
The Potential of Mesenchymal Stromal Cells in Neuroblastoma Therapy for Delivery of Anti-Cancer Agents and Hematopoietic Recovery

 ,
,  and
and
Abstract
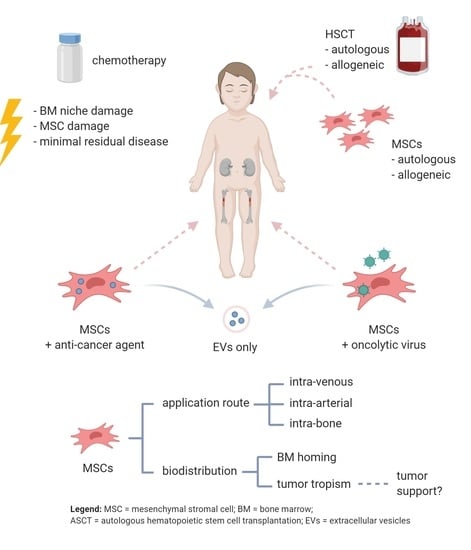
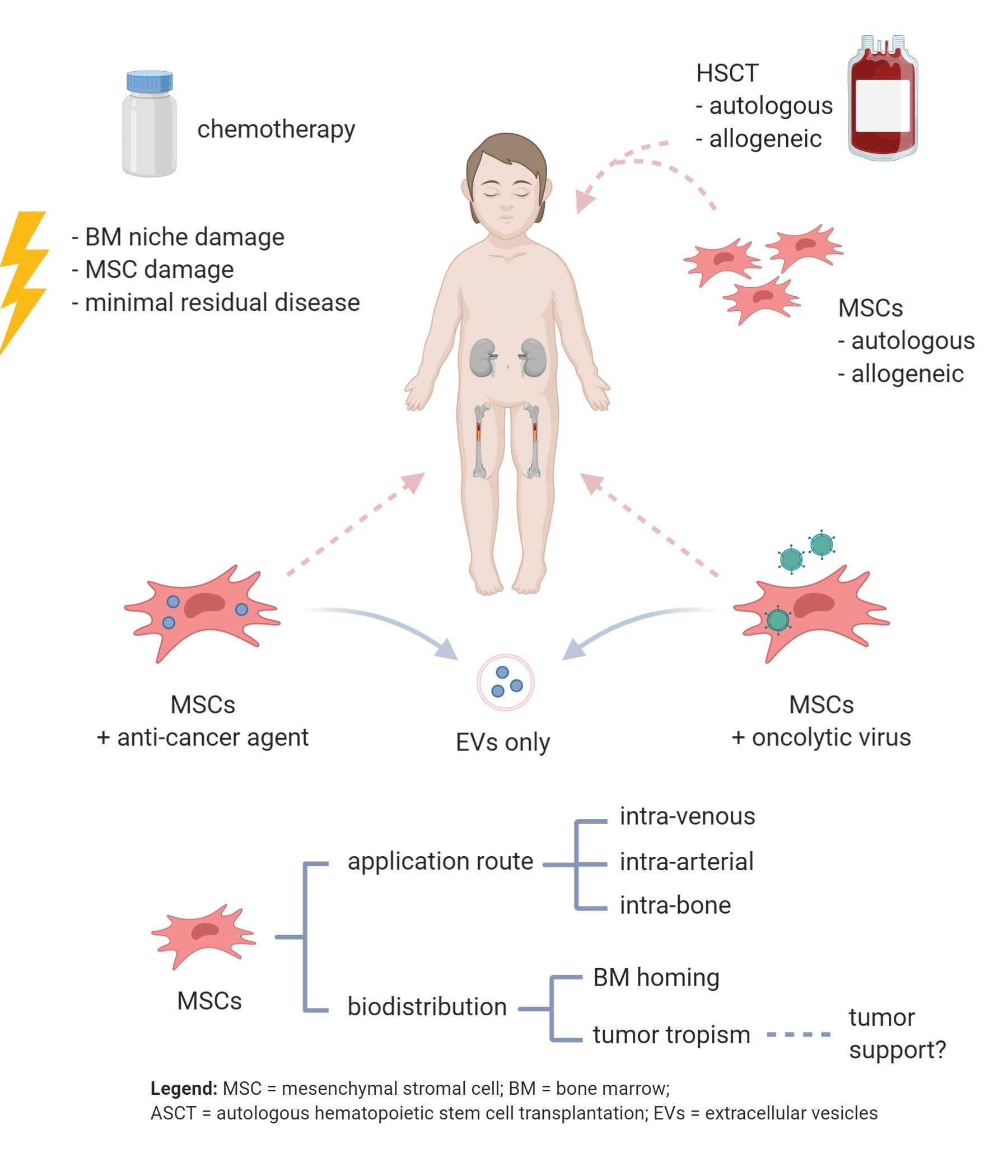
:
1. Introduction
2. MSCs as Delivery Vehicles
2.1. MSCs Delivering Anti-Cancer Agents
2.2. MSCs Delivering Oncolytic Viruses
3. MSCs in Hematopoietic Stem Cell Transplantation
3.1. Allogeneic MSCs
3.2. Autologous MSCs
4. Safety and Feasibility of MSC Therapy in NB
4.1. Safety of MSC (Co-) Infusion
4.2. Influence of MSCs on Tumor Progression
4.3. Cell-Free Approach Using Extracellular Vesicles
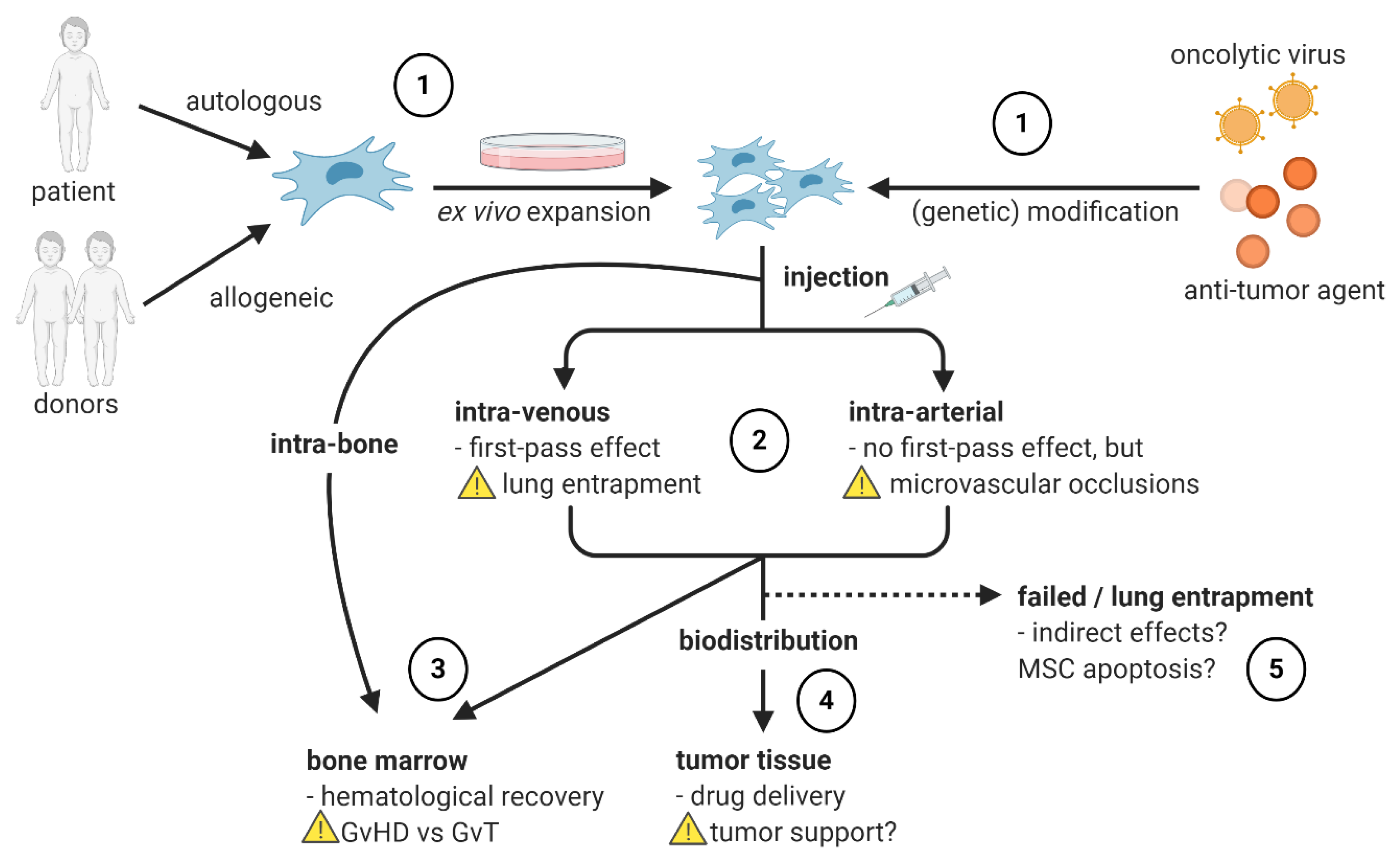
4.4. Influence of Administration Route on MSC Migration
4.5. MSC Engraftment in the BM after HSCT
4.6. Tumor-Tropism of MSCs upon Use as Delivery Vehicles
4.7. Influence of Ex Vivo Expansion of MSCs
5. Future Directions and Concluding Remarks
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Grupp, S.A.; Dvorak, C.C.; Nieder, M.L.; Levine, J.E.; Wall, D.A.; Langholz, B.; Pulsipher, M.A. Children’s Oncology Group’s 2013 blueprint for research: Stem cell transplantation. Pediatr. Blood Cancer 2013, 60, 1044–1047. [Google Scholar] [CrossRef] [Green Version]
- Mohanty, R.; Chowdhury, C.R.; Arega, S.; Sen, P.; Ganguly, P.; Ganguly, N. CAR T cell therapy: A new era for cancer treatment (Review). Oncol. Rep. 2019, 42, 2183–2195. [Google Scholar] [CrossRef] [PubMed]
- Brodeur, G.M. Neuroblastoma: Biological insights into a clinical enigma. Nat. Rev. Cancer 2003, 3, 203–216. [Google Scholar] [CrossRef]
- Park, J.R.; Bagatell, R.; London, W.B.; Maris, J.M.; Cohn, S.L.; Mattay, K.K.; Mattay, K.M.; Hogarty, M. Children’s Oncology Group’s 2013 blueprint for research: Neuroblastoma. Pediatr. Blood Cancer 2013, 60, 985–993. [Google Scholar] [CrossRef]
- Cheung, N.-K.V.; Dyer, M.A. Neuroblastoma: Developmental biology, cancer genomics and immunotherapy. Nat. Rev. Cancer 2013, 13, 397–411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maris, J.M. Recent advances in neuroblastoma. N. Engl. J. Med. 2010, 362, 2202–2211. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surveillance, Epidemiology, and End Results Program. Available online: https://seer.cancer.gov/ (accessed on 15 June 2020).
