
Histone Deacetylase Inhibitors in the Treatment of Hepatocellular Carcinoma: Current Evidence and Future Opportunities

 ,
,  ,
, 
 ,
,  ,
,  ,
,  ,
, 
Abstract
:
1. Introduction
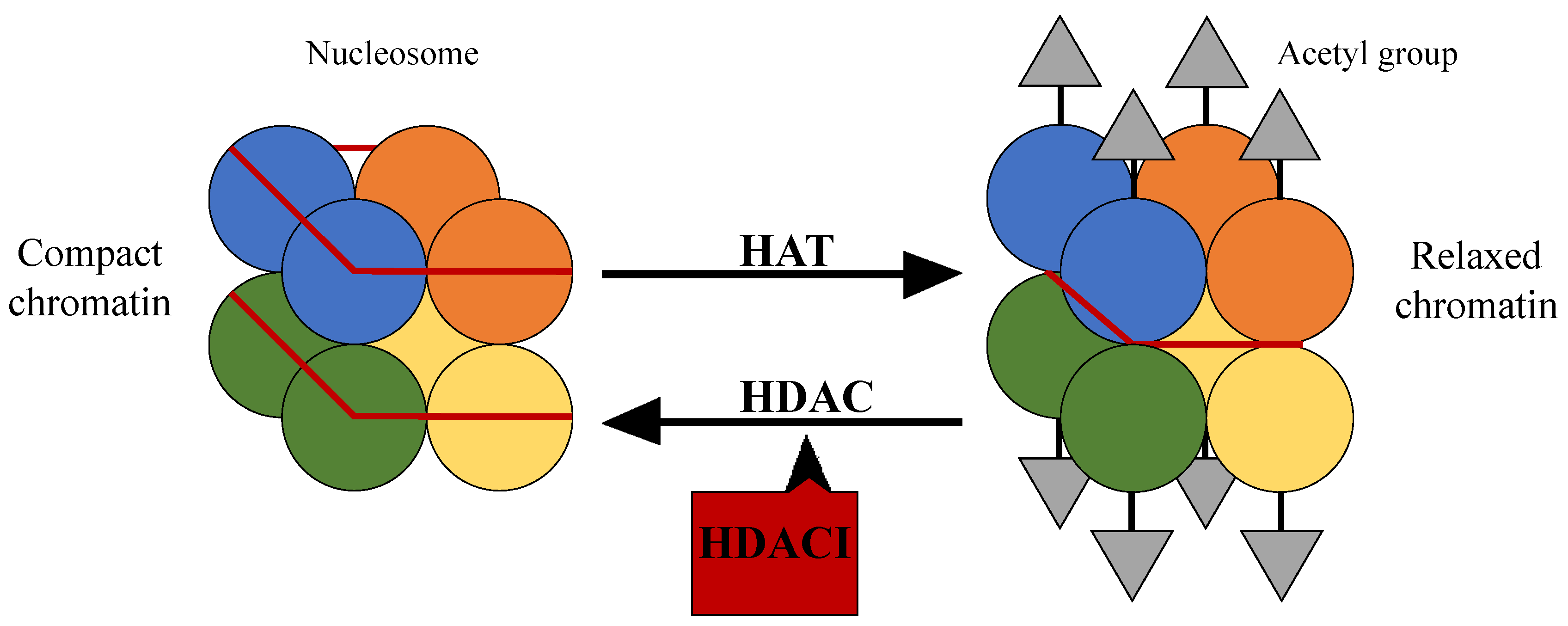
2. HDAC and Its Mechanism of Action
3. HDACIs as Anti-Cancer Agents in HCC
3.1. Panobinostat
3.2. Trichostatin A
3.3. Suberanilohydroxamic Acid
3.4. Valproic Acid
3.5. Resminostat
3.6. Other HDACIs
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2016. Cancer J. Clin. 2016, 66, 7–30. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dimitroulis, D.; Damaskos, C.; Valsami, S.; Davakis, S.; Garmpis, N.; Spartalis, E.; Athanasiou, A.; Moris, D.; Sakellariou, S.; Kykalos, S.; et al. From diagnosis to treatment of hepatocellular carcinoma: An epidemic problem for both developed and developing world. World J. Gastroenterol. 2017, 23, 5282–5294. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Soerjomataram, I.; Dikshit, R.; Eser, S.; Mathers, C.; Rebelo, M.; Parkin, D.M.; Forman, D.; Bray, F. Cancer incidence and mortality worldwide: Sources, methods and major patterns in GLOBOCAN 2012. Int. J. Cancer 2015, 136, E359–E386. [Google Scholar] [CrossRef] [PubMed]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef] [Green Version]
- Centers for Disease Control and Prevention (CDC). Hepatocellular carcinoma—United States, 2001–2006. Morb. Mortal. Wkly. Rep. 2010, 59, 517–520. [Google Scholar]
- Petrick, J.L.; Kelly, S.P.; Altekruse, S.F.; McGlynn, K.A.; Rosenberg, P.S. Future of hepatocellular carcinoma incidence in the United States forecast through 2030. J. Clin. Oncol. 2016, 34, 1787–1794. [Google Scholar] [CrossRef] [PubMed]
- Wong, R.; Corley, D.A. Racial and ethnic variations in hepatocellular carcinoma incidence within the United States. Am. J. Med. 2008, 121, 525–531. [Google Scholar] [CrossRef]
- Trichopoulos, D.; Bamia, C.; Lagiou, P.; Fedirko, V.; Trepo, E.; Jenab, M.; Pischon, T.; Nöthlings, U.; Overved, K.; Tjønneland, A.; et al. Hepatocellular carcinoma risk factors and disease burden in a European cohort: A nested case-control study. J. Natl. Cancer Inst. 2011, 103, 1686–1695. [Google Scholar] [CrossRef] [PubMed]
- Hamed, M.A.; Ali, S.A. Non-viral factors contributing to hepatocellular carcinoma. World J. Hepatol. 2013, 5, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Dragani, T.A. Risk of HCC: Genetic heterogeneity and complex genetics. J. Hepatol. 2020, 52, 252–257. [Google Scholar] [CrossRef] [Green Version]
- Makarova-Rusher, O.V.; Altekruse, S.F.; McNeel, T.S.; Ulahannan, S.; Duffy, A.G.; Graubard, B.I.; Greten, T.F.; McGlynn, K.A. Population attributable fractions of risk factors for hepatocellular carcinoma in the United States. Cancer 2016, 122, 1757–1765. [Google Scholar] [CrossRef]
- Wong, R.J.; Aguilar, M.; Cheung, R.; Perumpail, R.B.; Harrison, S.A.; Younossi, Z.M.; Ahmed, A. Nonalcoholic steatohepatitis is the second leading etiology of liver disease among adults awaiting liver transplantation in the United States. Gastroenterology 2015, 148, 547–555. [Google Scholar] [CrossRef]
- Kutsenko, A.; Ladenheim, M.R.; Kim, N.; Nguyen, P.; Chen, V.; Jayasekera, C.; Yang, J.D.; Kumari, R.; Roberts, L.; Nguyen, M.H. Increased prevalence of metabolic risk factors in Asian Americans with hepatocellular carcinoma. J. Clin. Gastroenterol. 2017, 51, 384–390. [Google Scholar] [CrossRef]
- Keating, G.M.; Santoro, A. Sorafenib: A review of its use in advanced hepatocellular carcinoma. Drugs 2009, 69, 223–240. [Google Scholar] [CrossRef] [PubMed]
- Kahn, F.S.; Ali, I.; Afridi, U.K.; Ishtiaq, M.; Mehmood, R. Epigenetic mechanisms regulating the development of hepatocellular carcinoma and their promise for therapeutics. Hepatol. Int. 2017, 11, 45–53. [Google Scholar] [CrossRef] [PubMed]
- Rando, O.J.; Ahmad, K. Rules and regulation in the primary structure of chromatin. Curr. Opin. Cell Biol. 2007, 19, 250–256. [Google Scholar] [CrossRef]
- Woo, Y.M. Epigenetic regulation in cystogenesis. Adv. Exp. Med. Biol. 2016, 933, 59–68. [Google Scholar]
- Kouzarides, T. Chromatin modifications and their function. Cell 2007, 128, 693–705. [Google Scholar] [CrossRef] [Green Version]
- Kuo, M.H.; Allis, C.D. Roles of histone acetyltransferases and deacetylases in gene regulation. Bioessays 1998, 20, 615–626. [Google Scholar] [CrossRef]
- Haberland, M.; Montgomery, R.L.; Olson, E.N. The many roles of histone deacetylases in development and physiology: Implications for disease and therapy. Nat. Rev. Genet. 2009, 10, 32–42. [Google Scholar] [CrossRef]
