
Selective Targeting of Epigenetic Readers and Histone Deacetylases in Autoimmune and Inflammatory Diseases: Recent Advances and Future Perspectives

 ,
, 
 , and
, and
Abstract
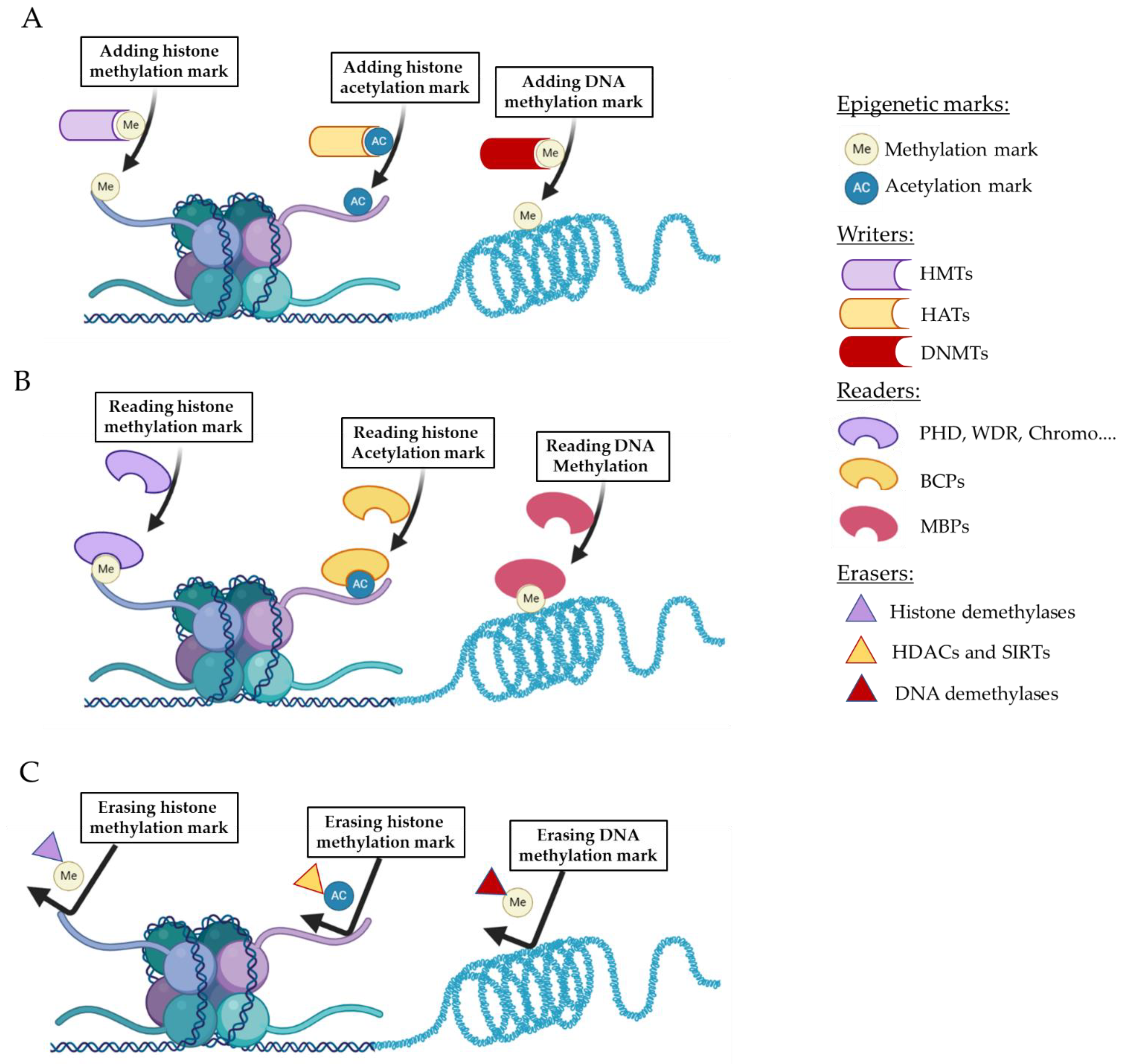
:1. Introduction
2. Methods
3. Advances in HDAC Selective Targeting in Autoimmune and Inflammatory Diseases