- Maris, J.M.; Hogarty, M.D.; Bagatell, R.; Cohn, S.L. Neuroblastoma. Lancet 2007, 369, 2106–2120. [Google Scholar] [CrossRef]
- Berthold, F.; Spix, C.; Kaatsch, P.; Lampert, F. Incidence, Survival, and Treatment of Localized and Metastatic Neuroblastoma in Germany 1979–2015. Pediatr. Drugs 2017, 19, 577–593. [Google Scholar] [CrossRef] [Green Version]
- Pinto, N.R.; Applebaum, M.A.; Volchenboum, S.L.; Matthay, K.K.; London, W.B.; Ambros, P.F.; Nakagawara, A.; Berthold, F.; Schleiermacher, G.; Park, J.R.; et al. Advances in Risk Classification and Treatment Strategies for Neuroblastoma. J. Clin. Oncol. 2015, 33, 3008–3017. [Google Scholar] [CrossRef]
- Strother, D.R.; London, W.B.; Schmidt, M.L.; Brodeur, G.M.; Shimada, H.; Thorner, P.; Collins, M.H.; Tagge, E.; Adkins, S.; Reynolds, C.P.; et al. Outcome After Surgery Alone or With Restricted Use of Chemotherapy for Patients With Low-Risk Neuroblastoma: Results of Children’s Oncology Group Study P9641. J. Clin. Oncol. 2012, 30, 1842–1848. [Google Scholar] [CrossRef]
- Simon, T.; Hero, B.; Schulte, J.; Deubzer, H.; Hundsdoerfer, P.; von Schweinitz, D.; Fuchs, J.; Schmidt, M.; Prasad, V.; Krug, B.; et al. 2017 GPOH Guidelines for Diagnosis and Treatment of Patients with Neuroblastic Tumors. Klin. Pädiatrie 2017, 229, 147–167. [Google Scholar] [CrossRef] [PubMed]
- Cohn, S.L.; Pearson, A.D.J.; London, W.B.; Monclair, T.; Ambros, P.F.; Brodeur, G.M.; Faldum, A.; Hero, B.; Iehara, T.; Machin, D.; et al. The International Neuroblastoma Risk Group (INRG) Classification System: An INRG Task Force Report. J. Clin. Oncol. 2009, 27, 289–297. [Google Scholar] [CrossRef]
- Park, J.R.; Kreissman, S.G.; London, W.B.; Naranjo, A.; Cohn, S.L.; Hogarty, M.D.; Tenney, S.C.; Haas-Kogan, D.; Shaw, P.J.; Kraveka, J.M.; et al. Effect of Tandem Autologous Stem Cell Transplant vs. Single Transplant on Event-Free Survival in Patients With High-Risk Neuroblastoma. JAMA 2019, 322, 746. [Google Scholar] [CrossRef] [PubMed]
- Ren, N.; Atyah, M.; Chen, W.-Y.; Zhou, C.-H. The various aspects of genetic and epigenetic toxicology: Testing methods and clinical applications. J. Transl. Med. 2017, 15, 110. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Horwitz, E.M.; Le Blanc, K.; Dominici, M.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.C.; Deans, R.J.; Krause, D.S.; Keating, A. Clarification of the nomenclature for MSC: The International Society for Cellular Therapy position statement. Cytotherapy 2005, 7, 393–395. [Google Scholar] [CrossRef] [PubMed]
- Viswanathan, S.; Shi, Y.; Galipeau, J.; Krampera, M.; Leblanc, K.; Martin, I.; Nolta, J.; Phinney, D.G.; Sensebe, L. Mesenchymal stem versus stromal cells: International Society for Cell & Gene Therapy (ISCT®) Mesenchymal Stromal Cell committee position statement on nomenclature. Cytotherapy 2019, 21, 1019–1024. [Google Scholar] [CrossRef] [PubMed]
- Dominici, M.; Le Blanc, K.; Mueller, I.; Slaper-Cortenbach, I.; Marini, F.; Krause, D.; Deans, R.; Keating, A.; Prockop, D.; Horwitz, E. Minimal criteria for defining multipotent mesenchymal stromal cells. The International Society for Cellular Therapy position statement. Cytotherapy 2006, 8, 315–317. [Google Scholar] [CrossRef] [PubMed]
- Murphy, M.B.; Moncivais, K.; Caplan, A.I. Mesenchymal stem cells: Environmentally responsive therapeutics for regenerative medicine. Exp. Mol. Med. 2013, 45, e54. [Google Scholar] [CrossRef] [Green Version]
- Lv, F.; Lu, M.; MC Cheung, K.; YL Leung, V.; Zhou, G. Intrinsic Properties of Mesemchymal Stem Cells from Human Bone Marrow, Umbilical Cord and Umbilical Cord Blood Comparing the Different Sources of MSC. Curr. Stem Cell Res. Ther. 2012, 7, 389–399. [Google Scholar] [CrossRef] [Green Version]
- Maijenburg, M.W.; Noort, W.A.; Kleijer, M.; Kompier, C.J.A.; Weijer, K.; Van Buul, J.D.; Van Der Schoot, C.E.; Voermans, C. Cell cycle and tissue of origin contribute to the migratory behaviour of human fetal and adult mesenchymal stromal cells. Br. J. Haematol. 2010, 148, 428–440. [Google Scholar] [CrossRef] [PubMed]
- Melief, S.M.; Zwaginga, J.J.; Fibbe, W.E.; Roelofs, H. Adipose Tissue-Derived Multipotent Stromal Cells Have a Higher Immunomodulatory Capacity Than Their Bone Marrow-Derived Counterparts. Stem Cells Transl. Med. 2013, 2, 455–463. [Google Scholar] [CrossRef] [PubMed]
- Batouli, S.; Miura, M.; Brahim, J.; Tsutsui, T.W.; Fisher, L.W.; Gronthos, S.; Robey, P.G.; Shi, S. Comparison of Stem-cell-mediated Osteogenesis and Dentinogenesis. J. Dent. Res. 2003, 82, 976–981. [Google Scholar] [CrossRef] [PubMed]
- Berebichez-Fridman, R.; Montero-Olvera, P.R. Sources and Clinical Applications of Mesenchymal Stem Cells: State-of-the-art review. Sultan Qaboos Univ. Med. J. 2018, 18, e264–e277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strioga, M.; Viswanathan, S.; Darinskas, A.; Slaby, O.; Michalek, J. Same or Not the Same? Comparison of Adipose Tissue-Derived Versus Bone Marrow-Derived Mesenchymal Stem and Stromal Cells. Stem Cells Dev. 2012, 21, 2724–2752. [Google Scholar] [CrossRef] [PubMed]
- Shiozawa, Y.; Eber, M.R.; Berry, J.E.; Taichman, R.S. Bone marrow as a metastatic niche for disseminated tumor cells from solid tumors. Bonekey Rep. 2015, 4. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Méndez-Ferrer, S.; Michurina, T.V.; Ferraro, F.; Mazloom, A.R.; MacArthur, B.D.; Lira, S.A.; Scadden, D.T.; Ma’ayan, A.; Enikolopov, G.N.; Frenette, P.S. Mesenchymal and haematopoietic stem cells form a unique bone marrow niche. Nature 2010, 466, 829–834. [Google Scholar] [CrossRef]
- Ehninger, A.; Trumpp, A. The bone marrow stem cell niche grows up: Mesenchymal stem cells and macrophages move in. J. Exp. Med. 2011, 208, 421–428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the Hematopoietic Stem Cell Pool by CXCL12-CXCR4 Chemokine Signaling in Bone Marrow Stromal Cell Niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Keller, J.; Ortiz, M.; Ruscetti, F. Steel factor (c-kit ligand) promotes the survival of hematopoietic stem/progenitor cells in the absence of cell division. Blood 1995, 86, 1757–1764. [Google Scholar] [CrossRef] [Green Version]
- Lento, W.; Congdon, K.; Voermans, C.; Kritzik, M.; Reya, T. Wnt Signaling in Normal and Malignant Hematopoiesis. Cold Spring Harb. Perspect. Biol. 2013, 5, a008011. [Google Scholar] [CrossRef] [Green Version]
- Yoshihara, H.; Arai, F.; Hosokawa, K.; Hagiwara, T.; Takubo, K.; Nakamura, Y.; Gomei, Y.; Iwasaki, H.; Matsuoka, S.; Miyamoto, K.; et al. Thrombopoietin/MPL Signaling Regulates Hematopoietic Stem Cell Quiescence and Interaction with the Osteoblastic Niche. Cell Stem Cell 2007, 1, 685–697. [Google Scholar] [CrossRef] [Green Version]
- Arai, F.; Hirao, A.; Ohmura, M.; Sato, H.; Matsuoka, S.; Takubo, K.; Ito, K.; Koh, G.Y.; Suda, T. Tie2/Angiopoietin-1 Signaling Regulates Hematopoietic Stem Cell Quiescence in the Bone Marrow Niche. Cell 2004, 118, 149–161. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reagan, M.R.; Rosen, C.J. Navigating the bone marrow niche: Translational insights and cancer-driven dysfunction. Nat. Rev. Rheumatol. 2016, 12, 154–168. [Google Scholar] [CrossRef] [PubMed]