- Garmpis, N.; Damaskos, C.; Garmpi, A.; Dimitroulis, D.; Spartalis, E.; Margonis, G.A.; Schizas, D.; Deskou, I.; Doula, C.; Magkouti, E. Targeting histone deacetylases in malignant melanoma: A future therapeutic agent or just great expectations? Anticancer Res. 2017, 37, 5355–5362. [Google Scholar] [PubMed]
- Trapp, J.; Jung, M. The role of NAD+-dependent histone deacetylases (sirtuins) in ageing. Curr. Drug Targets 2006, 7, 1553–1560. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.J.; Seto, E. Collaborative spirit of histone deacetylases in regulating chromatin structure and gene expression. Curr. Opin. Genet. Dev. 2003, 13, 143–153. [Google Scholar] [CrossRef]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nakagawa, M.; Oda, Y.; Eguchi, T.; Aishima, S.I.; Yao, T.; Hosoi, F.; Basaki, Y.; Ono, M.; Kuwano, M.; Tanaka, M.; et al. Expression profile of class I histone deacetylases in human cancer tissues. Oncol. Rep. 2007, 18, 769–774. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.W.; Dong, L.D.; Shang, W.J.; Pang, X.L.; Li, J.F.; Liu, L.; Wang, Y. HDAC5 promotes cell proliferation in human hepatocellular carcinoma by up-regulating Six1 expression. Eur. Rev. Med. Pharmacol. Sci. 2014, 18, 811–816. [Google Scholar]
- Quint, K.; Agaimy, A.; Di Fazio, P.; Montalbano, R.; Steindorf, C.; Jung, R.; Hellerbrand, C.; Hartmann, A.; Sitter, H.; Neureiter, D.; et al. Clinical significance of histone deacetylases 1, 2, 3, and 7: HDAC2 is an independent predictor of survival in HCC. Virchows Arch. 2011, 459, 129–139. [Google Scholar] [CrossRef] [PubMed]
- Rikimaru, T.; Taketomi, A.; Yamashita, Y.I.; Shirabe, K.; Hamatsu, T.; Shimada, M.; Maehara, Y. Clinical significance of histone deacetylase 1 expression in patients with hepatocellular carcinoma. Oncology 2007, 72, 69–74. [Google Scholar] [CrossRef]
- Wu, L.M.; Yang, Z.; Zhou, L.; Zhang, F.; Xie, H.Y.; Feng, X.W.; Wu, J.; Zheng, S.S. Identification of histone deacetylase 3 as a biomarker for tumor recurrence following liver transplantation in HBV-associated hepatocellular carcinoma. PLoS ONE 2010, 5, e14460. [Google Scholar] [CrossRef]
- Garmpi, A.; Garmpis, N.; Damaskos, C.; Valsami, S.; Spartalis, E.; Lavaris, A.; Patelis, N.; Margonis, G.A.; Apostolou, K.G.; Spartalis, M.; et al. Histone deacetylase inhibitors as a new anticancer option: How far can we go with expectations? J. BUON 2018, 23, 846–861. [Google Scholar]
- Marks, P.A. The clinical development of histone deacetylase inhibitors as targeted anticancer drugs. Expert Opin. Investig. Drugs 2010, 19, 1049–1066. [Google Scholar] [CrossRef]
- Damaskos, C.; Karatzas, T.; Nikolidakis, L.; Kostakis, I.D.; Karamaroudis, S.; Boutsikos, G.; Damaskou, Z.; Kostakis, A.; Kouraklis, G. Histone deacetylase (HDAC) inhibitors: Current evidence for therapeutic activities in pancreatic cancer. Anticancer Res. 2015, 35, 3129–3135. [Google Scholar]
- Giaginis, C.; Damaskos, C.; Koutsounas, I.; Zizi-Serbetzoglou, A.; Tsoukalas, N.; Patsouris, E.; Kouraklis, G.; Theocharis, S. Histone deacetylase (HDAC)-1, -2, -4 and -6 expression in human pancreatic adenocarcinoma: Associations with clinicopathological parameters, tumor proliferative capacity and patients’ survival. BMC Gastroenterol. 2015, 15, 148. [Google Scholar] [CrossRef] [Green Version]
- Damaskos, C.; Valsami, S.; Kontos, M.; Spartalis, E.; Kalampokas, T.; Kalampokas, E.; Athanasiou, A.; Moris, D.; Daskalopoulou, A.; Davakis, S.; et al. Histone deacetylase inhibitors: An attractive therapeutic strategy against breast cancer. Anticancer Res. 2017, 37, 35–46. [Google Scholar] [CrossRef] [Green Version]
- Garmpis, N.; Damaskos, C.; Garmpi, A.; Kalampokas, E.; Kalampokas, T.; Spartalis, E.; Daskalopoulou, A.; Valsami, S.; Kontos, M.; Nonni, A.; et al. Histone deacetylases as new therapeutic targets in triple-negative breast cancer: Progress and promises. Cancer Genom. Proteom. 2017, 14, 299–313. [Google Scholar]
- Damaskos, C.; Valsami, S.; Spartalis, E.; Antoniou, E.A.; Tomos, P.; Karamaroudis, S.; Zoumpou, T.; Pergialiotis, V.; Stergios, K.; Michaelides, C.; et al. Histone deacetylase inhibitors: A novel therapeutic weapon against medullary thyroid cancer? Anticancer Res. 2016, 36, 5019–5024. [Google Scholar] [CrossRef] [PubMed]
- Damaskos, C.; Tomos, I.; Garmpis, N.; Karakatsani, A.; Dimitroulis, D.; Garmpi, A.; Spartalis, E.; Kampolis, C.F.; Tsagkari, E.; Loukeri, A.A.; et al. Histone deacetylase inhibitors as a novel targeted therapy against non-small cell lung cancer: Where are we now and what should we expect? Anticancer Res. 2018, 38, 37–43. [Google Scholar] [PubMed]
