
Harnessing Extracellular Matrix Biology for Tumor Drug Delivery

Abstract
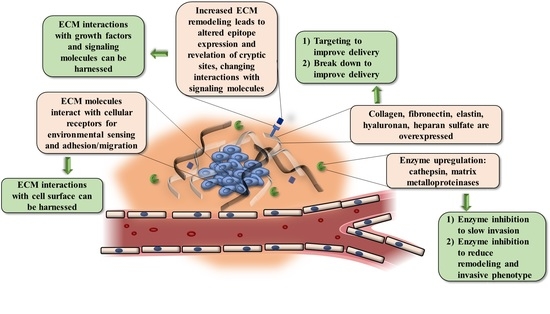
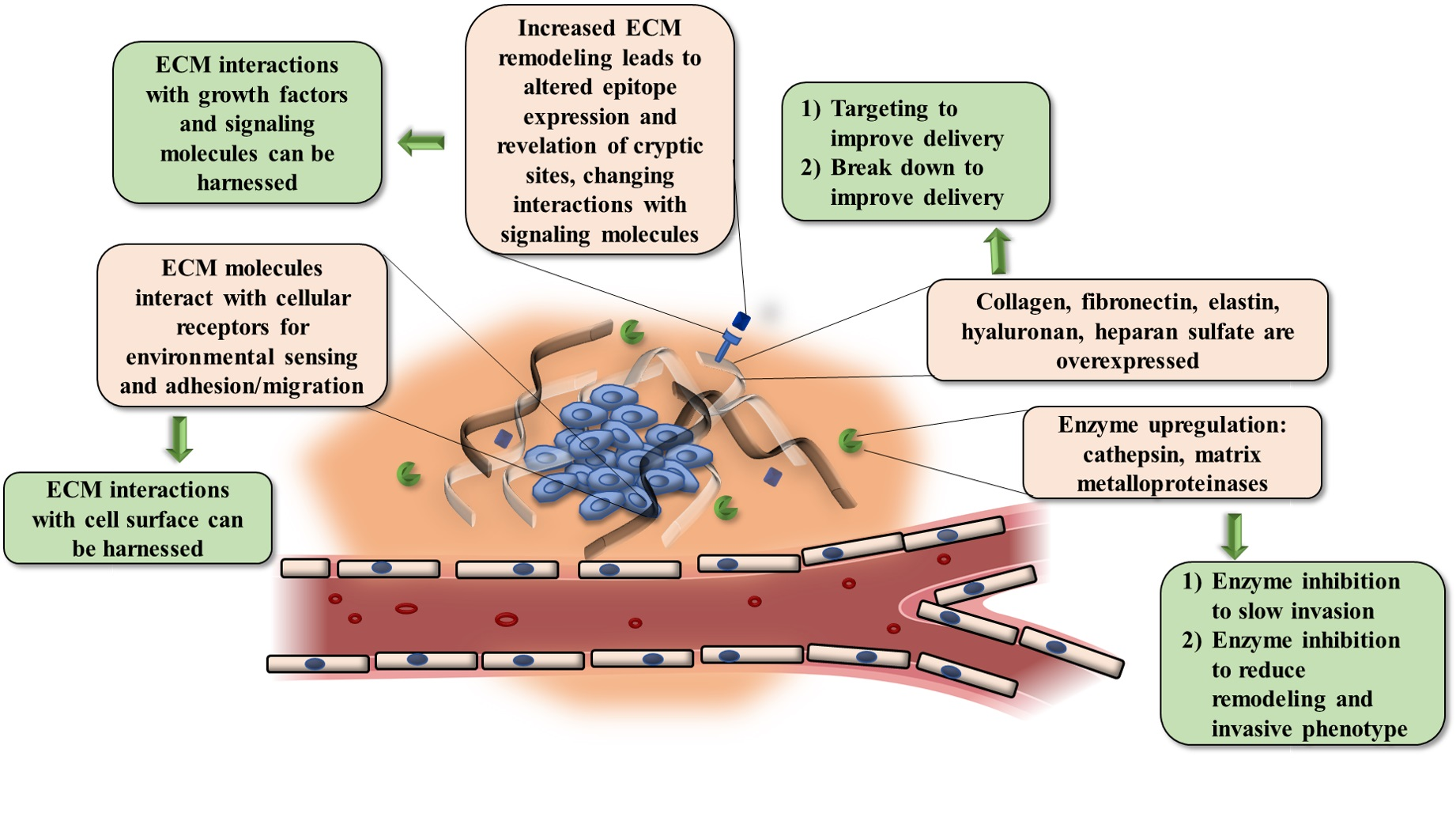
:
1. Introduction
2. Extracellular Matrix: Structure, Function, and Involvement in Cancer Etiology
2.1. Function and Role of the ECM
2.2. Composition of the ECM
2.3. Pathological ECM
2.3.1. Enzyme Upregulation
2.3.2. Weakening of Basement Membrane
2.3.3. Increased ECM Deposition and Stiffness
2.3.4. Causative Factors and Pathways
2.4. In Summary
3. Harnessing ECM Biology
3.1. Priming the ECM to Enhance Drug Delivery
3.2. ECM Molecules as a Therapeutic Target
3.3. ECM as a Prognostic and Diagnostic Biomarker
3.4. In Summary
4. ECM Targeting and Drug Delivery
4.1. ECM Targeting
4.1.1. Advantages of ECM Targeting
4.1.2. Limitations and Barriers to ECM Targeting
4.1.3. ECM Components as Targeting Peptides and as Delivery Targets
Fibronectin as a Target
Collagen as a Target
Hyaluronan as a Target
Tenascin-C as a Target
Heparan Sulfate as a Target
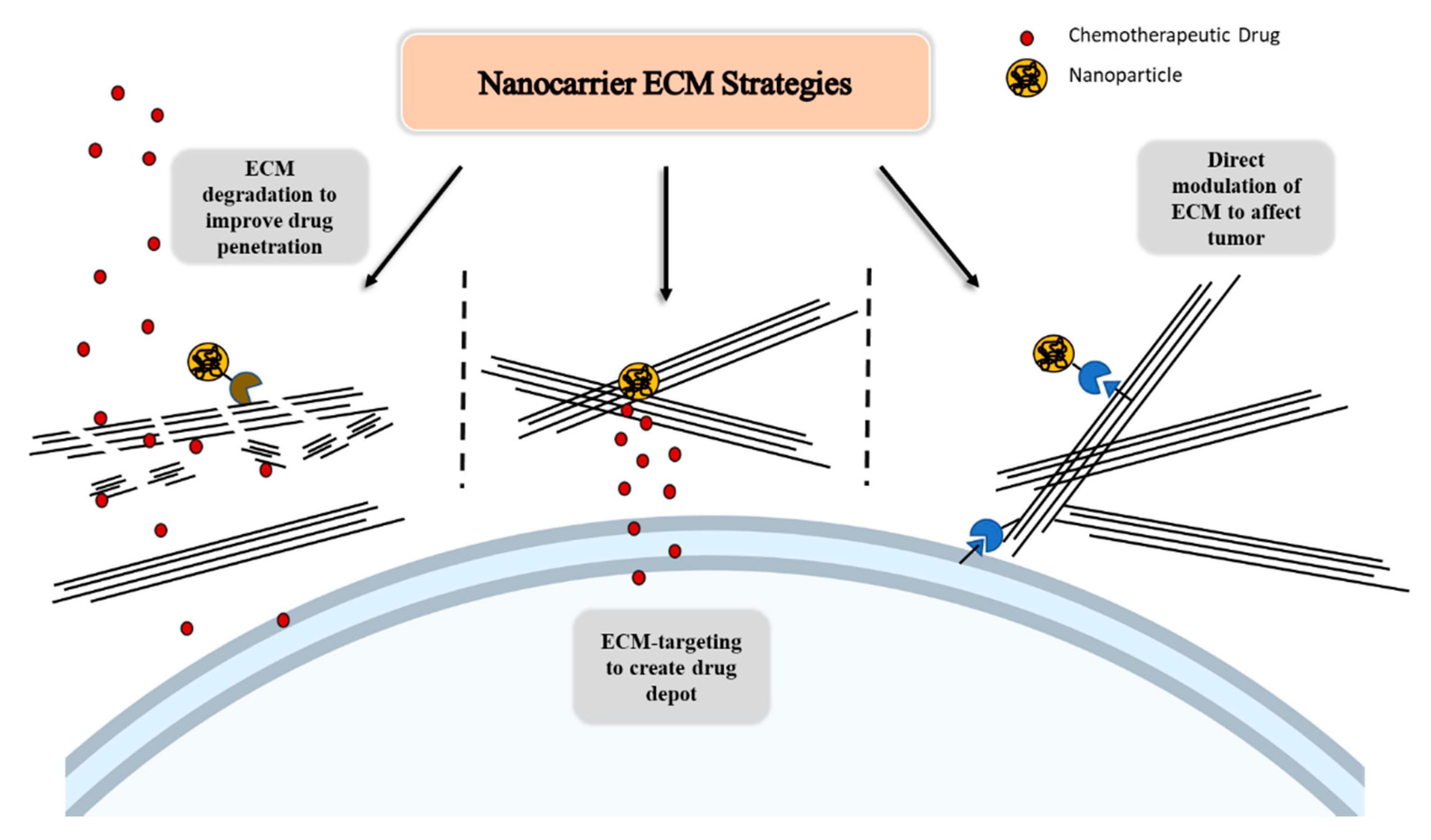
4.2. ECM Strategies to Improve Drug Delivery and Modulate Tumor Growth and Invasion
4.2.1. ECM-Based Strategies to Enhance Drug Penetration through Stroma Modulation
4.2.2. Utilizing the ECM as an Attachment Site for a Drug Delivery System
4.2.3. Modulating the ECM to Reduce Tumor Growth and Invasion
5. Discussion and Future Directions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Theocharis, A.D.; Skandalis, S.S.; Gialeli, C.; Karamanos, N.K. Extracellular matrix structure. Adv. Drug Deliv. Rev. 2016, 97, 4–27. [Google Scholar] [CrossRef] [PubMed]
- Frantz, C.; Stewart, K.M.; Weaver, V.M. The extracellular matrix at a glance. J. Cell Sci. 2010, 123, 4195–4200. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Humphrey, J.D.; Dufresne, E.R.; Schwartz, M.A. Mechanotransduction and extracellular matrix homeostasis. Nat. Rev. Mol. Cell Biol. 2014, 15, 802–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Insua-Rodriguez, J.; Oskarsson, T. The extracellular matrix in breast cancer. Adv. Drug Deliv. Rev. 2016, 97, 41–55. [Google Scholar] [CrossRef] [PubMed]
- Stylianopoulos, T.; Munn, L.L.; Jain, R.K. Reengineering the Physical Microenvironment of Tumors to Improve Drug Delivery and Efficacy: From Mathematical Modeling to Bench to Bedside. Trends Cancer 2018, 4, 292–319. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conklin, M.W.; Eickhoff, J.C.; Riching, K.M.; Pehlke, C.A.; Eliceiri, K.W.; Provenzano, P.P.; Friedl, A.; Keely, P.J. Aligned collagen is a prognostic signature for survival in human breast carcinoma. Am. J. Pathol. 2011, 178, 1221–1232. [Google Scholar] [CrossRef] [PubMed]
- Walker, R.A. The complexities of breast cancer desmoplasia. Breast Cancer Res. 2001, 3, 143–145. [Google Scholar] [CrossRef] [Green Version]
- Hoshiba, T.; Yamaoka, T. Chapter 1: Extracellular Matrix Scaffolds for Tissue Engineering and Biological Research. In Decellularized Extracellular Matrix: Characterization, Fabrication and Applications; The Royal Society of Chemistry: London, UK, 2019; pp. 1–14. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Rozario, T.; DeSimone, D.W. The extracellular matrix in development and morphogenesis: A dynamic view. Dev. Biol. 2010, 341, 126–140. [Google Scholar] [CrossRef] [Green Version]
- Geiger, B.; Yamada, K.M. Molecular architecture and function of matrix adhesions. Cold Spring Harb. Perspect. Biol. 2011, 3, a005033. [Google Scholar] [CrossRef] [Green Version]
- Zaidel-Bar, R.; Geiger, B. The switchable integrin adhesome. J. Cell Sci. 2010, 123, 1385–1388. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Costa, P.; Parsons, M. Chapter Two—New Insights into the Dynamics of Cell Adhesions. In International Review of Cell and Molecular Biology; Jeon, K., Ed.; Academic Press: Cambridge, MA, USA, 2010; Volume 283, pp. 57–91. [Google Scholar]
- Worth, D.C.; Parsons, M. Adhesion dynamics: Mechanisms and measurements. Int. J. Biochem. Cell Biol. 2008, 40, 2397–2409. [Google Scholar] [CrossRef] [PubMed]
- Filipe, E.C.; Chitty, J.L.; Cox, T.R. Charting the unexplored extracellular matrix in cancer. Int J. Exp. Pathol 2018, 99, 58–76. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kai, F.; Drain, A.P.; Weaver, V.M. The Extracellular Matrix Modulates the Metastatic Journey. Dev. Cell 2019, 49, 332–346. [Google Scholar] [CrossRef] [PubMed]