- Weiss, A.R.R.; Dahlke, M.H. Immunomodulation by Mesenchymal Stem Cells (MSCs): Mechanisms of Action of Living, Apoptotic, and Dead MSCs. Front. Immunol. 2019, 10, 1191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kidd, S.; Spaeth, E.; Dembinski, J.L.; Dietrich, M.; Watson, K.; Klopp, A.; Battula, V.L.; Weil, M.; Andreeff, M.; Marini, F.C. Direct evidence of mesenchymal stem cell tropism for tumor and wounding microenvironments using in vivo bioluminescent imaging. Stem Cells 2009, 27, 2614–2623. [Google Scholar] [CrossRef] [Green Version]
- Corradi, G.; Baldazzi, C.; Očadlíková, D.; Marconi, G.; Parisi, S.; Testoni, N.; Finelli, C.; Cavo, M.; Curti, A.; Ciciarello, M. Mesenchymal stromal cells from myelodysplastic and acute myeloid leukemia patients display in vitro reduced proliferative potential and similar capacity to support leukemia cell survival. Stem Cell Res. Ther. 2018, 9, 271. [Google Scholar] [CrossRef] [Green Version]
- Geyh, S.; Rodríguez-Paredes, M.; Jäger, P.; Khandanpour, C.; Cadeddu, R.-P.; Gutekunst, J.; Wilk, C.M.; Fenk, R.; Zilkens, C.; Hermsen, D.; et al. Functional inhibition of mesenchymal stromal cells in acute myeloid leukemia. Leukemia 2016, 30, 683–691. [Google Scholar] [CrossRef]
- Desbourdes, L.; Javary, J.; Charbonnier, T.; Ishac, N.; Bourgeais, J.; Iltis, A.; Chomel, J.-C.; Turhan, A.; Guilloton, F.; Tarte, K.; et al. Alteration Analysis of Bone Marrow Mesenchymal Stromal Cells from De Novo Acute Myeloid Leukemia Patients at Diagnosis. Stem Cells Dev. 2017, 26, 709–722. [Google Scholar] [CrossRef]
- Karnoub, A.E.; Dash, A.B.; Vo, A.P.; Sullivan, A.; Brooks, M.W.; Bell, G.W.; Richardson, A.L.; Polyak, K.; Tubo, R.; Weinberg, R.A. Mesenchymal stem cells within tumour stroma promote breast cancer metastasis. Nature 2007, 449, 557–563. [Google Scholar] [CrossRef]
- Chowdhury, R.; Webber, J.P.; Gurney, M.; Mason, M.D.; Tabi, Z.; Clayton, A. Cancer exosomes trigger mesenchymal stem cell differentiation into pro-angiogenic and pro-invasive myofibroblasts. Oncotarget 2015, 6, 715–731. [Google Scholar] [CrossRef]
- Bergfeld, S.A.; Blavier, L.; DeClerck, Y.A. Bone Marrow–Derived Mesenchymal Stromal Cells Promote Survival and Drug Resistance in Tumor Cells. Mol. Cancer Ther. 2014, 13, 962–975. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Borriello, L.; Nakata, R.; Sheard, M.A.; Fernandez, G.E.; Sposto, R.; Malvar, J.; Blavier, L.; Shimada, H.; Asgharzadeh, S.; Seeger, R.C.; et al. Cancer-Associated Fibroblasts Share Characteristics and Protumorigenic Activity with Mesenchymal Stromal Cells. Cancer Res. 2017, 77, 5142–5157. [Google Scholar] [CrossRef] [Green Version]
- Hochheuser, C.; Windt, L.J.; Kunze, N.Y.; de Vos, D.L.; Tytgat, G.A.M.; Voermans, C.; Timmerman, I. Mesenchymal Stromal Cells in Neuroblastoma: Exploring crosstalk and therapeutic implications. Stem Cells Dev. 2020, 30, 59–78. [Google Scholar] [CrossRef] [PubMed]
- Galland, S.; Stamenkovic, I. Mesenchymal stromal cells in cancer: A review of their immunomodulatory functions and dual effects on tumor progression. J. Pathol. 2020, 250, 555–572. [Google Scholar] [CrossRef] [Green Version]
- Nieddu, V.; Piredda, R.; Bexell, D.; Barton, J.; Anderson, J.; Sebire, N.; Kolluri, K.; Janes, S.M.; Karteris, E.; Sala, A. Engineered human mesenchymal stem cells for neuroblastoma therapeutics. Oncol. Rep. 2019, 42, 35–42. [Google Scholar] [CrossRef]
- Mueller, L.P.; Luetzkendorf, J.; Widder, M.; Nerger, K.; Caysa, H.; Mueller, T. TRAIL-transduced multipotent mesenchymal stromal cells (TRAIL-MSC) overcome TRAIL resistance in selected CRC cell lines in vitro and in vivo. Cancer Gene Ther. 2011, 18, 229–239. [Google Scholar] [CrossRef]
- Galleu, A.; Riffo-Vasquez, Y.; Trento, C.; Lomas, C.; Dolcetti, L.; Cheung, T.S.; von Bonin, M.; Barbieri, L.; Halai, K.; Ward, S.; et al. Apoptosis in mesenchymal stromal cells induces in vivo recipient-mediated immunomodulation. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [Green Version]
- Wong, S.H.M.; Kong, W.Y.; Fang, C.-M.; Loh, H.-S.; Chuah, L.-H.; Abdullah, S.; Ngai, S.C. The TRAIL to cancer therapy: Hindrances and potential solutions. Crit. Rev. Oncol. Hematol. 2019, 143, 81–94. [Google Scholar] [CrossRef]
- Golinelli, G.; Grisendi, G.; Prapa, M.; Bestagno, M.; Spano, C.; Rossignoli, F.; Bambi, F.; Sardi, I.; Cellini, M.; Horwitz, E.M.; et al. Targeting GD2-positive glioblastoma by chimeric antigen receptor empowered mesenchymal progenitors. Cancer Gene Ther. 2018, 27, 558–570. [Google Scholar] [CrossRef] [Green Version]
- Prapa, M.; Caldrer, S.; Spano, C.; Bestagno, M.; Golinelli, G.; Grisendi, G.; Petrachi, T.; Conte, P.; Horwitz, E.M.; Campana, D.; et al. A novel anti-GD2/4-1BB chimeric antigen receptor triggers neuroblastoma cell killing. Oncotarget 2015, 6, 24884–24894. [Google Scholar] [CrossRef] [Green Version]
- Shamili, F.H.; Bayegi, H.R.; Salmasi, Z.; Sadri, K.; Mahmoudi, M.; Kalantari, M.; Ramezani, M.; Abnous, K. Exosomes derived from TRAIL-engineered mesenchymal stem cells with effective anti-tumor activity in a mouse melanoma model. Int. J. Pharm. 2018, 549, 218–229. [Google Scholar] [CrossRef]
- Studeny, M.; Marini, F.C.; Champlin, R.E.; Zompetta, C.; Fidler, I.J.; Andreeff, M. Bone marrow-derived mesenchymal stem cells as vehicles for interferon-beta delivery into tumors. Cancer Res. 2002, 62, 3603–3608. [Google Scholar]
- Le Bon, A.; Schiavoni, G.; D’Agostino, G.; Gresser, I.; Belardelli, F.; Tough, D.F. Type I Interferons Potently Enhance Humoral Immunity and Can Promote Isotype Switching by Stimulating Dendritic Cells In Vivo. Immunity 2001, 14, 461–470. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Wang, A.; Hu, Q.; Lu, S.; Dong, Z. Adenovirus-mediated interferon-beta gene transfer inhibits angiogenesis in and progression of orthotopic tumors of human prostate cancer cells in nude mice. Int. J. Oncol. 2006, 29, 1405–1412. [Google Scholar]
- Johns, T.G.; Mackay, I.R.; Callister, K.A.; Hertzog, P.J.; Devenish, R.J.; Linnance, A.W. Antiproliferative Potencies of Interferons on Melanoma Cell Lines and Xenografts: Higher Efficacy of Interferon β. JNCI J. Natl. Cancer Inst. 1992, 84, 1185–1190. [Google Scholar] [CrossRef]
- Maniwa, J.; Fumino, S.; Kimura, K.; Tanaka, T.; Higashi, M.; Kishida, T.; Mazda, O.; Tajiri, T. Novel mesenchymal stem cell delivery system as targeted therapy against neuroblastoma using the TH-MYCN mouse model. J. Pediatr. Surg. 2019, 54, 2600–2605. [Google Scholar] [CrossRef]
- Ikeda, H.; Old, L.J.; Schreiber, R.D. The roles of IFNγ in protection against tumor development and cancer immunoediting. Cytokine Growth Factor Rev. 2002, 13, 95–109. [Google Scholar] [CrossRef]
- Relation, T.; Yi, T.; Guess, A.J.; La Perle, K.; Otsuru, S.; Hasgur, S.; Dominici, M.; Breuer, C.; Horwitz, E.M. Intratumoral Delivery of Interferonγ-Secreting Mesenchymal Stromal Cells Repolarizes Tumor-Associated Macrophages and Suppresses Neuroblastoma Proliferation In Vivo. Stem Cells 2018, 36, 915–924. [Google Scholar] [CrossRef] [Green Version]
- Goedhart, M.; Cornelissen, A.S.; Kuijk, C.; Geerman, S.; Kleijer, M.; van Buul, J.D.; Huveneers, S.; Raaijmakers, M.H.G.P.; Young, H.A.; Wolkers, M.C.; et al. Interferon-Gamma Impairs Maintenance and Alters Hematopoietic Support of Bone Marrow Mesenchymal Stromal Cells. Stem Cells Dev. 2018, 27, 579–589. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Q.; Jin, J.; Zhu, L.; Chen, M.; Xu, H.; Wang, H.; Feng, X.; Zhu, X. The optimal choice of medication administration route regarding intravenous, intramuscular, and subcutaneous injection. Patient Prefer. Adherence 2015, 923. [Google Scholar] [CrossRef] [Green Version]