- Schizas, D.; Mastoraki, A.; Naar, L.; Spartalis, E.; Tsilimigras, D.I.; Karachaliou, G.S.; Bagias, G.; Moris, D. Concept of histone deacetylases in cancer: Reflections on esophageal carcinogenesis and treatment. World J. Gastroenterol. 2018, 24, 4635–4642. [Google Scholar] [CrossRef] [PubMed]
- Garmpis, N.; Damaskos, C.; Garmpi, A.; Spartalis, E.; Kalampokas, E.; Kalampokas, T.; Margonis, G.A.; Schizas, D.; Andreatos, N.; Angelou, A.; et al. Targeting histone deacetylases in endometrial cancer: A paradigm-shifting therapeutic strategy? Eur. Rev. Med. Pharmacol. Sci. 2018, 22, 950–960. [Google Scholar]
- Tampakis, A.; Tampaki, E.C.; Nebiker, C.A.; Kouraklis, G. Histone deacetylase inhibitors and colorectal cancer: What is new? Anticancer Agents Med. Chem. 2014, 14, 1220–1227. [Google Scholar] [CrossRef]
- Moschos, M.M.; Dettoraki, M.; Androudi, S.; Kalogeropoulos, D.; Lavaris, A.; Garmpis, N.; Damaskos, C.; Garmpi, A.; Tsatsos, M. The role of histone deacetylase inhibitors in uveal melanoma: Current evidence. Anticancer Res. 2018, 38, 3817–3824. [Google Scholar] [CrossRef]
- Marks, P.A.; Breslow, R. Dimethyl sulfoxide to vorinostat: Development of this histone deacetylase inhibitor as an anticancer drug. Nat. Biotechnol. 2007, 25, 84–90. [Google Scholar] [CrossRef] [PubMed]
- Barbarotta, L.; Hurley, K. Romidepsin for the treatment of peripheral T-cell lymphoma. J. Adv. Pract. Oncol. 2015, 6, 22–36. [Google Scholar] [PubMed]
- Zhou, H.; Cai, Y.; Liu, D.; Li, M.; Sha, Y.; Zhang, W.; Wang, K.; Gong, J.; Tang, N.; Huang, A.; et al. Pharmacological or transcriptional inhibition of both HDAC1 and 2 leads to cell cycle blockage and apoptosis via p21Waf1/Cip1 and p19INK4d upregulation in hepatocellular carcinoma. Cell Prolif. 2018, 51, e12447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Warburg, O. On the origin of cancer cells. Science 1956, 123, 309–314. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, X.; Zhang, J.; Lam, E.K.; Shin, V.Y.; Cheng, A.S.; Yu, J.; Chan, F.K.; Sung, J.J.; Jin, H.C. Warburg effect revisited: An epigenetic link between glycolysis and gastric carcinogenesis. Oncogene 2010, 29, 442–450. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, M.; Zhang, J.; Li, N.; Qian, Z.; Zhu, M.; Li, Q.; Zheng, J.; Wang, X.; Shi, G. Promoter hypermethylation mediated downregulation of FBP1 in human hepatocellular carcinoma and colon cancer. PLoS ONE 2011, 6, e25564. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.; Jin, X.; Yan, Y.; Shao, Y.; Pan, Y.; Roberts, L.R.; Zhang, J.; Huang, H.; Jiang, J. Inhibiting histone deacetylases suppresses glucose metabolism and hepatocellular carcinoma growth by restoring FBP1 expression. Sci. Rep. 2017, 7, 1–13. [Google Scholar] [CrossRef]
- Prince, H.M.; Bishton, M.J.; Johnstone, R.W. Panobinostat (LBH589): A potentpan-deacetylase inhibitor with promising activity against hematologic and solid tumors. Future Oncol. 2009, 5, 601–612. [Google Scholar] [CrossRef]
- Edwards, A.; Li, J.; Atadja, P.; Bhalla, K.; Haura, E.B. Effect of the histone deacetylase inhibitor LBH589 against epidermal growth factor receptor-dependent human lung cancer cells. Mol. Cancer Ther. 2007, 6, 2515–2524. [Google Scholar] [CrossRef] [Green Version]
- Giles, F.; Fischer, T.; Cortes, J.; Garcia-Manero, G.; Beck, J.; Ravandi, F.; Masson, E.; Rae, P.; Laird, G.; Sharma, S.; et al. A phase I study of intravenous LBH589, a novel cinnamic hydroxamic acid analogue histone deacetylase inhibitor, in patients with refractory hematologic malignancies. Clin. Cancer Res. 2006, 12, 4628–4635. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Di Fazio, P.; Schneider-Stock, R.; Neureiter, D.; Okamoto, K.; Wissniowski, T.; Gahr, S.; Quint, K.; Meissnitzer, M.; Alinger, B.; Montalbano, R.; et al. The pan-deacetylase inhibitor panobinostat inhibits growth of hepatocellular carcinoma models by alternative pathways of apoptosis. Cell Oncol. 2010, 32, 285–300. [Google Scholar] [PubMed]
- Gnyszka, A.; Jastrzebski, Z.; Flis, S. DNA methyltransferase inhibitors and their emerging role in epigenetic therapy of cancer. Anticancer Res. 2013, 33, 2989–2996. [Google Scholar] [PubMed]
- Oh, B.K.; Kim, H.; Park, H.J.; Shim, Y.H.; Choi, J.; Park, C.; Park, Y.N. DNA methyltransferase expression and DNA methylation in human hepatocellular carcinoma and their clinicopathological correlation. Int. J. Mol. Med. 2007, 20, 65–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zopf, S.; Ocker, M.; Neureiter, D.; Alinger, B.; Gahr, S.; Neurath, M.F.; Di Fazio, P. Inhibition of DNA methyltransferase activity and expression by treatment with the pan-deacetylase inhibitor panobinostat in hepatocellular carcinoma cell lines. BMC Cancer 2012, 12, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ocker, M. Deacetylase inhibitors—Focus on non-histone targets and effects. World J. Biol. Chem. 2010, 1, 55–61. [Google Scholar] [CrossRef]