- Walker, C.; Mojares, E.; Del Rio Hernandez, A. Role of Extracellular Matrix in Development and Cancer Progression. Int. J. Mol. Sci. 2018, 19, 3028. [Google Scholar] [CrossRef] [Green Version]
- Humphries, J.D.; Chastney, M.R.; Askari, J.A.; Humphries, M.J. Signal transduction via integrin adhesion complexes. Curr. Opin. Cell Biol. 2019, 56, 14–21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dunehoo, A.L.; Anderson, M.; Majumdar, S.; Kobayashi, N.; Berkland, C.; Siahaan, T.J. Cell Adhesion Molecules for Targeted Drug Delivery. J. Pharm. Sci. 2006, 95, 1856–1872. [Google Scholar] [CrossRef]
- Raab-Westphal, S.; Marshall, J.F.; Goodman, S.L. Integrins as Therapeutic Targets: Successes and Cancers. Cancers 2017, 9, 110. [Google Scholar] [CrossRef]
- Goodman, S.L.; Picard, M. Integrins as therapeutic targets. Trends Pharmacol. Sci. 2012, 33, 405–412. [Google Scholar] [CrossRef]
- Sainio, A.; Järveläinen, H. Extracellular matrix-cell interactions: Focus on therapeutic applications. Cell. Signal. 2020, 66, 109487. [Google Scholar] [CrossRef]
- Hastings, J.F.; Skhinas, J.N.; Fey, D.; Croucher, D.R.; Cox, T.R. The extracellular matrix as a key regulator of intracellular signalling networks. Br. J. Pharmacol. 2019, 176, 82–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taipale, J.; Keski-Oja, J. Growth factors in the extracellular matrix. Faseb J. 1997, 11, 51–59. [Google Scholar] [CrossRef] [PubMed]
- Isaacson, K.J.; Martin Jensen, M.; Subrahmanyam, N.B.; Ghandehari, H. Matrix-metalloproteinases as targets for controlled delivery in cancer: An analysis of upregulation and expression. J. Control. Release 2017, 259, 62–75. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O. The extracellular matrix: Not just pretty fibrils. Science 2009, 326, 1216–1219. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. 2016, 49, 10–24. [Google Scholar] [CrossRef]
- Ricard-Blum, S. The collagen family. Cold Spring Harb. Perspect. Biol. 2011, 3, a004978. [Google Scholar] [CrossRef] [Green Version]
- Karsdal, M.A.; Nielsen, S.H.; Leeming, D.J.; Langholm, L.L.; Nielsen, M.J.; Manon-Jensen, T.; Siebuhr, A.; Gudmann, N.S.; Rønnow, S.; Sand, J.M.; et al. The good and the bad collagens of fibrosis—Their role in signaling and organ function. Adv. Drug Deliv. Rev. 2017, 121, 43–56. [Google Scholar] [CrossRef]
- Kalluri, R. Basement membranes: Structure, assembly and role in tumour angiogenesis. Nat. Rev. Cancer 2003, 3, 422–433. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. The hallmarks of cancer. Cell 2000, 100, 57–70. [Google Scholar] [CrossRef] [Green Version]
- Pickup, M.W.; Mouw, J.K.; Weaver, V.M. The extracellular matrix modulates the hallmarks of cancer. EMBO Rep. 2014, 15, 1243–1253. [Google Scholar] [CrossRef] [Green Version]
- Decock, J.; Thirkettle, S.; Wagstaff, L.; Edwards, D.R. Matrix metalloproteinases: Protective roles in cancer. J. Cell Mol. Med. 2011, 15, 1254–1265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- López-Otín, C.; Palavalli, L.H.; Samuels, Y. Protective roles of matrix metalloproteinases: From mouse models to human cancer. Cell Cycle 2009, 8, 3657–3662. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duong, L.T.; Leung, A.T.; Langdahl, B. Cathepsin K Inhibition: A New Mechanism for the Treatment of Osteoporosis. Calcif. Tissue Int. 2016, 98, 381–397. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.; Hao, S.; Young, P.; Zhang, H. Targeting Cathepsin B for Cancer Therapies. Horiz. Cancer Res. 2015, 56, 23–40. [Google Scholar] [PubMed]
- Vincenti, M.P.; Brinckerhoff, C.E. Transcriptional regulation of collagenase (MMP-1, MMP-13) genes in arthritis: Integration of complex signaling pathways for the recruitment of gene-specific transcription factors. Arthr. Res. Ther. 2001, 4, 157. [Google Scholar] [CrossRef]
- Li, H.; Qiu, Z.; Li, F.; Wang, C. The relationship between MMP-2 and MMP-9 expression levels with breast cancer incidence and prognosis. Oncol. Lett. 2017, 14, 5865–5870. [Google Scholar] [CrossRef] [Green Version]
- Mirastschijski, U.; Lupše, B.; Maedler, K.; Sarma, B.; Radtke, A.; Belge, G.; Dorsch, M.; Wedekind, D.; McCawley, L.J.; Boehm, G.; et al. Matrix Metalloproteinase-3 is Key Effector of TNF-α-Induced Collagen Degradation in Skin. Int. J. Mol. Sci. 2019, 20, 5234. [Google Scholar] [CrossRef] [Green Version]