- Pessina, A.; Bonomi, A.; Coccè, V.; Invernici, G.; Navone, S.; Cavicchini, L.; Sisto, F.; Ferrari, M.; Viganò, L.; Locatelli, A.; et al. Mesenchymal Stromal Cells Primed with Paclitaxel Provide a New Approach for Cancer Therapy. PLoS ONE 2011, 6, e28321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alessandri, G.; Coccè, V.; Pastorino, F.; Paroni, R.; Dei Cas, M.; Restelli, F.; Pollo, B.; Gatti, L.; Tremolada, C.; Berenzi, A.; et al. Microfragmented human fat tissue is a natural scaffold for drug delivery: Potential application in cancer chemotherapy. J. Control. Release 2019, 302, 2–18. [Google Scholar] [CrossRef]
- Zhou, Y.; Zhou, W.; Chen, X.; Wang, Q.; Li, C.; Chen, Q.; Zhang, Y.; Lu, Y.; Ding, X.; Jiang, C. Bone marrow mesenchymal stem cells-derived exosomes for penetrating and targeted chemotherapy of pancreatic cancer. Acta Pharm. Sin. B 2020, 10, 1563–1575. [Google Scholar] [CrossRef]
- Sharif, S.; Ghahremani, M.H.; Soleimani, M. Differentiation Induction and Proliferation Inhibition by A Cell-Free Approach for Delivery of Exogenous miRNAs to Neuroblastoma Cells Using Mesenchymal Stem Cells. Cell J. 2021, 22, 556–564. [Google Scholar] [CrossRef]
- Mortara, L.; Balza, E.; Bruno, A.; Poggi, A.; Orecchia, P.; Carnemolla, B. Anti-cancer Therapies Employing IL-2 Cytokine Tumor Targeting: Contribution of Innate, Adaptive and Immunosuppressive Cells in the Anti-tumor Efficacy. Front. Immunol. 2018, 9. [Google Scholar] [CrossRef] [Green Version]
- Chulpanova, D.S.; Solovyeva, V.V.; James, V.; Arkhipova, S.S.; Gomzikova, M.O.; Garanina, E.E.; Akhmetzyanova, E.R.; Tazetdinova, L.G.; Khaiboullina, S.F.; Rizvanov, A.A. Human Mesenchymal Stem Cells Overexpressing Interleukin 2 Can Suppress Proliferation of Neuroblastoma Cells in Co-Culture and Activate Mononuclear Cells In Vitro. Bioengineering 2020, 7, 59. [Google Scholar] [CrossRef]
- Fukuhara, H.; Ino, Y.; Todo, T. Oncolytic virus therapy: A new era of cancer treatment at dawn. Cancer Sci. 2016, 107, 1373–1379. [Google Scholar] [CrossRef]
- Qiao, J.; Kottke, T.; Willmon, C.; Galivo, F.; Wongthida, P.; Diaz, R.M.; Thompson, J.; Ryno, P.; Barber, G.N.; Chester, J.; et al. Purging metastases in lymphoid organs using a combination of antigen-nonspecific adoptive T cell therapy, oncolytic virotherapy and immunotherapy. Nat. Med. 2008, 14, 37–44. [Google Scholar] [CrossRef]
- Ruano, D.; López-Martín, J.A.; Moreno, L.; Lassaletta, Á.; Bautista, F.; Andión, M.; Hernández, C.; González-Murillo, Á.; Melen, G.; Alemany, R.; et al. First-in-Human, First-in-Child Trial of Autologous MSCs Carrying the Oncolytic Virus Icovir-5 in Patients with Advanced Tumors. Mol. Ther. 2020, 28, 1–10. [Google Scholar] [CrossRef]
- García-Castro, J.; Alemany, R.; Cascalló, M.; Martínez-Quintanilla, J.; del Mar Arriero, M.; Lassaletta, Á.; Madero, L.; Ramírez, M. Treatment of metastatic neuroblastoma with systemic oncolytic virotherapy delivered by autologous mesenchymal stem cells: An exploratory study. Cancer Gene Ther. 2010, 17, 476–483. [Google Scholar] [CrossRef]
- Mader, E.K.; Butler, G.; Dowdy, S.C.; Mariani, A.; Knutson, K.L.; Federspiel, M.J.; Russell, S.J.; Galanis, E.; Dietz, A.B.; Peng, K.-W. Optimizing patient derived mesenchymal stem cells as virus carriers for a phase I clinical trial in ovarian cancer. J. Transl. Med. 2013, 11, 20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stoff-Khalili, M.A.; Rivera, A.A.; Mathis, J.M.; Banerjee, N.S.; Moon, A.S.; Hess, A.; Rocconi, R.P.; Numnum, T.M.; Everts, M.; Chow, L.T.; et al. Mesenchymal stem cells as a vehicle for targeted delivery of CRAds to lung metastases of breast carcinoma. Breast Cancer Res. Treat. 2007, 105, 157–167. [Google Scholar] [CrossRef]
- Melen, G.J.; Franco-Luzón, L.; Ruano, D.; González-Murillo, Á.; Alfranca, A.; Casco, F.; Lassaletta, Á.; Alonso, M.; Madero, L.; Alemany, R.; et al. Influence of carrier cells on the clinical outcome of children with neuroblastoma treated with high dose of oncolytic adenovirus delivered in mesenchymal stem cells. Cancer Lett. 2016, 371, 161–170. [Google Scholar] [CrossRef]
- Franco-Luzón, L.; García-Mulero, S.; Sanz-Pamplona, R.; Melen, G.; Ruano, D.; Lassaletta, Á.; Madero, L.; González-Murillo, Á.; Ramírez, M. Genetic and Immune Changes Associated with Disease Progression under the Pressure of Oncolytic Therapy in A Neuroblastoma Outlier Patient. Cancers 2020, 12, 1104. [Google Scholar] [CrossRef]
- Morales-Molina, Á.; Gambera, S.; Cejalvo, T.; Moreno, R.; Rodríguez-Milla, M.Á.; Perisé-Barrios, A.J.; García-Castro, J. Antitumor virotherapy using syngeneic or allogeneic mesenchymal stem cell carriers induces systemic immune response and intratumoral leukocyte infiltration in mice. Cancer Immunol. Immunother. 2018, 67, 1589–1602. [Google Scholar] [CrossRef]
- Matthay, K.K.; Villablanca, J.G.; Seeger, R.C.; Stram, D.O.; Harris, R.E.; Ramsay, N.K.; Swift, P.; Shimada, H.; Black, C.T.; Brodeur, G.M.; et al. Treatment of high-risk neuroblastoma with intensive chemotherapy, radiotherapy, autologous bone marrow transplantation, and 13-cis-retinoic acid. Children’s Cancer Group. N. Engl. J. Med. 1999, 341, 1165–1173. [Google Scholar] [CrossRef] [PubMed]
- Battiwalla, M.; Hematti, P. Mesenchymal stem cells in hematopoietic stem cell transplantation. Cytotherapy 2009, 11, 503–515. [Google Scholar] [CrossRef]
- Noort, W.A.; Kruisselbrink, A.B.; in’t Anker, P.S.; Kruger, M.; van Bezooijen, R.L.; de Paus, R.A.; Heemskerk, M.H.M.; Löwik, C.W.G.M.; Falkenburg, J.H.F.; Willemze, R.; et al. Mesenchymal stem cells promote engraftment of human umbilical cord blood--derived CD34+ cells in NOD/SCID mice. Exp. Hematol. 2002, 30, 870–878. [Google Scholar] [CrossRef]
- Crippa, S.; Santi, L.; Bosotti, R.; Porro, G.; Bernardo, M.E. Bone Marrow-Derived Mesenchymal Stromal Cells: A Novel Target to Optimize Hematopoietic Stem Cell Transplantation Protocols in Hematological Malignancies and Rare Genetic Disorders. J. Clin. Med. 2019, 9, 2. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munneke, J.M.; Spruit, M.J.A.; Cornelissen, A.S.; van Hoeven, V.; Voermans, C.; Hazenberg, M.D. The Potential of Mesenchymal Stromal Cells as Treatment for Severe Steroid-Refractory Acute Graft-Versus-Host Disease: A Critical Review of the Literature. Transplantation 2016, 100, 2309–2314. [Google Scholar] [CrossRef]
- Toporski, J.; Garkavij, M.; Tennvall, J.; Ora, I.; Gleisner, K.S.; Dykes, J.H.; Lenhoff, S.; Juliusson, G.; Scheding, S.; Turkiewicz, D.; et al. High-dose iodine-131-metaiodobenzylguanidine with haploidentical stem cell transplantation and posttransplant immunotherapy in children with relapsed/refractory neuroblastoma. Biol. Blood Marrow Transplant. 2009, 15, 1077–1085. [Google Scholar] [CrossRef] [Green Version]
- Illhardt, T.; Toporski, J.; Feuchtinger, T.; Turkiewicz, D.; Teltschik, H.-M.; Ebinger, M.; Schwarze, C.-P.; Holzer, U.; Lode, H.N.; Albert, M.H.; et al. Haploidentical Stem Cell Transplantation for Refractory/Relapsed Neuroblastoma. Biol. Blood Marrow Transplant. 2018, 24, 1005–1012. [Google Scholar] [CrossRef] [Green Version]