- Kanno, S.; Oda, N.; Abe, M.; Terai, Y.; Ito, M.; Shitara, K.; Tabayashi, K.; Shibuya, M.; Sato, Y. Roles of two VEGF receptors, Flt-1 and KDR, in the signal transduction of VEGF effects in human vascular endothelial cells. Oncogene 2000, 19, 2138–2146. [Google Scholar] [CrossRef] [Green Version]
- Schoenleber, S.J.; Kurtz, D.M.; Talwalkar, J.A.; Roberts, L.R.; Gores, G.J. Prognostic role of vascular endothelial growth factor in hepatocellular carcinoma: Systematic review and meta-analysis. Br. J. Cancer 2009, 100, 1385–1392. [Google Scholar] [CrossRef] [Green Version]
- Greten, T.F.; Korangy, F.; Manns, M.P.; Malek, N.P. Molecular therapy for the treatment of hepatocellular carcinoma. Br. J. Cancer 2009, 100, 19–23. [Google Scholar] [CrossRef]
- Shimo, T.; Nakanishi, T.; Nishida, T.; Asano, M.; Kanyama, M.; Kuboki, T.; Tamatani, T.; Tezuka, K.; Takemura, M.; Matsumura, T.; et al. Connective tissue growth factor induces the proliferation, migration, and tube formation of vascular endothelial cells in vitro, and angiogenesis in vivo. J. Biochem. 1999, 126, 137–145. [Google Scholar] [CrossRef]
- Urtasun, R.; Latasa, M.U.; Demartis, M.I.; Balzani, S.; Goñi, S.; Garcia-Irigoyen, O.; Elizalde, M.; Azcona, M.; Pascale, R.M.; Feo, F.; et al. Connective tissue growth factor autocriny in human hepatocellular carcinoma: Oncogenic role and regulation by epidermal growth factor receptor/yes-associated protein-mediated activation. Hepatology 2011, 54, 2149–2158. [Google Scholar] [CrossRef]
- Gahr, S.; Mayr, C.; Kiesslich, T.; Illig, R.; Neureiter, D.; Alinger, B.; Ganslmayer, M.; Wissniowski, T.; Fazio, P.D.; Montalbano, R.; et al. The pan-deacetylase inhibitor panobinostat affects angiogenesis in hepatocellular carcinoma models via modulation of CTGF expression. Int. J. Oncol. 2015, 47, 963–970. [Google Scholar] [CrossRef] [Green Version]
- Lachenmayer, A.; Toffanin, S.; Cabellos, L.; Alsinet, C.; Hoshida, Y.; Villanueva, A.; Minguez, B.; Tsai, H.W.; Ward, S.C.; Thung, S.; et al. Combination therapy for hepatocellular carcinoma: Additive preclinical efficacy of the HDAC inhibitor panobinostat with sorafenib. J. Hepatol. 2012, 56, 1343–1350. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Finnin, M.S.; Donigian, J.R.; Cohen, A.; Richon, V.M.; Rifkind, R.A.; Marks, P.A.; Breslow, R.; Pavletich, N.P. Structures of a histone deacetylase homologue bound to the TSA and SAHA inhibitors. Nature 1999, 401, 188–193. [Google Scholar] [CrossRef] [PubMed]
- Yoshida, M.; Horinouchi, S.; Beppu, T. Trichostatin A and trapoxin: Novel chemical probes for the role of histone acetylation in chromatin structure and function. Bioessays 1995, 17, 423–430. [Google Scholar] [CrossRef] [PubMed]
- Buurman, R.; Gürlevik, E.; Schäffer, V.; Eilers, M.; Sandbothe, M.; Kreipe, H.; Wilkens, L.; Schlegelberger, B.; Kühnel, F.; Skawran, B. Histone deacetylases activate hepatocyte growth factor signaling by repressing microRNA-449 in hepatocellular carcinoma cells. Gastroenterology 2012, 143, 811–820. [Google Scholar] [CrossRef]
- Chiba, T.; Yokosuka, O.; Arai, M.; Tada, M.; Fukai, K.; Imazeki, F.; Kato, M.; Seki, N.; Saisho, H. Identification of genes up-regulated by histone deacetylase inhibition with cDNA microarray and exploration of epigenetic alterations on hepatoma cells. J. Hepatol. 2004, 41, 436–445. [Google Scholar] [CrossRef]
- Chiba, T.; Yokosuka, O.; Fukai, K.; Kojima, H.; Tada, M.; Arai, M.; Imazeki, F.; Saisho, H. Cell growth inhibition and gene expression induced by the histone deacetylase inhibitor, trichostatin A, on human hepatoma cells. Oncology 2004, 66, 481–491. [Google Scholar] [CrossRef]
- Buurman, R.; Sandbothe, M.; Schlegelberger, B.; Skawran, B. HDAC inhibition activates the apoptosome via Apaf1 upregulation in hepatocellular carcinoma. Eur. J. Med. Res. 2016, 21, 1–6. [Google Scholar] [CrossRef] [Green Version]
- Shin, S.; Kim, M.; Lee, S.J.; Park, K.S.; Lee, C.H. Trichostatin A sensitizes hepatocellular carcinoma cells to enhanced NK cell-mediated killing by regulating immune-related genes. Cancer Genom. Proteom. 2017, 14, 349–362. [Google Scholar]
- Yoshihama, S.; Roszik, J.; Downs, I.; Meissner, T.B.; Vijayan, S.; Chapuy, B.; Sidiq, T.; Shipp, M.A.; Lizee, G.A.; Kobayashi, K.S. NLRC5/MHC class I transactivator is a target for immune evasion in cancer. Proc. Natl. Acad. Sci. USA 2016, 113, 5999–6004. [Google Scholar] [CrossRef] [Green Version]