- Han, J.-C.; Li, X.-D.; Du, J.; Xu, F.; Wei, Y.-J.; Li, H.-B.; Zhang, Y.-J. Elevated matrix metalloproteinase-7 expression promotes metastasis in human lung carcinoma. World J. Surg. Oncol. 2015, 13, 5. [Google Scholar] [CrossRef] [Green Version]
- Balbín, M.; Fueyo, A.; Tester, A.M.; Pendás, A.M.; Pitiot, A.S.; Astudillo, A.; Overall, C.M.; Shapiro, S.D.; López-Otín, C. Loss of collagenase-2 confers increased skin tumor susceptibility to male mice. Nat. Genet. 2003, 35, 252–257. [Google Scholar] [CrossRef]
- Jabłońska-Trypuć, A.; Matejczyk, M.; Rosochacki, S. Matrix metalloproteinases (MMPs), the main extracellular matrix (ECM) enzymes in collagen degradation, as a target for anticancer drugs. J. Enzyme Inhib. Med. Chem. 2016, 31, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Sudhan, D.R.; Siemann, D.W. Cathepsin L targeting in cancer treatment. Pharmacol. Ther. 2015, 155, 105–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Richard, D.A.W.; Rich, W.; Christopher, J.S.; Roberta, E.B. Cathepsin S: Therapeutic, diagnostic, and prognostic potential. Biol. Chem. 2015, 396, 867–882. [Google Scholar] [CrossRef]
- Smith-Mungo, L.I.; Kagan, H.M. Lysyl oxidase: Properties, regulation and multiple functions in biology. Matrix Biol. 1998, 16, 387–398. [Google Scholar] [CrossRef]
- Planche, A.; Bacac, M.; Provero, P.; Fusco, C.; Delorenzi, M.; Stehle, J.-C.; Stamenkovic, I. Identification of prognostic molecular features in the reactive stroma of human breast and prostate cancer. PLoS ONE 2011, 6, e18640. [Google Scholar] [CrossRef]
- Cox, T.R.; Gartland, A.; Erler, J.T. Lysyl Oxidase, a Targetable Secreted Molecule Involved in Cancer Metastasis. Cancer Res. 2016, 76, 188–192. [Google Scholar] [CrossRef] [Green Version]
- Miles, F.L.; Sikes, R.A. Insidious changes in stromal matrix fuel cancer progression. Mol. Cancer Res. 2014, 12, 297–312. [Google Scholar] [CrossRef] [Green Version]
- Reiland, J.; Kempf, D.; Roy, M.; Denkins, Y.; Marchetti, D. FGF2 binding, signaling, and angiogenesis are modulated by heparanase in metastatic melanoma cells. Neoplasia 2006, 8, 596–606. [Google Scholar] [CrossRef] [Green Version]
- Mierke, C.T. The matrix environmental and cell mechanical properties regulate cell migration and contribute to the invasive phenotype of cancer cells. Rep. Prog. Phys. 2019, 82, 064602. [Google Scholar] [CrossRef]
- Giavazzi, R.; Garofalo, A.; Ferri, C.; Lucchini, V.; Bone, E.A.; Chiari, S.; Brown, P.D.; Nicoletti, M.I.; Taraboletti, G. Batimastat, a synthetic inhibitor of matrix metalloproteinases, potentiates the antitumor activity of cisplatin in ovarian carcinoma xenografts. Clin. Cancer Res. 1998, 4, 985. [Google Scholar]
- Xiao, L.; Wang, M. Batimastat Nanoparticles Associated with Transcatheter Arterial Chemoembolization Decrease Hepatocellular Carcinoma Recurrence. Cell Biochem. Biophys. 2014, 70, 269–272. [Google Scholar] [CrossRef]
- Sinno, M.; Biagioni, S.; Ajmone-Cat, M.A.; Pafumi, I.; Caramanica, P.; Medda, V.; Tonti, G.; Minghetti, L.; Mannello, F.; Cacci, E. The matrix metalloproteinase inhibitor marimastat promotes neural progenitor cell differentiation into neurons by gelatinase-independent TIMP-2-dependent mechanisms. Stem Cells Dev. 2013, 22, 345–358. [Google Scholar] [CrossRef] [PubMed]
- Cathcart, J.; Pulkoski-Gross, A.; Cao, J. Targeting matrix metalloproteinases in cancer: Bringing new life to old ideas. Genes Dis. 2015, 2, 26–34. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Levin, M.; Udi, Y.; Solomonov, I.; Sagi, I. Next generation matrix metalloproteinase inhibitors—Novel strategies bring new prospects. BBA Mol. Cell Res. 2017, 1864, 1927–1939. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G. Tissue inhibitors of metalloproteinases. Genome Biol. 2011, 12, 233. [Google Scholar] [CrossRef] [PubMed]
- Sela-Passwell, N.; Kikkeri, R.; Dym, O.; Rozenberg, H.; Margalit, R.; Arad-Yellin, R.; Eisenstein, M.; Brenner, O.; Shoham, T.; Danon, T.; et al. Antibodies targeting the catalytic zinc complex of activated matrix metalloproteinases show therapeutic potential. Nat. Med. 2012, 18, 143–147. [Google Scholar] [CrossRef] [PubMed]
- Fields, G.B. Mechanisms of Action of Novel Drugs Targeting Angiogenesis-Promoting Matrix Metalloproteinases. Front. Immunol. 2019, 10, 1278. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bissett, D.; O’Byrne, K.J.; Von Pawel, J.; Gatzemeier, U.; Price, A.; Nicolson, M.; Mercier, R.; Mazabel, E.; Penning, C.; Zhang, M.H.; et al. Phase III Study of Matrix Metalloproteinase Inhibitor Prinomastat in Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2005, 23, 842–849. [Google Scholar] [CrossRef]
