrossRef] [PubMed]
- Conlon, M.A.; Bird, A.R. The impact of diet and lifestyle on gut microbiota and human health. Nutrients 2014, 7, 17–44. [Google Scholar] [CrossRef] [PubMed]
- Berding, K.; Donovan, S.M. Diet can impact microbiota composition in children with autism spectrum disorder. Front. Neurosci. 2018, 12, 515. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldsmith, J.R.; Sartor, R.B. The role of diet on intestinal microbiota metabolism: Downstream impacts on host immune function and health, and therapeutic implications. J. Gastroenterol. 2014, 49, 785–798. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Murphy, K.J.; Meyer, B.J.; Mori, T.A.; Burke, V.; Mansour, J.; Patch, C.S.; Tapsell, L.C.; Noakes, M.; Clifton, P.A.; Barden, A.; et al. Impact of foods enriched with n-3 long-chain polyunsaturated fatty acids on erythrocyte n-3 levels and cardiovascular risk factors. Br. J. Nutr. 2007, 97, 749–757. [Google Scholar] [CrossRef] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Panisi, C.; Guerini, F.R.; Abruzzo, P.M.; Balzola, F.; Biava, P.M.; Bolotta, A.; Brunero, M.; Burgio, E.; Chiara, A.; Clerici, M.; et al. Autism Spectrum Disorder from the Womb to Adulthood: Suggestions for a Paradigm Shift. J. Pers. Med. 2021, 11, 70. https://doi.org/10.3390/jpm11020070
Panisi C, Guerini FR, Abruzzo PM, Balzola F, Biava PM, Bolotta A, Brunero M, Burgio E, Chiara A, Clerici M, et al. Autism Spectrum Disorder from the Womb to Adulthood: Suggestions for a Paradigm Shift. Journal of Personalized Medicine. 2021; 11(2):70. https://doi.org/10.3390/jpm11020070
Chicago/Turabian StylePanisi, Cristina, Franca Rosa Guerini, Provvidenza Maria Abruzzo, Federico Balzola, Pier Mario Biava, Alessandra Bolotta, Marco Brunero, Ernesto Burgio, Alberto Chiara, Mario Clerici, and et al. 2021. "Autism Spectrum Disorder from the Womb to Adulthood: Suggestions for a Paradigm Shift" Journal of Personalized Medicine 11, no. 2: 70. https://doi.org/10.3390/jpm11020070
APA StylePanisi, C., Guerini, F. R., Abruzzo, P. M., Balzola, F., Biava, P. M., Bolotta, A., Brunero, M., Burgio, E., Chiara, A., Clerici, M., Croce, L., Ferreri, C., Giovannini, N., Ghezzo, A., Grossi, E., Keller, R., Manzotti, A., Marini, M., Migliore, L., ... Fanos, V. (2021). Autism Spectrum Disorder from the Womb to Adulthood: Suggestions for a Paradigm Shift. Journal of Personalized Medicine, 11(2), 70. https://doi.org/10.3390/jpm11020070