- Hishida, M.; Nomoto, S.; Inokawa, Y.; Hayashi, M.; Kanda, M.; Okamura, Y.; Nishikawa, Y.; Tanaka, C.; Kobayashi, D.; Yamada, S.; et al. Estrogen receptor 1 gene as a tumor suppressor gene in hepatocellular carcinoma detected by triple-combination array analysis. Int. J. Oncol. 2013, 43, 88–94. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Phillips, D.L.; Ferguson, A.T.; Nelson, W.G.; Herman, J.G.; Davidson, N.E. Synergistic activation of functional estrogen receptor (ER)-alpha by DNA methyltransferase and histone deacetylase inhibition in human ER-alpha-negative breast cancer cells. Cancer Res. 2011, 61, 7025–7029. [Google Scholar]
- Sanaei, M.; Kavoosi, F.; Salehi, H. Genistein and trichostatin A induction of estrogen receptor alpha gene expression, apoptosis and cell growth inhibition in hepatocellular carcinoma HepG 2 cells. Asian Pac. J. Cancer Prev. 2017, 18, 3445–3450. [Google Scholar]
- Kim, S.H.; Kang, H.J.; Na, H.; Lee, M.O. Trichostatin A enhances acetylation as well as protein stability of ERα through induction of p300 protein. Breast Cancer Res. 2010, 12, 1–8. [Google Scholar] [CrossRef] [Green Version]
- Sanaei, M.; Kavoosi, F.; Arabloo, M. Effect of curcumin in comparison with trichostatin A on the reactivation of estrogen receptor alpha gene expression, cell growth inhibition and apoptosis induction in hepatocellular carcinoma Hepa 1-6 cell lline. Asian Pac. J. Cancer Prev. 2020, 21, 1045–1050. [Google Scholar] [CrossRef]
- Slingerland, M.; Guchelaar, H.J.; Gelderblom, H. Histone deacetylase inhibitors: An overview of the clinical studies in solid tumors. Anticancer Drugs 2014, 25, 140–149. [Google Scholar] [CrossRef]
- Marks, P.; Rifkind, R.A.; Richon, V.M.; Breslow, R.; Miller, T.; Kelly, W.K. Histone deacetylases and cancer: Causes and therapies. Nat. Rev. Cancer 2001, 1, 194–202. [Google Scholar] [CrossRef] [PubMed]
- Kunnimalaiyaan, S.; Sokolowski, K.; Gamblin, T.C.; Kunnimalaiyaan, M. Suberoylanilide hydroxamic acid, a histone deacetylase inhibitor, alters multiple signaling pathways in hepatocellular carcinoma cell lines. Am. J. Surg. 2017, 213, 645–651. [Google Scholar] [CrossRef] [PubMed]
- Domingo-Domenech, J.; Pippa, R.; Tapia, M.; Gascon, P.; Bachs, O.; Bosch, M. Inactivation of NF-kappaB by proteasome inhibition contributes to increased apoptosis induced by histone deacetylase inhibitors in human breast cancer cells. Breast Cancer Res. Treat. 2008, 112, 53–62. [Google Scholar] [CrossRef] [PubMed]
- Dai, Y.; Guzman, M.L.; Chen, S.; Wang, L.; Yeung, S.K.; Pei, X.Y.; Dent, P.; Jordan, C.T.; Grant, S. The NF (nuclear factor)-kappaB inhibitor parthenolide interacts with histone deacetylase inhibitors to induce MKK7/JNK1-dependent apoptosis in human acute myeloid leukaemia cells. Br. J. Haematol. 2010, 151, 70–83. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, I.T.; Liu, Y.C.; Wang, W.H.; Hsu, F.T.; Chen, H.W.; Lin, W.J.; Chang, W.Y.; Hwang, J.J. Sorafenib inhibits TPA-induced MMP-9 and VEGF expression via suppression of ERK/NF-kappaB pathway in hepatocellular carcinoma cells. In Vivo 2012, 26, 671–681. [Google Scholar] [PubMed]
- Hsu, F.T.; Liu, Y.C.; Chiang, I.; Liu, R.S.; Wang, H.E.; Lin, W.J.; Hwang, J.J. Sorafenib increases efficacy of vorinostat against human hepatocellular carcinoma through transduction inhibition of vorinostat-induced ERK/NF-κB signaling. Int. J. Oncol. 2014, 45, 177–188. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yuan, H.; Li, A.J.; Ma, S.L.; Cui, L.J.; Wu, B.; Yin, L.; Wu, M.C. Inhibition of autophagy significantly enhances combination therapy with sorafenib and HDAC inhibitors for human hepatoma cells. World J. Gastroenterol. 2014, 20, 4953–4962. [Google Scholar] [CrossRef] [PubMed]
- Freese, K.; Seitz, T.; Dietrich, P.; Lee, S.M.; Thasler, W.E.; Bosserhoff, A.; Hellerbrand, C. Histone deacetylase expressions in hepatocellular carcinoma and functional effects of histone deacetylase inhibitors on liver cancer cells in vitro. Cancers 2019, 11, 1587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, H.A.; Chu, K.B.; Moon, E.K.; Kim, S.S.; Quan, F.S. Sensitization to oxidative stress and G2/M cell cycle arrest by histone deacetylase inhibition in hepatocellular carcinoma cells. Free Radic. Biol. Med. 2020, 147, 129–138. [Google Scholar] [CrossRef]
- Gordon, S.W.; McGuire, W.P.; Shafer, D.A.; Sterling, R.K.; Lee, H.M.; Matherly, S.C.; Roberts, J.D.; Bose, P.; Tombes, M.B.; Shrader, E.E.; et al. Phase I study of Sorafenib and Vorinostat in advanced hepatocellular carcinoma. Am. J. Clin. Oncol. 2019, 42, 649–654. [Google Scholar] [CrossRef] [PubMed]