- Fouillard, L.; Chapel, A.; Bories, D.; Bouchet, S.; Costa, J.-M.; Rouard, H.; Hervé, P.; Gourmelon, P.; Thierry, D.; Lopez, M.; et al. Infusion of allogeneic-related HLA mismatched mesenchymal stem cells for the treatment of incomplete engraftment following autologous haematopoietic stem cell transplantation. Leukemia 2007, 21, 568–570. [Google Scholar] [CrossRef]
- Xiong, Y.-Y.; Fan, Q.; Huang, F.; Zhang, Y.; Wang, Y.; Chen, X.-Y.; Fan, Z.-P.; Zhou, H.-S.; Xiao, Y.; Xu, X.-J.; et al. Mesenchymal stem cells versus mesenchymal stem cells combined with cord blood for engraftment failure after autologous hematopoietic stem cell transplantation: A pilot prospective, open-label, randomized trial. Biol. Blood Marrow Transplant. 2014, 20, 236–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sung, K.W. Allogeneic stem cell transplantation for neuroblastoma. Korean J. Hematol. 2012, 47, 3–5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ladenstein, R.; Pötschger, U.; Hartman, O.; Pearson, A.D.J.; Klingebiel, T.; Castel, V.; Yaniv, I.; Demirer, T.; Dini, G. 28 years of high-dose therapy and SCT for neuroblastoma in Europe: Lessons from more than 4000 procedures. Bone Marrow Transplant. 2008, 41, S118–S127. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Le Blanc, K.; Ringdén, O. Mesenchymal stem cells: Properties and role in clinical bone marrow transplantation. Curr. Opin. Immunol. 2006, 18, 586–591. [Google Scholar] [CrossRef]
- Lazarus, H.M.; Koc, O.N.; Devine, S.M.; Curtin, P.; Maziarz, R.T.; Holland, H.K.; Shpall, E.J.; McCarthy, P.; Atkinson, K.; Cooper, B.W.; et al. Cotransplantation of HLA-identical sibling culture-expanded mesenchymal stem cells and hematopoietic stem cells in hematologic malignancy patients. Biol. Blood Marrow Transplant. 2005, 11, 389–398. [Google Scholar] [CrossRef] [Green Version]
- Macmillan, M.L.; Blazar, B.R.; DeFor, T.E.; Wagner, J.E. Transplantation of ex-vivo culture-expanded parental haploidentical mesenchymal stem cells to promote engraftment in pediatric recipients of unrelated donor umbilical cord blood: Results of a phase I-II clinical trial. Bone Marrow Transplant. 2009, 43, 447–454. [Google Scholar] [CrossRef] [Green Version]
- Bernardo, M.E.; Ball, L.M.; Cometa, A.M.; Roelofs, H.; Zecca, M.; Avanzini, M.A.; Bertaina, A.; Vinti, L.; Lankester, A.; Maccario, R.; et al. Co-infusion of ex vivo-expanded, parental MSCs prevents life-threatening acute GVHD, but does not reduce the risk of graft failure in pediatric patients undergoing allogeneic umbilical cord blood transplantation. Bone Marrow Transplant. 2011, 46, 200–207. [Google Scholar] [CrossRef] [Green Version]
- Ning, H.; Yang, F.; Jiang, M.; Hu, L.; Feng, K.; Zhang, J.; Yu, Z.; Li, B.; Xu, C.; Li, Y.; et al. The correlation between cotransplantation of mesenchymal stem cells and higher recurrence rate in hematologic malignancy patients: Outcome of a pilot clinical study. Leukemia 2008, 22, 593–599. [Google Scholar] [CrossRef] [Green Version]
- Wu, K.-H.; Sheu, J.-N.; Wu, H.-P.; Tsai, C.; Sieber, M.; Peng, C.-T.; Chao, Y.-H. Cotransplantation of umbilical cord-derived mesenchymal stem cells promote hematopoietic engraftment in cord blood transplantation: A pilot study. Transplantation 2013, 95, 773–777. [Google Scholar] [CrossRef]
- Wu, K.-H.; Tsai, C.; Wu, H.-P.; Sieber, M.; Peng, C.-T.; Chao, Y.-H. Human application of ex vivo expanded umbilical cord-derived mesenchymal stem cells: Enhance hematopoiesis after cord blood transplantation. Cell Transplant. 2013, 22, 2041–2051. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.H.; Lee, M.W.; Yoo, K.H.; Kim, D.S.; Son, M.H.; Sung, K.W.; Cheuh, H.; Choi, S.J.; Oh, W.; Yang, Y.S.; et al. Co-transplantation of third-party umbilical cord blood-derived MSCs promotes engraftment in children undergoing unrelated umbilical cord blood transplantation. Bone Marrow Transplant. 2013, 48, 1040–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goto, T.; Murata, M.; Terakura, S.; Nishida, T.; Adachi, Y.; Ushijima, Y.; Shimada, K.; Ishikawa, Y.; Hayakawa, F.; Nishio, N.; et al. Phase I study of cord blood transplantation with intrabone marrow injection of mesenchymal stem cells: A clinical study protocol. Medicine 2018, 97, e0449. [Google Scholar] [CrossRef]
- Yang, H.; Robinson, S.N.; Nieto, Y.; Jones, R.J.; Gocke, C.D.; Lu, J.; Giralt, S.A.; Jones, R.B.; Decker, W.K.; Xing, D.; et al. Ex vivo graft purging and expansion of autologous blood progenitor cell products from patients with multiple myeloma. Cancer Res. 2011, 71, 5040–5049. [Google Scholar] [CrossRef] [Green Version]
- Paciejewska, M.M.; Maijenburg, M.W.; Gilissen, C.; Kleijer, M.; Vermeul, K.; Weijer, K.; Veltman, J.A.; Von Lindern, M.; Van Der Schoot, C.E.; Voermans, C. Different balance of Wnt signaling in adult and fetal bone marrow-derived mesenchymal stromal cells. Stem Cells Dev. 2016, 25, 934–947. [Google Scholar] [CrossRef] [PubMed]
- De Lima, M.; McNiece, I.; Robinson, S.N.; Munsell, M.; Eapen, M.; Horowitz, M.; Alousi, A.; Saliba, R.; McMannis, J.D.; Kaur, I.; et al. Cord-blood engraftment with ex vivo mesenchymal-cell coculture. N. Engl. J. Med. 2012, 367, 2305–2315. [Google Scholar] [CrossRef] [Green Version]
- Ghebes, C.A.; Morhayim, J.; Kleijer, M.; Koroglu, M.; Erkeland, S.J.; Hoogenboezem, R.; Bindels, E.; van Alphen, F.P.J.; van den Biggelaar, M.; Nolte, M.A.; et al. Extracellular vesicles derived from adult and fetal bone marrow mesenchymal stromal cells differentially promote ex vivo expansion of hematopoietic stem and progenitor cells. Front. Bioeng. Biotechnol. 2021. accepted. [Google Scholar]
- Budgude, P.; Kale, V.; Vaidya, A. Mesenchymal stromal cell-derived extracellular vesicles as cell-free biologics for the ex vivo expansion of hematopoietic stem cells. Cell Biol. Int. 2020, 44, 1078–1102. [Google Scholar] [CrossRef] [PubMed]
- Kemp, K.; Morse, R.; Wexler, S.; Cox, C.; Mallam, E.; Hows, J.; Donaldson, C. Chemotherapy-induced mesenchymal stem cell damage in patients with hematological malignancy. Ann. Hematol. 2010, 89, 701–713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Somaiah, C.; Kumar, A.; Sharma, R.; Sharma, A.; Anand, T.; Bhattacharyya, J.; Das, D.; Deka Talukdar, S.; Jaganathan, B.G. Mesenchymal stem cells show functional defect and decreased anti-cancer effect after exposure to chemotherapeutic drugs. J. Biomed. Sci. 2018, 25. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münz, F.; Lopez Perez, R.; Trinh, T.; Sisombath, S.; Weber, K.J.; Wuchter, P.; Debus, J.; Saffrich, R.; Huber, P.E.; Nicolay, N.H. Human mesenchymal stem cells lose their functional properties after paclitaxel treatment. Sci. Rep. 2018, 8, 312. [Google Scholar] [CrossRef] [Green Version]
- Hochheuser, C.; van Zogchel, L.M.J.; Kleijer, M.; Kuijk, C.; Tol, S.; van der Schoot, C.E.; Voermans, C.; Tytgat, G.A.M.; Timmerman, I. The Metastatic Bone Marrow Niche in Neuroblastoma: Altered Phenotype and Function of Mesenchymal Stromal Cells. Cancers 2020, 12, 3231. [Google Scholar] [CrossRef]
- Koç, O.N.; Gerson, S.L.; Cooper, B.W.; Dyhouse, S.M.; Haynesworth, S.E.; Caplan, A.I.; Lazarus, H.M. Rapid hematopoietic recovery after coinfusion of autologous-blood stem cells and culture-expanded marrow mesenchymal stem cells in advanced breast cancer patients receiving high-dose chemotherapy. J. Clin. Oncol. 2000, 18, 307–316. [Google Scholar] [CrossRef]