- Duong, L.T.; Wesolowski, G.A.; Leung, P.; Oballa, R.; Pickarski, M. Efficacy of a cathepsin K inhibitor in a preclinical model for prevention and treatment of breast cancer bone metastasis. Mol. Cancer Ther. 2014, 13, 2898–2909. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Dare, L.; Vasko-Moser, J.A.; James, I.E.; Blake, S.M.; Rickard, D.J.; Hwang, S.M.; Tomaszek, T.; Yamashita, D.S.; Marquis, R.W.; et al. A highly potent inhibitor of cathepsin K (relacatib) reduces biomarkers of bone resorption both in vitro and in an acute model of elevated bone turnover in vivo in monkeys. Bone 2007, 40, 122–131. [Google Scholar] [CrossRef]
- Jensen, A.; Wynne, C.; Ramirez, G.; He, W.; Song, Y.; Berd, Y.; Wang, H.; Mehta, A.; Lombardi, A. The Cathepsin K Inhibitor Odanacatib Suppresses Bone Resorption in Women With Breast Cancer and Established Bone Metastases: Results of a 4-Week, Double-Blind, Randomized, Controlled Trial. Clin. Breast Cancer 2010, 10, 452–458. [Google Scholar] [CrossRef]
- Alberca, L.N.; Chuguransky, S.R.; Álvarez, C.L.; Talevi, A.; Salas-Sarduy, E. In silico Guided Drug Repurposing: Discovery of New Competitive and Non-competitive Inhibitors of Falcipain-2. Front Chem. 2019, 7, 534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilder, C.L.; Walton, C.; Watson, V.; Stewart, F.A.A.; Johnson, J.; Peyton, S.R.; Payne, C.K.; Odero-Marah, V.; Platt, M.O. Differential cathepsin responses to inhibitor-induced feedback: E-64 and cystatin C elevate active cathepsin S and suppress active cathepsin L in breast cancer cells. Int. J. Biochem. Cell Biol. 2016, 79, 199–208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fleming, J.M.; Yeyeodu, S.T.; McLaughlin, A.; Schuman, D.; Taylor, D.K. In Situ Drug Delivery to Breast Cancer-Associated Extracellular Matrix. ACS Chem. Biol. 2018, 13, 2825–2840. [Google Scholar] [CrossRef] [PubMed]
- Doherty, G.J.; Tempero, M.; Corrie, P.G. HALO-109-301: A Phase III trial of PEGPH20 (with gemcitabine and nab-paclitaxel) in hyaluronic acid-high stage IV pancreatic cancer. Future Oncol. 2018, 14, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Goodman, T.T.; Olive, P.L.; Pun, S.H. Increased nanoparticle penetration in collagenase-treated multicellular spheroids. Int. J. Nanomed. 2007, 2, 265–274. [Google Scholar]
- Lee, S.; Han, H.; Koo, H.; Na, J.H.; Yoon, H.Y.; Lee, K.E.; Lee, H.; Kim, H.; Kwon, I.C.; Kim, K. Extracellular matrix remodeling in vivo for enhancing tumor-targeting efficiency of nanoparticle drug carriers using the pulsed high intensity focused ultrasound. J. Control. Release 2017, 263, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Frazier, N.; Payne, A.; De Bever, J.; Dillon, C.; Panda, A.; Subrahmanyam, N.; Ghandehari, H. High intensity focused ultrasound hyperthermia for enhanced macromolecular delivery. J. Control. Release 2016, 241, 186–193. [Google Scholar] [CrossRef] [PubMed]
- Chronopoulos, A.; Robinson, B.; Sarper, M.; Cortes, E.; Auernheimer, V.; Lachowski, D.; Attwood, S.; García, R.; Ghassemi, S.; Fabry, B.; et al. ATRA mechanically reprograms pancreatic stellate cells to suppress matrix remodelling and inhibit cancer cell invasion. Nat. Commun. 2016, 7, 12630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Röhrig, F.; Vorlová, S.; Hoffmann, H.; Wartenberg, M.; Escorcia, F.E.; Keller, S.; Tenspolde, M.; Weigand, I.; Gätzner, S.; Manova, K.; et al. VEGF-ablation therapy reduces drug delivery and therapeutic response in ECM-dense tumors. Oncogene 2017, 36, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sawyer, A.J.; Kyriakides, T.R. Matricellular proteins in drug delivery: Therapeutic targets, active agents, and therapeutic localization. Adv. Drug Deliv. Rev. 2016, 97, 56–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Winer, A.; Adams, S.; Mignatti, P. Matrix Metalloproteinase Inhibitors in Cancer Therapy: Turning Past Failures Into Future Successes. Mol. Cancer Ther. 2018, 17, 1147–1155. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, H.-Y.; Gu, W.-J.; Wang, C.-Z.; Ji, X.-J.; Mu, Y.-M. Matrix metalloproteinase-9 and -2 and tissue inhibitor of matrix metalloproteinase-2 in invasive pituitary adenomas: A systematic review and meta-analysis of case-control trials. Medicine 2016, 95, e3904. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Wang, G.; Gu, W.; Mu, Y. Cathepsin K: The association between Cathepsin K expression and sphenoid sinus invasion of pituitary adenomas. Med. Hypotheses 2016, 97, 88–89. [Google Scholar] [CrossRef] [PubMed]