- Sanoei, M.; Kavoosi, F.; Esmi, Z. The effect of 5-aza-2’-deoxycytidine in combination to and in comparison with vorinostat on DNA methyltransferases, histone deacetylase 1, glutathione S-transferase 1 and suppressor of cytokine signaling 1 genes expression, cell growth inhibition and apoptotic induction in hepatocellular LCL-PI 11 cell line. Int. J. Hematol. Oncol. Stem Cell Res. 2020, 14, 45–55. [Google Scholar]
- Perucca, E. Pharmacological and therapeutic properties of valproate: A summary after 35 years of clinical experience. CNS Drugs 2002, 16, 695–714. [Google Scholar] [CrossRef]
- Tsai, C.; Leslie, J.S.; Franko-Tobin, L.G.; Prasnal, M.C.; Yang, T.; Mackey, L.V.; Fuselier, J.A.; Coy, D.H.; Liu, M.; Yu, C.; et al. Valproic acid suppresses cervical cancer tumor progression possibly via activating Notch1 signaling and enhances receptor-targeted cancer chemotherapeutic via activating somatostatin receptor type II. Arch. Gynecol. Obstet. 2013, 288, 393–400. [Google Scholar] [CrossRef]
- Platta, C.S.; Greenblatt, D.Y.; Kunnimalaiyaan, M.; Chen, H. Valproic acid induces Notch1 signaling in small cell lung cancer cells. J. Surg. Res. 2008, 148, 31–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmid, M.M.; Freudenmann, R.W.; Keller, F.; Connemann, B.J.; Hiemke, C.; Gahr, M.; Kratzer, W.; Fuchs, M.; Schoenfeldt-Lecuona, C. Non-fatal and fatal liver failure associated with valproic acid. Pharmacopsychiatry 2013, 46, 63–68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cesar Machado, M.C.; Bellodi-Privato, M.; Kubrusly, M.S.; Trindade Molan, N.A.; Junior, T.T.; de Oliveira, E.R.; Carneiro D’Albuquerque, L.A. Valproic acid inhibits human hepatocellular cancer cells growth in vitro and in vivo. J. Exp. Ther. Oncol. 2011, 9, 85–92. [Google Scholar]
- Sanaei, M.; Kavoosi, F.; Roustazadeh, A.; Shahsavani, H. In vitro effect of the histone deacetylase inhibitor valproic acid on viability and apoptosis of the PLC/PRF5 human hepatocellular harcinoma cell line. Asian Pac. J. Cancer Prev. 2018, 19, 2507–2510. [Google Scholar] [PubMed]
- Morell, C.M.; Strazzabosco, M. Notch signaling and new therapeutic options in liver disease. J. Hepatol. 2014, 60, 885–890. [Google Scholar] [CrossRef] [Green Version]
- Bogaerts, E.; Heindryckx, F.; Vandewynckel, Y.P.; Van Grunsven, L.A.; Van Vlierberghe, H. The roles of transforming growth factor-beta, Wnt, Notch and hypoxia on liver progenitor cells in primary liver tumours (Review). Int. J. Oncol. 2014, 44, 1015–1022. [Google Scholar] [CrossRef] [Green Version]
- Gramantieri, L.; Giovannini, C.; Lanzi, A.; Chieco, P.; Ravaioli, M.; Venturi, A.; Grazi, G.L.; Bolondi, L. Aberrant Notch3 and Notch4 expression in human hepatocellular carcinoma. Liver Int. 2007, 27, 997–1007. [Google Scholar] [CrossRef]
- Viatour, P.; Ehmer, U.; Saddic, L.A.; Dorrell, C.; Andersen, J.B.; Lin, C.; Zmoos, A.F.; Mazur, P.K.; Schaffer, B.E.; Ostermeier, A.; et al. Notch signaling inhibits hepatocellular carcinoma following inactivation of the RB pathway. J. Exp. Med. 2011, 208, 1963–1976. [Google Scholar] [CrossRef] [Green Version]
- Qi, R.; An, H.; Yu, Y.; Zhang, M.; Liu, S.; Xu, H.; Guo, Z.; Cheng, T.; Cao, X. Notch1 signaling inhibits growth of human hepatocellular carcinoma through induction of cell cycle arrest and apoptosis. Cancer Res. 2003, 63, 8323–8329. [Google Scholar]
- Sun, G.; Mackey, L.V.; Coy, D.H.; Yu, C.Y.; Sun, L. The histone deacetylase inhibitor vaproic acid induces cell growth arrest in hepatocellular carcinoma cells via suppressing Notch signaling. J. Cancer 2015, 6, 996–1004. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Liu, J.; Liang, Q.; Sun, G. Valproic acid reverses sorafenib resistance through inhibiting activated Notch/Akt signaling pathway in HCC. Fundam. Clin. Pharmacol. 2020. [Google Scholar] [CrossRef]
- Liu, J.; Yang, X.; Liang, Q.; Yu, Y.; Shen, X.; Sun, G. Valproic acid overcomes sorafenib resistance by reducing the migration of Jagged2-mediated Notch1 signaling pathway in hepatocellular carcinoma cells. Int. J. Biochem. Cell Biol. 2020, 126, 105820. [Google Scholar] [CrossRef]
- Yu, J.I.; Choi, C.; Shin, S.W.; Son, A.; Lee, G.H.; Kim, S.Y.; Park, H.C. Valproic acid sensitizes hepatocellular carcinoma cells to proton therapy by suppressing NRF2 activation. Sci. Rep. 2017, 7, 14986. [Google Scholar] [CrossRef]