- Batorov, E.V.; Shevela, E.Y.; Tikhonova, M.A.; Batorova, D.S.; Ushakova, G.Y.; Sizikova, S.A.; Sergeevicheva, V.V.; Gilevich, A.V.; Kryuchkova, I.V.; Ostanin, A.A.; et al. Mesenchymal stromal cells improve early lymphocyte recovery and T cell reconstitution after autologous hematopoietic stem cell transplantation in patients with malignant lymphomas. Cell. Immunol. 2015, 297, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Modak, S.; Cheung, I.Y.; Kushner, B.H.; Kramer, K.; Reich, L.; Cheung, N.K.V. Plerixafor plus granulocyte-colony stimulating factor for autologous hematopoietic stem cell mobilization in patients with metastatic neuroblastoma. Pediatr. Blood Cancer 2012, 58, 469–471. [Google Scholar] [CrossRef]
- Masuda, S.; Ageyama, N.; Shibata, H.; Obara, Y.; Ikeda, T.; Takeuchi, K.; Ueda, Y.; Ozawa, K.; Hanazono, Y. Cotransplantation with MSCs improves engraftment of HSCs after autologous intra-bone marrow transplantation in nonhuman primates. Exp. Hematol. 2009, 37, 1250–1257.e1. [Google Scholar] [CrossRef]
- Lalu, M.M.; McIntyre, L.; Pugliese, C.; Fergusson, D.; Winston, B.W.; Marshall, J.C.; Granton, J.; Stewart, D.J. Safety of Cell Therapy with Mesenchymal Stromal Cells (SafeCell): A Systematic Review and Meta-Analysis of Clinical Trials. PLoS ONE 2012, 7, e47559. [Google Scholar] [CrossRef]
- Klopp, A.H.; Gupta, A.; Spaeth, E.; Andreeff, M.; Marini, F. Concise review: Dissecting a discrepancy in the literature: Do mesenchymal stem cells support or suppress tumor growth? Stem Cells 2011, 29, 11–19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Christodoulou, I.; Goulielmaki, M.; Devetzi, M.; Panagiotidis, M.; Koliakos, G.; Zoumpourlis, V. Mesenchymal stem cells in preclinical cancer cytotherapy: A systematic review. Stem Cell Res. Ther. 2018, 9, 336. [Google Scholar] [CrossRef]
- Bruna, F.; Plaza, A.; Arango, M.; Espinoza, I.; Conget, P. Systemically administered allogeneic mesenchymal stem cells do not aggravate the progression of precancerous lesions: A new biosafety insight. Stem cell Res. Ther. 2018, 9, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Baumann, I.; Testa, N.G.; Lange, C.; De Wynter, E.; Luft, T.; Dexter, T.M.; Van Hoef, M.E.H.M.; Howell, A. Haemopoietic cells mobilised into the circulation by lenograstim as alternative to bone marrow for allogeneic transplants. Lancet 1993, 341, 369. [Google Scholar] [CrossRef]
- Anasetti, C.; Logan, B.R.; Lee, S.J.; Waller, E.K.; Weisdorf, D.J.; Wingard, J.R.; Cutler, C.S.; Westervelt, P.; Woolfrey, A.; Couban, S.; et al. Peripheral-Blood Stem Cells versus Bone Marrow from Unrelated Donors. N. Engl. J. Med. 2012, 367, 1487–1496. [Google Scholar] [CrossRef] [Green Version]
- Stefani, F.R.; Eberstål, S.; Vergani, S.; Kristiansen, T.A.; Bengzon, J. Low-dose irradiated mesenchymal stromal cells break tumor defensive properties in vivo. Int. J. Cancer 2018, 143, 2200–2212. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waterman, R.S.; Tomchuck, S.L.; Henkle, S.L.; Betancourt, A.M. A new mesenchymal stem cell (MSC) paradigm: Polarization into a pro-inflammatory MSC1 or an Immunosuppressive MSC2 phenotype. PLoS ONE 2010, 5, e10088. [Google Scholar] [CrossRef] [PubMed]
- Waterman, R.S.; Henkle, S.L.; Betancourt, A.M. Mesenchymal stem cell 1 (MSC1)-based therapy attenuates tumor growth whereas MSC2-treatment promotes tumor growth and metastasis. PLoS ONE 2012, 7, e45590. [Google Scholar] [CrossRef] [Green Version]
- Abels, E.R.; Breakefield, X.O. Introduction to Extracellular Vesicles: Biogenesis, RNA Cargo Selection, Content, Release, and Uptake. Cell. Mol. Neurobiol. 2016, 36, 301–312. [Google Scholar] [CrossRef]
- Wen, S.; Dooner, M.; Cheng, Y.; Papa, E.; Del Tatto, M.; Pereira, M.; Deng, Y.; Goldberg, L.; Aliotta, J.; Chatterjee, D.; et al. Mesenchymal stromal cell-derived extracellular vesicles rescue radiation damage to murine marrow hematopoietic cells. Leukemia 2016, 30, 2221–2231. [Google Scholar] [CrossRef] [PubMed]
- Wen, S.; Dooner, M.; Papa, E.; Del Tatto, M.; Pereira, M.; Borgovan, T.; Cheng, Y.; Goldberg, L.; Liang, O.; Camussi, G.; et al. Biodistribution of Mesenchymal Stem Cell-Derived Extracellular Vesicles in a Radiation Injury Bone Marrow Murine Model. Int. J. Mol. Sci. 2019, 20. [Google Scholar] [CrossRef] [Green Version]
- Grange, C.; Tapparo, M.; Bruno, S.; Chatterjee, D.; Quesenberry, P.J.; Tetta, C.; Camussi, G. Biodistribution of mesenchymal stem cell-derived extracellular vesicles in a model of acute kidney injury monitored by optical imaging. Int. J. Mol. Med. 2014, 33, 1055–1063. [Google Scholar] [CrossRef] [Green Version]
- Garofalo, M.; Saari, H.; Somersalo, P.; Crescenti, D.; Kuryk, L.; Aksela, L.; Capasso, C.; Madetoja, M.; Koskinen, K.; Oksanen, T.; et al. Antitumor effect of oncolytic virus and paclitaxel encapsulated in extracellular vesicles for lung cancer treatment. J. Control. Release 2018, 283, 223–234. [Google Scholar] [CrossRef]
- Garofalo, M.; Villa, A.; Rizzi, N.; Kuryk, L.; Rinner, B.; Cerullo, V.; Yliperttula, M.; Mazzaferro, V.; Ciana, P. Extracellular vesicles enhance the targeted delivery of immunogenic oncolytic adenovirus and paclitaxel in immunocompetent mice. J. Control. Release 2019, 294, 165–175. [Google Scholar] [CrossRef] [PubMed]
- Kordelas, L.; Rebmann, V.; Ludwig, A.-K.; Radtke, S.; Ruesing, J.; Doeppner, T.R.; Epple, M.; Horn, P.A.; Beelen, D.W.; Giebel, B. MSC-derived exosomes: A novel tool to treat therapy-refractory graft-versus-host disease. Leukemia 2014, 28, 970–973. [Google Scholar] [CrossRef]
- Cussó, L.; Mirones, I.; Peña-Zalbidea, S.; García-Vázquez, V.; García-Castro, J.; Desco, M. Combination of single-photon emission computed tomography and magnetic resonance imaging to track 111in-oxine-labeled human mesenchymal stem cells in neuroblastoma-bearing mice. Mol. Imaging 2014, 13, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Kimura, K.; Kishida, T.; Wakao, J.; Tanaka, T.; Higashi, M.; Fumino, S.; Aoi, S.; Furukawa, T.; Mazda, O.; Tajiri, T. Tumor-homing effect of human mesenchymal stem cells in a TH-MYCN mouse model of neuroblastoma. J. Pediatr. Surg. 2016, 51, 2068–2073. [Google Scholar] [CrossRef] [PubMed]
- Barbash, I.M.; Chouraqui, P.; Baron, J.; Feinberg, M.S.; Etzion, S.; Tessone, A.; Miller, L.; Guetta, E.; Zipori, D.; Kedes, L.H.; et al. Systemic delivery of bone marrow-derived mesenchymal stem cells to the infarcted myocardium: Feasibility, cell migration, and body distribution. Circulation 2003, 108, 863–868. [Google Scholar] [CrossRef] [PubMed]
- Lee, R.H.; Pulin, A.A.; Seo, M.J.; Kota, D.J.; Ylostalo, J.; Larson, B.L.; Semprun-Prieto, L.; Delafontaine, P.; Prockop, D.J. Intravenous hMSCs improve myocardial infarction in mice because cells embolized in lung are activated to secrete the anti-inflammatory protein TSG-6. Cell Stem Cell 2009, 5, 54–63. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Furlani, D.; Ugurlucan, M.; Ong, L.L.; Bieback, K.; Pittermann, E.; Westien, I.; Wang, W.; Yerebakan, C.; Li, W.; Gaebel, R.; et al. Is the intravascular administration of mesenchymal stem cells safe? Mesenchymal stem cells and intravital microscopy. Microvasc. Res. 2009, 77, 370–376. [Google Scholar] [CrossRef]
- Dorland, Y.L.; Cornelissen, A.S.; Kuijk, C.; Tol, S.; Hoogenboezem, M.; van Buul, J.D.; Nolte, M.A.; Voermans, C.; Huveneers, S. Nuclear shape, protrusive behaviour and in vivo retention of human bone marrow mesenchymal stromal cells is controlled by Lamin-A/C expression. Sci. Rep. 2019, 9, 14401. [Google Scholar] [CrossRef] [PubMed]