- Lu, J.; Wang, M.; Wang, Z.; Fu, Z.; Lu, A.; Zhang, G. Advances in the discovery of cathepsin K inhibitors on bone resorption. J. Enzyme Inhib. Med. Chem. 2018, 33, 890–904. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, K.; Iwatake, M.; Okamoto, K.; Yamada, S.-I.; Umeda, M.; Tsukuba, T. Cathepsin K modulates invasion, migration and adhesion of oral squamous cell carcinomas in vitro. Oral Diseases 2017, 23, 518–525. [Google Scholar] [CrossRef] [PubMed]
- Le Gall, C.; Bellahcène, A.; Bonnelye, E.; Gasser, J.A.; Castronovo, V.; Green, J.; Zimmermann, J.; Clézardin, P. A Cathepsin K Inhibitor Reduces Breast Cancer–Induced Osteolysis and Skeletal Tumor Burden. Cancer Res. 2007, 67, 9894–9902. [Google Scholar] [CrossRef] [Green Version]
- Wojtowicz-Praga, S.M.; Dickson, R.B.; Hawkins, M.J. Matrix metalloproteinase inhibitors. Invest. New Drugs 1997, 15, 61–75. [Google Scholar] [CrossRef]
- Rasmussen, H.S.; McCann, P.P. Matrix Metalloproteinase Inhibition as a Novel Anticancer Strategy: A Review with Special Focus on Batimastat and Marimastat. Pharmacol. Ther. 1997, 75, 69–75. [Google Scholar] [CrossRef]
- Shevde, L.A.; Samant, R.S. Role of osteopontin in the pathophysiology of cancer. Matrix Biol. 2014, 37, 131–141. [Google Scholar] [CrossRef]
- Zhao, H.; Chen, Q.; Alam, A.; Cui, J.; Suen, K.C.; Soo, A.P.; Eguchi, S.; Gu, J.; Ma, D. The role of osteopontin in the progression of solid organ tumour. Cell Death Disease 2018, 9, 356. [Google Scholar] [CrossRef]
- Ramadan, A.; Afifi, N.; Yassin, N.Z.; Abdel-Rahman, R.F.; Abd El-Rahman, S.S.; Fayed, H.M. Mesalazine, an osteopontin inhibitor: The potential prophylactic and remedial roles in induced liver fibrosis in rats. Chem. Biol. Interact. 2018, 289, 109–118. [Google Scholar] [CrossRef] [PubMed]
- Wei, R.; Wong, J.P.C.; Kwok, H.F. Osteopontin—A promising biomarker for cancer therapy. J. Cancer 2017, 8, 2173–2183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yee, K.O.; Connolly, C.M.; Duquette, M.; Kazerounian, S.; Washington, R.; Lawler, J. The effect of thrombospondin-1 on breast cancer metastasis. Breast Cancer Res. Treat. 2009, 114, 85–96. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Daubon, T.; Léon, C.; Clarke, K.; Andrique, L.; Salabert, L.; Darbo, E.; Pineau, R.; Guérit, S.; Maitre, M.; Dedieu, S.; et al. Deciphering the complex role of thrombospondin-1 in glioblastoma development. Nat. Commun. 2019, 10, 1146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xie, X.-S.; Li, F.-Y.; Liu, H.-C.; Deng, Y.; Li, Z.; Fan, J.-M. LSKL, a peptide antagonist of thrombospondin-1, attenuates renal interstitial fibrosis in rats with unilateral ureteral obstruction. Arch. Pharmacal Res. 2010, 33, 275–284. [Google Scholar] [CrossRef] [PubMed]
- Chuay-Yeng, K.; Yin-Ping, S.; Boon-Huat, B.; George, W.Y. Targeting Heparan Sulfate Proteoglycans in Breast Cancer Treatment. Recent Pat. Anti Cancer Drug Discov. 2008, 3, 151–158. [Google Scholar] [CrossRef]
- Knelson, E.H.; Nee, J.C.; Blobe, G.C. Heparan sulfate signaling in cancer. Trends Biochem. Sci. 2014, 39, 277–288. [Google Scholar] [CrossRef] [Green Version]
- Xu, S.; Xu, H.; Wang, W.; Li, S.; Li, H.; Li, T.; Zhang, W.; Yu, X.; Liu, L. The role of collagen in cancer: From bench to bedside. J. Transl. Med. 2019, 17, 309. [Google Scholar] [CrossRef] [Green Version]
- Han, X.; Wang, W.; He, J.; Jiang, L.; Li, X. Osteopontin as a biomarker for osteosarcoma therapy and prognosis. Oncol. Lett. 2019, 17, 2592–2598. [Google Scholar] [CrossRef] [Green Version]
- Obonai, T.; Fuchigami, H.; Furuya, F.; Kozuka, N.; Yasunaga, M.; Matsumura, Y. Tumour imaging by the detection of fibrin clots in tumour stroma using an anti-fibrin Fab fragment. Sci. Rep. 2016, 6, 23613. [Google Scholar] [CrossRef]
- Zhou, Z.; Qutaish, M.; Han, Z.; Schur, R.M.; Liu, Y.; Wilson, D.L.; Lu, Z.-R. MRI detection of breast cancer micrometastases with a fibronectin-targeting contrast agent. Nat. Commun. 2015, 6, 7984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, R.K. Transport of molecules in the tumor interstitium: A review. Cancer Res. 1987, 47, 3039–3051. [Google Scholar] [PubMed]