- Saha, S.K.; Yin, Y.; Kim, K.; Yang, G.M.; Dayem, A.A.; Choi, H.Y.; Cho, S.G. Valproic acid induces endocytosis-mediated doxorubicin internalization and shows synergistic cytotoxic effects in hepatocellular carcinoma cells. Int. J. Mol. Sci. 2017, 18, 1048. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.C.; Su, C.W.; Ko, P.S.; Lee, R.C.; Liu, C.J.; Huang, Y.H.; Gau, J.P.; Liu, J.H. A clinical trial with valproic acid and hydralazine in combination with gemcitabine and cisplatin followed by doxorubicin and dacarbazine for advanced hepatocellular carcinoma. Asia Pac. J. Clin. Oncol. 2020. [Google Scholar] [CrossRef]
- Mandl-Weber, S.; Meinel, F.; Jankowsky, R.; Oduncu, F.; Schmidmaier, R.; Baumann, P. The novel inhibitor of histone deacetylase resminostat (RAS2410) inhibits proliferation and induces apoptosis in multiple myeloma (MM) cells. Br. J. Haematol. 2010, 149, 518–528. [Google Scholar] [CrossRef] [PubMed]
- Fu, M.; Shi, W.; Li, Z.; Liu, H. Activation of mPTP-dependent mitochondrial apoptosis pathway by a novel pan HDAC inhibitor resminostat in hepatocellular carcinoma cells. Biochem. Biophys. Res. Commun. 2016, 477, 527–533. [Google Scholar] [CrossRef] [PubMed]
- Llovet, J.M.; Ricci, S.; Mazzaferro, V.; Hilgard, P.; Gane, E.; Blanc, J.F.; De Oliveira, A.C.; Santoro, A.; Raoul, J.L.; Forner, A.; et al. Sorafenib in advanced hepatocellular carcinoma. N. Engl. J. Med. 2008, 359, 378–390. [Google Scholar] [CrossRef] [PubMed]
- Bitzer, M.; Horger, M.; Giannini, E.G.; Ganten, T.M.; Wörns, M.A.; Siveke, J.T.; Dollinger, M.M.; Gerken, G.; Scheulen, M.E.; Wege, H.; et al. Resminostat plus sorafenib as second-line therapy of advanced hepatocellular carcinoma—The SHELTER study. J. Hepatol. 2016, 65, 280–288. [Google Scholar] [CrossRef]
- Fernando, J.; Malfettone, A.; Cepeda, E.B.; Vilarrasa-Blasi, R.; Bertran, E.; Raimondi, G.; Fabra, À.; Alvarez-Barrientos, A.; Fernández-Salguero, P.; Fernández-Rodríguez, C.M.; et al. A mesenchymal-like phenotype and expression of CD44 predict lack of apoptotic response to sorafenib in liver tumor cells. Int. J. Cancer 2015, 136, E161–E172. [Google Scholar] [CrossRef] [PubMed]
- Soukupova, J.; Bertran, E.; Peñuelas-Haro, I.; Urdiroz-Urricelqui, U.; Borgman, M.; Kohlhof, H.; Fabregat, I. Resminostat induces changes in epithelial plasticity of hepatocellular carcinoma cells and sensitizes them to sorafenib-induced apoptosis. Oncotarget 2017, 8, 110367–110379. [Google Scholar] [CrossRef] [Green Version]
- Kulp, S.K.; Chen, C.S.; Wang, D.S.; Chen, C.Y.; Chen, C.S. Antitumor effects of a novel phenylbutyrate-based histone deacetylase inhibitor, (S)-HDAC-42, in prostate cancer. Clin. Cancer Res. 2006, 12, 5199–5206. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.S.; Chou, C.H.; Tzen, K.Y.; Gao, M.; Cheng, A.L.; Kulp, S.K.; Cheng, J.C. Radiosensitizing effect of a phenylbutyrate-derived histone deacetylase inhibitor in hepatocellular carcinoma. Int. J. Radiat. Oncol. Biol. Phys. 2012, 83, E181–E189. [Google Scholar] [CrossRef]
- Wood, T.E.; Dalili, S.; Simpson, C.D.; Sukhai, M.A.; Hurren, R.; Anyiwe, K.; Mao, X.; Saiz, F.S.; Gronda, M.; Eberhard, Y.; et al. Selective inhibition of histone deacetylases sensitizes malignant cells to death receptor ligands. Mol. Cancer Ther. 2010, 9, 246–256. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mawji, I.A.; Simpson, C.D.; Hurren, R.; Gronda, M.; Williams, M.A.; Filmus, J.; Jonkman, J.; Da Costa, R.S.; Wilson, B.C.; Thomas, M.P.; et al. Critical role for Fas-associated death domain-like interleukin-1-converting enzyme-like inhibitory protein in anoikis resistance and distant tumor formation. J. Natl. Cancer Inst. 2007, 99, 811–812. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schimmer, A.D.; Thomas, M.P.; Hurren, R.; Gronda, M.; Pellecchia, M.; Pond, G.R.; Konopleva, M.; Gurfinkel, D.; Mawji, I.A.; Brown, E.; et al. Identification of small molecules that sensitize resistant tumor cells to tumor necrosis factor-family death receptors. Cancer Res. 2006, 66, 2367–2375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liu, J.; Li, G.; Wang, X.; Wang, L.; Zhao, R.; Wang, J.; Kong, Y.; Ding, J.; Li, J.; Zhang, L. Droxinostat, a histone deacetylase inhibitor, induces apoptosis in hepatocellular carcinoma cell lines via activation of the mitochondrial pathway and downregulation of FLIP. Transl. Oncol. 2016, 9, 70–78. [Google Scholar] [CrossRef] [Green Version]
- Yu, P.; Petrus, M.N.; Ju, W.; Zhang, M.; Conlon, K.C.; Nakagawa, M.; Maeda, M.; Bamford, R.N.; Waldmann, T.A. Augmented efficacy with the combination of blockade of the Notch-1 pathway, bortezomib and romidepsin in a murine MT-1 adult T-cell leukemia model. Leukemia 2015, 29, 556–566. [Google Scholar] [CrossRef] [Green Version]