- Deak, E.; Rüster, B.; Keller, L.; Eckert, K.; Fichtner, I.; Seifried, E.; Henschler, R. Suspension medium influences interaction of mesenchymal stromal cells with endothelium and pulmonary toxicity after transplantation in mice. Cytotherapy 2010, 12, 260–264. [Google Scholar] [CrossRef]
- Cui, L.; Kerkelä, E.; Bakreen, A.; Nitzsche, F.; Andrzejewska, A.; Nowakowski, A.; Janowski, M.; Walczak, P.; Boltze, J.; Lukomska, B.; et al. The cerebral embolism evoked by intra-arterial delivery of allogeneic bone marrow mesenchymal stem cells in rats is related to cell dose and infusion velocity. Stem Cell Res. Ther. 2015, 6, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- François, M.; Romieu-Mourez, R.; Li, M.; Galipeau, J. Human MSC suppression correlates with cytokine induction of indoleamine 2,3-dioxygenase and bystander M2 macrophage differentiation. Mol. Ther. 2012, 20, 187–195. [Google Scholar] [CrossRef] [PubMed]
- Giri, J.; Das, R.; Nylen, E.; Chinnadurai, R.; Galipeau, J. CCL2 and CXCL12 Derived from Mesenchymal Stromal Cells Cooperatively Polarize IL-10+ Tissue Macrophages to Mitigate Gut Injury. Cell Rep. 2020, 30, 1923–1934. [Google Scholar] [CrossRef] [Green Version]
- De Witte, S.F.H.; Luk, F.; Sierra Parraga, J.M.; Gargesha, M.; Merino, A.; Korevaar, S.S.; Shankar, A.S.; O’Flynn, L.; Elliman, S.J.; Roy, D.; et al. Immunomodulation By Therapeutic Mesenchymal Stromal Cells (MSC) Is Triggered Through Phagocytosis of MSC By Monocytic Cells. Stem Cells 2018, 36, 602–615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lombaert, I.M.A.; Wierenga, P.K.; Kok, T.; Kampinga, H.H.; deHaan, G.; Coppes, R.P. Mobilization of bone marrow stem cells by granulocyte colony-stimulating factor ameliorates radiation-induced damage to salivary glands. Clin. Cancer Res. 2006, 12, 1804–1812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Klopp, A.H.; Spaeth, E.L.; Dembinski, J.L.; Woodward, W.A.; Munshi, A.; Meyn, R.E.; Cox, J.D.; Andreeff, M.; Marini, F.C. Tumor Irradiation Increases the Recruitment of Circulating Mesenchymal Stem Cells into the Tumor Microenvironment. Cancer Res. 2007, 67, 11687–11695. [Google Scholar] [CrossRef] [Green Version]
- Simmons, P.J.; Przepiorka, D.; Thomas, E.D.; Torok-Storb, B. Host origin of marrow stromal cells following allogeneic bone marrow transplantation. Nature 1987, 328, 429–432. [Google Scholar] [CrossRef]
- Cilloni, D.; Carlo-Stella, C.; Falzetti, F.; Sammarelli, G.; Regazzi, E.; Colla, S.; Rizzoli, V.; Aversa, F.; Martelli, M.F.; Tabilio, A. Limited engraftment capacity of bone marrow-derived mesenchymal cells following T-cell-depleted hematopoietic stem cell transplantation. Blood 2000, 96, 3637–3643. [Google Scholar] [CrossRef]
- Horwitz, E.M.; Prockop, D.J.; Fitzpatrick, L.A.; Koo, W.W.; Gordon, P.L.; Neel, M.; Sussman, M.; Orchard, P.; Marx, J.C.; Pyeritz, R.E.; et al. Transplantability and therapeutic effects of bone marrow-derived mesenchymal cells in children with osteogenesis imperfecta. Nat. Med. 1999, 5, 309–313. [Google Scholar] [CrossRef]
- Fouillard, L.; Bensidhoum, M.; Bories, D.; Bonte, H.; Lopez, M.; Moseley, A.-M.; Smith, A.; Lesage, S.; Beaujean, F.; Thierry, D.; et al. Engraftment of allogeneic mesenchymal stem cells in the bone marrow of a patient with severe idiopathic aplastic anemia improves stroma. Leukemia 2003, 17, 474–476. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leibacher, J.; Henschler, R. Biodistribution, migration and homing of systemically applied mesenchymal stem/stromal cells. Stem Cell Res. Ther. 2016, 7, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rombouts, W.J.C.; Ploemacher, R.E. Primary murine MSC show highly efficient homing to the bone marrow but lose homing ability following culture. Leukemia 2003, 17, 160–170. [Google Scholar] [CrossRef] [Green Version]
- Cornelissen, A.S.; Maijenburg, M.W.; Nolte, M.A.; Voermans, C. Organ-specific migration of mesenchymal stromal cells: Who, when, where and why? Immunol. Lett. 2015, 168, 159–169. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Yang, Y.; Zhang, Y.; Cai, W. Non-Invasive Cell Tracking in Cancer and Cancer Therapy. Curr. Top. Med. Chem. 2010, 10, 1237–1248. [Google Scholar] [CrossRef]
- Schweizer, M.T.; Wang, H.; Bivalacqua, T.J.; Partin, A.W.; Lim, S.J.; Chapman, C.; Abdallah, R.; Levy, O.; Bhowmick, N.A.; Karp, J.M.; et al. A Phase I Study to Assess the Safety and Cancer-Homing Ability of Allogeneic Bone Marrow-Derived Mesenchymal Stem Cells in Men with Localized Prostate Cancer. Stem Cells Transl. Med. 2019, 8, 441–449. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krueger, T.E.G.; Thorek, D.L.J.; Denmeade, S.R.; Isaacs, J.T.; Brennen, W.N. Concise Review: Mesenchymal Stem Cell-Based Drug Delivery: The Good, the Bad, the Ugly, and the Promise. Stem Cells Transl. Med. 2018, 7, 651–663. [Google Scholar] [CrossRef] [Green Version]
- Kim, S.M.; Kim, D.-S.; Jeong, C.H.; Kim, D.H.; Kim, J.H.; Jeon, H.B.; Kwon, S.-J.; Jeun, S.-S.; Yang, Y.S.; Oh, W.; et al. CXC chemokine receptor 1 enhances the ability of human umbilical cord blood-derived mesenchymal stem cells to migrate toward gliomas. Biochem. Biophys. Res. Commun. 2011, 407, 741–746. [Google Scholar] [CrossRef]
- Ferrer, F.A.; Pantschenko, A.G.; Miller, L.J.; Anderson, K.; Grunnet, M.; Mckenna, P.H.; Kreutzer, D. Angiogenesis and neuroblastomas: Interleukin-8 and interleukin-8 receptor expression in human neuroblastoma. J. Urol. 2000, 1016–1020. [Google Scholar] [CrossRef]
- Nakata, R.; Shimada, H.; Fernandez, G.E.; Fanter, R.; Fabbri, M.; Malvar, J.; Zimmermann, P.; DeClerck, Y.A. Contribution of neuroblastoma-derived exosomes to the production of pro-tumorigenic signals by bone marrow mesenchymal stromal cells. J. Extracell. Vesicles 2017, 6, 1332941. [Google Scholar] [CrossRef]
- Mu, C.-F.; Shen, J.; Liang, J.; Zheng, H.-S.; Xiong, Y.; Wei, Y.-H.; Li, F. Targeted drug delivery for tumor therapy inside the bone marrow. Biomaterials 2018, 155, 191–202. [Google Scholar] [CrossRef] [PubMed]
- Johannsen, M.; Spitaleri, G.; Curigliano, G.; Roigas, J.; Weikert, S.; Kempkensteffen, C.; Roemer, A.; Kloeters, C.; Rogalla, P.; Pecher, G.; et al. The tumour-targeting human L19-IL2 immunocytokine: Preclinical safety studies, phase I clinical trial in patients with solid tumours and expansion into patients with advanced renal cell carcinoma. Eur. J. Cancer 2010, 46, 2926–2935. [Google Scholar] [CrossRef]
- Bobis-Wozowicz, S.; Miekus, K.; Wybieralska, E.; Jarocha, D.; Zawisz, A.; Madeja, Z.; Majka, M. Genetically modified adipose tissue−derived mesenchymal stem cells overexpressing CXCR4 display increased motility, invasiveness, and homing to bone marrow of NOD/SCID mice. Exp. Hematol. 2011, 39, 686–696.e4. [Google Scholar] [CrossRef] [PubMed]
- Chen, W.; Li, M.; Cheng, H.; Yan, Z.; Cao, J.; Pan, B.; Sang, W.; Wu, Q.; Zeng, L.; Li, Z.; et al. Overexpression of the mesenchymal stem cell Cxcr4 gene in irradiated mice increases the homing capacity of these cells. Cell Biochem. Biophys. 2013, 67, 1181–1191. [Google Scholar] [CrossRef] [PubMed]