- Jain, R.K.; Stylianopoulos, T. Delivering nanomedicine to solid tumors. Nat. Rev. Clin. Oncol. 2010, 7, 653–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stylianopoulos, T.; Poh, M.-Z.; Insin, N.; Bawendi, M.G.; Fukumura, D.; Munn, L.L.; Jain, R.K. Diffusion of particles in the extracellular matrix: The effect of repulsive electrostatic interactions. Biophys. J. 2010, 99, 1342–1349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tomasetti, L.; Breunig, M. Preventing Obstructions of Nanosized Drug Delivery Systems by the Extracellular Matrix. Adv. Healthcare Mater. 2018, 7, 1700739. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Yang, K.; Tuguntaev, R.G.; Mozhi, A.; Zhang, J.; Wang, P.C.; Liang, X.-J. Targeting tumor microenvironment with PEG-based amphiphilic nanoparticles to overcome chemoresistance. Nanomedicine 2016, 12, 269–286. [Google Scholar] [CrossRef] [Green Version]
- Hwang, J.; Sullivan, M.O.; Kiick, K.L. Targeted Drug Delivery via the Use of ECM-Mimetic Materials. Front. Bioeng. Biotechnol. 2020, 8, 69. [Google Scholar] [CrossRef] [Green Version]
- Raavé, R.; Van Kuppevelt, T.H.; Daamen, W.F. Chemotherapeutic drug delivery by tumoral extracellular matrix targeting. J. Control. Release 2018, 274, 1–8. [Google Scholar] [CrossRef]
- Han, Z.; Lu, Z.-R. Targeting Fibronectin for Cancer Imaging and Therapy. J. Mater. Chem. B 2017, 5, 639–654. [Google Scholar] [CrossRef] [Green Version]
- Kumra, H.; Reinhardt, D.P. Fibronectin-targeted drug delivery in cancer. Adv. Drug Deliv. Rev. 2016, 97, 101–110. [Google Scholar] [CrossRef]
- Mees, G.; Dierckx, R.; Mertens, K.; Vermeire, S.; Van Steenkiste, M.; Reutelingsperger, C.; D’Asseler, Y.; Peremans, K.; Van Damme, N.; Van de Wiele, C. 99mTc-Labeled Tricarbonyl His-CNA35 as an Imaging Agent for the Detection of Tumor Vasculature. J. Nucl. Med. 2012, 53, 464–471. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rothenfluh, D.A.; Bermudez, H.; O’Neil, C.P.; Hubbell, J.A. Biofunctional polymer nanoparticles for intra-articular targeting and retention in cartilage. Nat. Mater. 2008, 7, 248–254. [Google Scholar] [CrossRef] [PubMed]
- Bennink, L.L.; Li, Y.; Kim, B.; Shin, I.J.; San, B.H.; Zangari, M.; Yoon, D.; Yu, S.M. Visualizing collagen proteolysis by peptide hybridization: From 3D cell culture to in vivo imaging. Biomaterials 2018, 183, 67–76. [Google Scholar] [CrossRef] [PubMed]
- Wahyudi, H.; Reynolds, A.A.; Li, Y.; Owen, S.C.; Yu, S.M. Targeting collagen for diagnostic imaging and therapeutic delivery. J. Control. Release 2016, 240, 323–331. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goldbloom-Helzner, L.; Hao, D.; Wang, A. Developing Regenerative Treatments for Developmental Defects, Injuries, and Diseases Using Extracellular Matrix Collagen-Targeting Peptides. Int. J. Mol. Sci. 2019, 20, 4072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, M.Y.; Kim, O.R.; Choi, Y.S.; Lee, H.; Park, K.; Lee, C.-T.; Kang, K.W.; Jeong, S. Selection and characterization of tenascin C targeting peptide. Mol. Cells 2012, 33, 71–77. [Google Scholar] [CrossRef]
- Ikemoto, H.; Lingasamy, P.; Anton Willmore, A.-M.; Hunt, H.; Kurm, K.; Tammik, O.; Scodeller, P.; Simón-Gracia, L.; Kotamraju, V.R.; Lowy, A.M.; et al. Hyaluronan-binding peptide for targeting peritoneal carcinomatosis. Tumour. Biol. 2017, 39, 1010428317701628. [Google Scholar] [CrossRef] [Green Version]
- Depau, L.; Brunetti, J.; Falciani, C.; Scali, S.; Riolo, G.; Mandarini, E.; Pini, A.; Bracci, L. Coupling to a cancer-selective heparan-sulfate-targeted branched peptide can by-pass breast cancer cell resistance to methotrexate. Oncotarget 2017, 8, 76141–76152. [Google Scholar] [CrossRef] [Green Version]
- Jiang, K.; Song, X.; Yang, L.; Li, L.; Wan, Z.; Sun, X.; Gong, T.; Lin, Q.; Zhang, Z. Enhanced antitumor and anti-metastasis efficacy against aggressive breast cancer with a fibronectin-targeting liposomal doxorubicin. J. Control. Release 2018, 271, 21–30. [Google Scholar] [CrossRef]
- Romijn, R.A.P.; Bouma, B.; Wuyster, W.; Gros, P.; Kroon, J.; Sixma, J.J.; Huizinga, E.G. Identification of the Collagen-binding Site of the von Willebrand Factor A3-domain. J. Biol. Chem. 2001, 276, 9985–9991. [Google Scholar] [CrossRef] [Green Version]