- Afaloniati, H.; Angelopoulou, K.; Giakoustidis, A.; Hardas, A.; Pseftogas, A.; Makedou, K.; Gargavanis, A.; Goulopoulos, T.; Iliadis, S.; Papadopoulos, V.; et al. HDAC1/2 inhibitor romidepsin suppresses DEN-induced hepatocellular carcinogenesis in mice. Onco Targets Ther. 2020, 13, 5575–5588. [Google Scholar] [CrossRef]
- Sun, W.J.; Huang, H.; He, B.; Hu, D.H.; Li, P.H.; Yu, Y.J.; Zhou, X.H.; Lv, Z.; Zhou, L.; Hu, T.Y.; et al. Romidepsin induces G2/M phase arrest via Erk/cdc25C/cdc2/cyclin B pathway and apoptosis induction through JNK/c-Jun/caspase3 pathway in hepatocellular carcinoma cells. Biochem. Pharmacol. 2017, 127, 90–100. [Google Scholar] [CrossRef]
- Foss, F.; Advani, R.; Duvic, M.; Hymes, K.B.; Intragumtornchai, T.; Lekhakula, A.; Shpilberg, O.; Lerner, A.; Belt, R.J.; Jacobsen, E.D.; et al. A Phase II trial of Belinostat (PXD101) in patients with relapsed or refractory peripheral or cutaneous T-cell lymphoma. Br. J. Haematol. 2015, 168, 811–819. [Google Scholar] [CrossRef] [PubMed]
- Yeo, W.; Chung, H.C.; Chan, S.L.; Wang, L.Z.; Lim, R.; Picus, J.; Boyer, M.; Mo, F.K.; Koh, J.; Rha, S.Y.; et al. Epigenetic therapy using belinostat for patients with unresectable hepatocellular carcinoma: A multicenter phase I/II study with biomarker and pharmacokinetic analysis of tumors from patients in the Mayo Phase II Consortium and the Cancer Therapeutics Research Group. J. Clin. Oncol. 2012, 30, 3361–3367. [Google Scholar] [PubMed] [Green Version]
- Llopiz, D.; Ruiz, M.; Villanueva, L.; Iglesias, T.; Silva, L.; Egea, J.; Lasarte, J.J.; Pivette, P.; Trochon-Joseph, V.; Vasseur, B.; et al. Enhanced anti-tumor efficacy of checkpoint inhibitors in combination with the histone deacetylase inhibitor belinostat in a murine hepatocellular carcinoma model. Cancer Immunol. Immunother. 2019, 68, 379–393. [Google Scholar] [CrossRef]
- He, B.; Dai, L.; Zhang, X.; Chen, D.; Wu, J.; Feng, X.; Zhang, Y.; Xie, H.; Zhou, L.; Wu, J.; et al. The HDAC inhibitor quisinostat (JNJ-26481585) supresses hepatocellular carcinoma alone and synergistically in combination with sorafenib by G0/G1 phase arrest and apoptosis induction. Int. J. Biol. Sci. 2018, 14, 1845–1858. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
| Class | Dependence | ||||
|---|---|---|---|---|---|
| I | II | III | IV | ||
| A | B | ||||
| HDAC-1 HDAC-2 HDAC-3 HDAC-8 | HDAC-4 HDAC-5 HDAC-7 HDAC-9 | HDAC-6 HDAC-10 | HDAC-11 | Zn2+ | |
| SIRT-1 SIRT-2 SIRT-3 SIRT-4 SIRT-5 SIRT-6 SIRT-7 | NAD+ | ||||
| HDACI | Anti-HCC Effect |
|---|---|
| Panobinostat | Cell proliferation inhibition Cell cycle arrest Apoptosis |
| Trichostatin A (TSA) | Cell growth inhibition Apoptosis |
| Suberanilohydroxamic acid (SAHA) | Cell proliferation inhibition Cell cycle arrest Apoptosis |
| Valproic acid (VPA) | Cell growth inhibition Apoptosis |
| Resminostat | Apoptosis |
| AR-42 | Cell growth inhibition Apoptosis |
| Droxinostat | Apoptosis |
| Romidepsin | Cell cycle arrest Apoptosis |
| Belinostat | Antitumor immunity |
| Quisinostat | Cell cycle arrest Apoptosis |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Garmpis, N.; Damaskos, C.; Garmpi, A.; Georgakopoulou, V.E.; Sarantis, P.; Antoniou, E.A.; Karamouzis, M.V.; Nonni, A.; Schizas, D.; Diamantis, E.; et al. Histone Deacetylase Inhibitors in the Treatment of Hepatocellular Carcinoma: Current Evidence and Future Opportunities. J. Pers. Med. 2021, 11, 223. https://doi.org/10.3390/jpm11030223
Garmpis N, Damaskos C, Garmpi A, Georgakopoulou VE, Sarantis P, Antoniou EA, Karamouzis MV, Nonni A, Schizas D, Diamantis E, et al. Histone Deacetylase Inhibitors in the Treatment of Hepatocellular Carcinoma: Current Evidence and Future Opportunities. Journal of Personalized Medicine. 2021; 11(3):223. https://doi.org/10.3390/jpm11030223
Chicago/Turabian StyleGarmpis, Nikolaos, Christos Damaskos, Anna Garmpi, Vasiliki E. Georgakopoulou, Panagiotis Sarantis, Efstathios A. Antoniou, Michalis V. Karamouzis, Afroditi Nonni, Dimitrios Schizas, Evangelos Diamantis, and et al. 2021. "Histone Deacetylase Inhibitors in the Treatment of Hepatocellular Carcinoma: Current Evidence and Future Opportunities" Journal of Personalized Medicine 11, no. 3: 223. https://doi.org/10.3390/jpm11030223
APA StyleGarmpis, N., Damaskos, C., Garmpi, A., Georgakopoulou, V. E., Sarantis, P., Antoniou, E. A., Karamouzis, M. V., Nonni, A., Schizas, D., Diamantis, E., Koustas, E., Farmaki, P., Syllaios, A., Patsouras, A., Kontzoglou, K., Trakas, N., & Dimitroulis, D. (2021). Histone Deacetylase Inhibitors in the Treatment of Hepatocellular Carcinoma: Current Evidence and Future Opportunities. Journal of Personalized Medicine, 11(3), 223. https://doi.org/10.3390/jpm11030223