- Sackstein, R.; Merzaban, J.S.; Cain, D.W.; Dagia, N.M.; Spencer, J.A.; Lin, C.P.; Wohlgemuth, R. Ex vivo glycan engineering of CD44 programs human multipotent mesenchymal stromal cell trafficking to bone. Nat. Med. 2008, 14, 181–187. [Google Scholar] [CrossRef]
- Caplan, A.I. Adult mesenchymal stem cells for tissue engineering versus regenerative medicine. J. Cell. Physiol. 2007, 213, 341–347. [Google Scholar] [CrossRef]
- In’t Anker, P.S.; Noort, W.A.; Kruisselbrink, A.B.; Scherjon, S.A.; Beekhuizen, W.; Willemze, R.; Kanhai, H.H.H.; Fibbe, W.E. Nonexpanded primary lung and bone marrow--derived mesenchymal cells promote the engraftment of umbilical cord blood--derived CD34+ cells in NOD/SCID mice. Exp. Hematol. 2003, 31, 881–889. [Google Scholar] [CrossRef]
- Ghazanfari, R.; Zacharaki, D.; Li, H.; Ching Lim, H.; Soneji, S.; Scheding, S. Human Primary Bone Marrow Mesenchymal Stromal Cells and Their in vitro Progenies Display Distinct Transcriptional Profile Signatures. Sci. Rep. 2017, 7, 10338. [Google Scholar] [CrossRef]
- Kim, M.; Rhee, J.-K.; Choi, H.; Kwon, A.; Kim, J.; Lee, G.D.; Jekarl, D.W.; Lee, S.; Kim, Y.; Kim, T.-M. Passage-dependent accumulation of somatic mutations in mesenchymal stromal cells during in vitro culture revealed by whole genome sequencing. Sci. Rep. 2017, 7, 14508. [Google Scholar] [CrossRef] [Green Version]
- Bellagamba, B.C.; Grudzinski, P.B.; Ely, P.B.; Nader, P. de J.H.; Nardi, N.B.; da Silva Meirelles, L. Induction of Expression of CD271 and CD34 in Mesenchymal Stromal Cells Cultured as Spheroids. Stem Cells Int. 2018, 2018, 1–14. [Google Scholar] [CrossRef]
- Abbuehl, J.P.; Tatarova, Z.; Held, W.; Huelsken, J. Long-Term Engraftment of Primary Bone Marrow Stromal Cells Repairs Niche Damage and Improves Hematopoietic Stem Cell Transplantation. Cell Stem Cell 2017, 21, 241–255.e6. [Google Scholar] [CrossRef] [PubMed]
- Nakahara, F.; Borger, D.K.; Wei, Q.; Pinho, S.; Maryanovich, M.; Zahalka, A.H.; Suzuki, M.; Cruz, C.D.; Wang, Z.; Xu, C.; et al. Engineering a haematopoietic stem cell niche by revitalizing mesenchymal stromal cells. Nat. Cell Biol. 2019, 21, 560–567. [Google Scholar] [CrossRef] [PubMed]
- Seeger, R.C.; Reynolds, C.P.; Gallego, R.; Stram, D.O.; Gerbing, R.B.; Matthay, K.K. Quantitative Tumor Cell Content of Bone Marrow and Blood as a Predictor of Outcome in Stage IV Neuroblastoma: A Children’s Cancer Group Study. J. Clin. Oncol. 2000, 18, 4067–4076. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.G.; Parker Kerrigan, B.C.; Hossain, A.; Gumin, J.; Shinojima, N.; Nwajei, F.; Ezhilarasan, R.; Love, P.; Sulman, E.P.; Lang, F.F. Ionizing radiation augments glioma tropism of mesenchymal stem cells. J. Neurosurg. 2018, 128, 287–295. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
| Phase | Anti-Cancer Agent | Properties and Dose of MSCs | Administration Route | Nr. of Patients | Disease Context | Key Findings | Publication/Status | ClinicalTrials.gov Identifier | Year | |
|---|---|---|---|---|---|---|---|---|---|---|
| Delivery of Oncolytic Viruses in MSCs | ||||||||||
| A | n/a | ICOVIR-5, an oncolytic adenovirus | autologous irradiated BM-MSCs (“CELYVIR”), 2–4 doses of each 0.1–0.9 × 106 cells/kg | “infused through a central line” | n = 4, single-arm study | Therapy-resistant NB patients | CR (>3 years) in 1 out of 4 patients, virus detected in BM biopsy | García-Castro et al. [71] | n/a (exploratory study) | 2010 |
| Very low systemic toxicity | ||||||||||
| B | n/a | ICOVIR-5, an oncolytic adenovirus | autologous irradiated BM-MSCs (“CELYVIR”), 4–70 doses of each 150–2640 × 106 cells | “systemic infusion” | n = 12, single-arm study | NB | In vitro assays: adhesion molecules like CXCR1 and CCR1 significantly higher in MSCs of responders | Melen et al. [74] | n/a (compassionate use program) | 2016 |
| Mild and auto-limited virus-related toxicities; none had grade 3+ toxicities | ||||||||||
| Clinical response (SD, PR, CR) in 5 out of 12 patients | ||||||||||
| C | I/II | ICOVIR-5, an oncolytic adenovirus | autologous irradiated BM-MSCs (“CELYVIR”), 2 × 106 cells/kg (children) or 0.5–1 × 106 cells/kg (adults) | intra-venous | n = 9 (pediatric), n = 7 (adult); single-arm study | Metastatic and refractory tumors, including NB | Adenoviral replication detected by PCR in 7 out of 9 pediatric patients but in none of the adults | Ruano et al. [70] | NCT01844661 | 2013 |
| SD in 2 out of 4 patients | ||||||||||
| Increasingly higher numbers of circulating lymphocytes (B and T) in responders compared to non-responders | ||||||||||
| No grade 2–5 toxicities were reported. | ||||||||||
| Phase | Details | Properties and Median Dose of MSCs | Nr. of Patients | Disease Context | Key Findings | Publication/Status | ClinicalTrials.gov Identifier | Year | |
|---|---|---|---|---|---|---|---|---|---|
| allo-MSCs | |||||||||
| A | I | Allogeneic MSCs co-transplanted with haplo-HSCT and subsequent DLI | Allogeneic BM-MSCs, 0.75 × 106 MSC/kg | n = 5 (all received MSCs, no control) | NB (relapsed/refractory) | 2 of 5 patients achieved long-lasting remission (40 and 42 months) | Toporski et al. [82] | NCT00790413 | 2008 |
| Neutrophil recovery in all children (median 13 days), platelet recovery in 4/5 children (12 days) | |||||||||
| Rapid immune reconstitution of NK- and T cells | |||||||||
| No primary aGVHD, but 4/4 patients had secondary GvHD after DLI | |||||||||
| B | I | Allogeneic MSCs co-transplanted with haplo-HSCT and DLI | Allogeneic MSCs, no details or dose mentioned | MSC(+): n = 9, MSC(-): n = 17 | NB (relapsed/refractory) | Primary engraftment in 96% (25/26) of the patients | Illhardt et al. [83] | NCT00790413 | 2018 |
| GvHD: no significant differences between MSC and non-MSC group | |||||||||
| Model | MSC Origin | Labeling Method | Administration Route | Maximum Follow up | Detection in Tumor | Publication |
|---|---|---|---|---|---|---|
| NB xenograft model in NOD/SCID mice | human, BM | Radiolabeling | IP | 48 h | Yes | Cussó et al. [126] |
| TH-MYCN transgenic mouse | human, AT | Near-IR | IP, IV | 24 h | Only i.p. | Kimura et al. [127] |
| TH-MYCN transgenic mouse | mouse, BM | GFP | IP | 2 weeks | Yes | Maniwa et al. [57] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hochheuser, C.; Kunze, N.Y.; Tytgat, G.A.M.; Voermans, C.; Timmerman, I. The Potential of Mesenchymal Stromal Cells in Neuroblastoma Therapy for Delivery of Anti-Cancer Agents and Hematopoietic Recovery. J. Pers. Med. 2021, 11, 161. https://doi.org/10.3390/jpm11030161
Hochheuser C, Kunze NY, Tytgat GAM, Voermans C, Timmerman I. The Potential of Mesenchymal Stromal Cells in Neuroblastoma Therapy for Delivery of Anti-Cancer Agents and Hematopoietic Recovery. Journal of Personalized Medicine. 2021; 11(3):161. https://doi.org/10.3390/jpm11030161
Chicago/Turabian StyleHochheuser, Caroline, Nina Y. Kunze, Godelieve A. M. Tytgat, Carlijn Voermans, and Ilse Timmerman. 2021. "The Potential of Mesenchymal Stromal Cells in Neuroblastoma Therapy for Delivery of Anti-Cancer Agents and Hematopoietic Recovery" Journal of Personalized Medicine 11, no. 3: 161. https://doi.org/10.3390/jpm11030161
APA StyleHochheuser, C., Kunze, N. Y., Tytgat, G. A. M., Voermans, C., & Timmerman, I. (2021). The Potential of Mesenchymal Stromal Cells in Neuroblastoma Therapy for Delivery of Anti-Cancer Agents and Hematopoietic Recovery. Journal of Personalized Medicine, 11(3), 161. https://doi.org/10.3390/jpm11030161