- Sasaki, K.; Ishihara, J.; Ishihara, A.; Miura, R.; Mansurov, A.; Fukunaga, K.; Hubbell, J.A. Engineered collagen-binding serum albumin as a drug conjugate carrier for cancer therapy. Sci. Adv. 2019, 5, eaaw6081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platt, V.M.; Szoka, F.C. Anticancer Therapeutics: Targeting Macromolecules and Nanocarriers to Hyaluronan or CD44, a Hyaluronan Receptor. Mol. Pharm. 2008, 5, 474–486. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shuster, S.; Frost, G.I.; Csoka, A.B.; Formby, B.; Stern, R. Hyaluronidase reduces human breast cancer xenografts in SCID mice. Int. J. Cancer 2002, 102, 192–197. [Google Scholar] [CrossRef] [PubMed]
- Kultti, A.; Li, X.; Jiang, P.; Thompson, C.B.; Frost, G.I.; Shepard, H.M. Therapeutic Targeting of Hyaluronan in the Tumor Stroma. Cancers 2012, 4, 873–903. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stern, R.; Shuster, S.; Wiley, T.S.; Formby, B. Hyaluronidase can modulate expression of CD44. Exp. Cell Res. 2001, 266, 167–176. [Google Scholar] [CrossRef]
- Simon Davis, D.A.; Parish, C.R. Heparan Sulfate: A Ubiquitous Glycosaminoglycan with Multiple Roles in Immunity. Front. Immunol. 2013, 4. [Google Scholar] [CrossRef] [Green Version]
- Neesse, A.; Michl, P.; Frese, K.K.; Feig, C.; Cook, N.; Jacobetz, M.A.; Lolkema, M.P.; Buchholz, M.; Olive, K.P.; Gress, T.M.; et al. Stromal biology and therapy in pancreatic cancer. Gut 2011, 60, 861–868. [Google Scholar] [CrossRef]
- Pothula, S.P.; Xu, Z.; Goldstein, D.; Pirola, R.C.; Wilson, J.S.; Apte, M.V. Key role of pancreatic stellate cells in pancreatic cancer. Cancer Lett. 2016, 381, 194–200. [Google Scholar] [CrossRef]
- Looi, C.-K.; Chung, F.F.-L.; Leong, C.-O.; Wong, S.-F.; Rosli, R.; Mai, C.-W. Therapeutic challenges and current immunomodulatory strategies in targeting the immunosuppressive pancreatic tumor microenvironment. J. Exp. Clin. Cancer Res. 2019, 38, 162. [Google Scholar] [CrossRef]
- Feig, C.; Gopinathan, A.; Neesse, A.; Chan, D.S.; Cook, N.; Tuveson, D.A. The pancreas cancer microenvironment. Clin. Cancer Res. 2012, 18, 4266–4276. [Google Scholar] [CrossRef] [Green Version]
- Dolor, A.; Szoka, F.C. Digesting a Path Forward: The Utility of Collagenase Tumor Treatment for Improved Drug Delivery. Mol. Pharm. 2018, 15, 2069–2083. [Google Scholar] [CrossRef] [PubMed]
- Ji, T.; Lang, J.; Wang, J.; Cai, R.; Zhang, Y.; Qi, F.; Zhang, L.; Zhao, X.; Wu, W.; Hao, J.; et al. Designing Liposomes To Suppress Extracellular Matrix Expression To Enhance Drug Penetration and Pancreatic Tumor Therapy. ACS Nano 2017, 11, 8668–8678. [Google Scholar] [CrossRef] [PubMed]
- Zinger, A.; Koren, L.; Adir, O.; Poley, M.; Alyan, M.; Yaari, Z.; Noor, N.; Krinsky, N.; Simon, A.; Gibori, H.; et al. Collagenase Nanoparticles Enhance the Penetration of Drugs into Pancreatic Tumors. ACS Nano 2019, 13, 11008–11021. [Google Scholar] [CrossRef] [PubMed]
- Han, X.; Li, Y.; Xu, Y.; Zhao, X.; Zhang, Y.; Yang, X.; Wang, Y.; Zhao, R.; Anderson, G.J.; Zhao, Y.; et al. Reversal of pancreatic desmoplasia by re-educating stellate cells with a tumour microenvironment-activated nanosystem. Nat. Commun. 2018, 9, 3390. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, T.; Hu, H.; Wang, H.; Li, J.; Wang, Z.; Wang, J.; Wang, S.; Zhang, Z.; Li, Y. Bioinspired lipoproteins-mediated photothermia remodels tumor stroma to improve cancer cell accessibility of second nanoparticles. Nat. Commun. 2019, 10, 3322. [Google Scholar] [CrossRef] [Green Version]
- Van der Steen, S.C.; Raavé, R.; Langerak, S.; Van Houdt, L.; Van Duijnhoven, S.M.; Van Lith, S.A.; Massuger, L.F.; Daamen, W.F.; Leenders, W.P.; Van Kuppevelt, T.H. Targeting the extracellular matrix of ovarian cancer using functionalized, drug loaded lyophilisomes. Eur. J. Pharm. Biopharm. 2017, 113, 229–239. [Google Scholar] [CrossRef]
- Stras, S.; Howe, A.; Prasad, A.; Salerno, D.; Bhatavdekar, O.; Sofou, S. Growth of Metastatic Triple-Negative Breast Cancer Is Inhibited by Deep Tumor-Penetrating and Slow Tumor-Clearing Chemotherapy: The Case of Tumor-Adhering Liposomes with Interstitial Drug Release. Mol. Pharm. 2020, 17, 118–131. [Google Scholar] [CrossRef]
- Stras, S.; Holleran, T.; Howe, A.; Sofou, S. Interstitial Release of Cisplatin from Triggerable Liposomes Enhances Efficacy against Triple Negative Breast Cancer Solid Tumor Analogues. Mol. Pharm. 2016, 13, 3224–3233. [Google Scholar] [CrossRef]
- Lyu, Y.; Xiao, Q.; Yin, L.; Yang, L.; He, W. Potent delivery of an MMP inhibitor to the tumor microenvironment with thermosensitive liposomes for the suppression of metastasis and angiogenesis. Signal. Transduct. Target. Ther. 2019, 4, 26. [Google Scholar] [CrossRef]
- Mushtaq, M.U.; Papadas, A.; Pagenkopf, A.; Flietner, E.; Morrow, Z.; Chaudhary, S.G.; Asimakopoulos, F. Tumor matrix remodeling and novel immunotherapies: The promise of matrix-derived immune biomarkers. J. Immunother. Cancer 2018, 6, 65. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
{kind=link}
| Enzyme Family | Enzyme | Substrate | Ref |
|---|---|---|---|
| MMPs | MMP-1 | Type I and II collagen | [37] |
| MMP-2 | Gelatin, Type IV collagen | [38] | |
| MMP-3 | E-cadherin, laminin, Type IV collagen; activates cytokines and growth factors; activates MMP-1, -8, -13, -9 | [39] | |
| MMP-7 | Type IV collagen, fibronectin, vitronectin, elastin, aggrecan | [40] | |
| MMP-8 | Type I, II, and III collagens, gelatin, aggrecan, fibronectin | [34,41,42] | |
| MMP-9 | Gelatin, Type IV collagen | [38] | |
| Cathepsins | Cat B | Type IV collagen, laminin, fibronectin | [36] |
| Cat K | Type I collagen, particularly bone | [35] | |
| Cat L | Type I and IV collagen, laminin, fibronectin, and elastin | [43] | |
| Cat S | Collagen, Elastin, E-cadherin | [44] | |
| ADAMs | ADAMTS-18 | Chondroitin sulfate | |
| Other Proteinases | Lysyl oxidase | Crosslinks elastins and collagens through conversion of lysines | [45] |
| Inhibitor Family | Inhibitor | Type of Inhibitor | Enzyme | Clinical Use/Translation | Ref. |
|---|---|---|---|---|---|
| MMP Inhibitors | Batimastat | Small molecule | Broad spectrum | Ended at Phase III | [51,52] |
| Marimastat | Small molecule | Broad spectrum | Ended at Phase III | [53] | |
| Tanomastat | Small molecule | MMP-2, -3, -9 | Ended at Phase III | [54] | |
| Doxycycline | Small molecule | Broad spectrum | Ongoing | [54] | |
| TIMP-1, -2, -3, -4 | Endogenous inhibitor | Broad spectrum | n/a | [55,56] | |
| SDS3 | Antibody | MMP-2, MMP-9 | n/a | [55,57,58] | |
| Prinomastat | Small molecule | Broad spectrum | Ended at Phase III | [59] | |
| Cathepsin Inhibitors | L-235 | Small molecule | Cat K | n/a | [60] |
| Relacatib | Small molecule | Cat K, B, L, S V | Ended at Phase I | [35,61] | |
| Odanacatib | Small molecule | Cat K, B, L, S, V | Ended at Phase III | [35,62,63] | |
| E64 | Small molecule | Cat B, Cat L | n/a | [64] |
| Matrix Targets | Targeting Peptides/Antibodies | Reference |
|---|---|---|
| Fibronectin | CREKA | [101] |
| CLT1 | [101] | |
| CLT2 | [101] | |
| F8 antibody | [102] | |
| L19 antibody | [102] | |
| Collagen | CNA35 | [103] |
| WYRGRL | [104] | |
| Collagen mimetic peptides | [105] | |
| TKKTLRT | [106] | |
| WREPSFMALS | [107] | |
| Tenascin-C | FHKHKSPALSPVGGG | [108] |
| F16 antibody | [108] | |
| Hyaluronan | CKRDLSRRC (IP3) | [109] |
| Heparan Sulfate | NT4 | [110] |
| CGKRK | [100] |
| Types of Nanocarrier Delivery Systems | Examples and Remarks | |
|---|---|---|
| Targeting ECM Components | Polymer nanoparticles [52] | Matricellular targets include: Collagen [112,113], Fibronectin [101,102]. Laminin, Hyaluronan [114], Tenascin-C [114], Heparan sulfate [100] |
| Antibodies [114] | ||
| Liposomes [114] | ||
| Modulating ECM to reduce barrier to delivery | Liposomes [124] | Breakdown of matrix through direct breakdown or reduction of matrix expression |
| Gold nanoparticles [126] | ||
| Lipoprotein nanoparticles [127] | ||
| Using ECM as a local drug depot | Liposomes [128,129,130] | |
| Lyophilosomes [129] | ||
| Modulating ECM to directly alter tumor growth and invasion | Liposomes [131] | MMP inhibitors [131] |
| Polymer nanoparticles [52] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Subrahmanyam, N.; Ghandehari, H. Harnessing Extracellular Matrix Biology for Tumor Drug Delivery. J. Pers. Med. 2021, 11, 88. https://doi.org/10.3390/jpm11020088
Subrahmanyam N, Ghandehari H. Harnessing Extracellular Matrix Biology for Tumor Drug Delivery. Journal of Personalized Medicine. 2021; 11(2):88. https://doi.org/10.3390/jpm11020088
Chicago/Turabian StyleSubrahmanyam, Nithya, and Hamidreza Ghandehari. 2021. "Harnessing Extracellular Matrix Biology for Tumor Drug Delivery" Journal of Personalized Medicine 11, no. 2: 88. https://doi.org/10.3390/jpm11020088
APA StyleSubrahmanyam, N., & Ghandehari, H. (2021). Harnessing Extracellular Matrix Biology for Tumor Drug Delivery. Journal of Personalized Medicine, 11(2), 88. https://doi.org/10.3390/jpm11020088