3.1. Class-Specific HDACi
3.1.1. Class I-Specific HDACi
3.1.2. Class II-Specific HDACi
3.2. Isoform-Specific HDACi
3.2.1. HDAC3 Inhibitors
3.2.2. HDAC6 Inhibitors
3.2.3. HDAC8 Inhibitors
3.2.4. Other Isoform-Specific Inhibitors
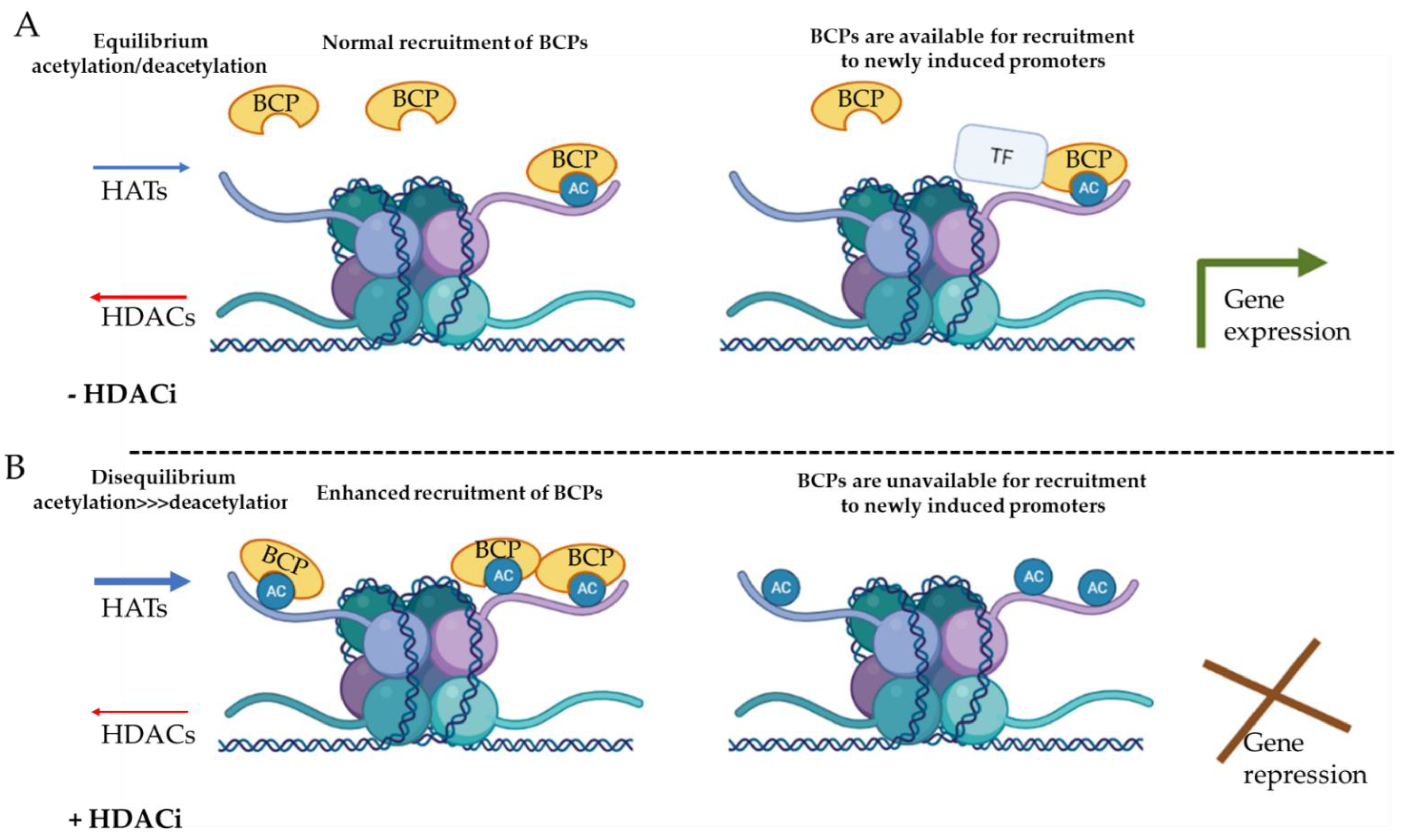
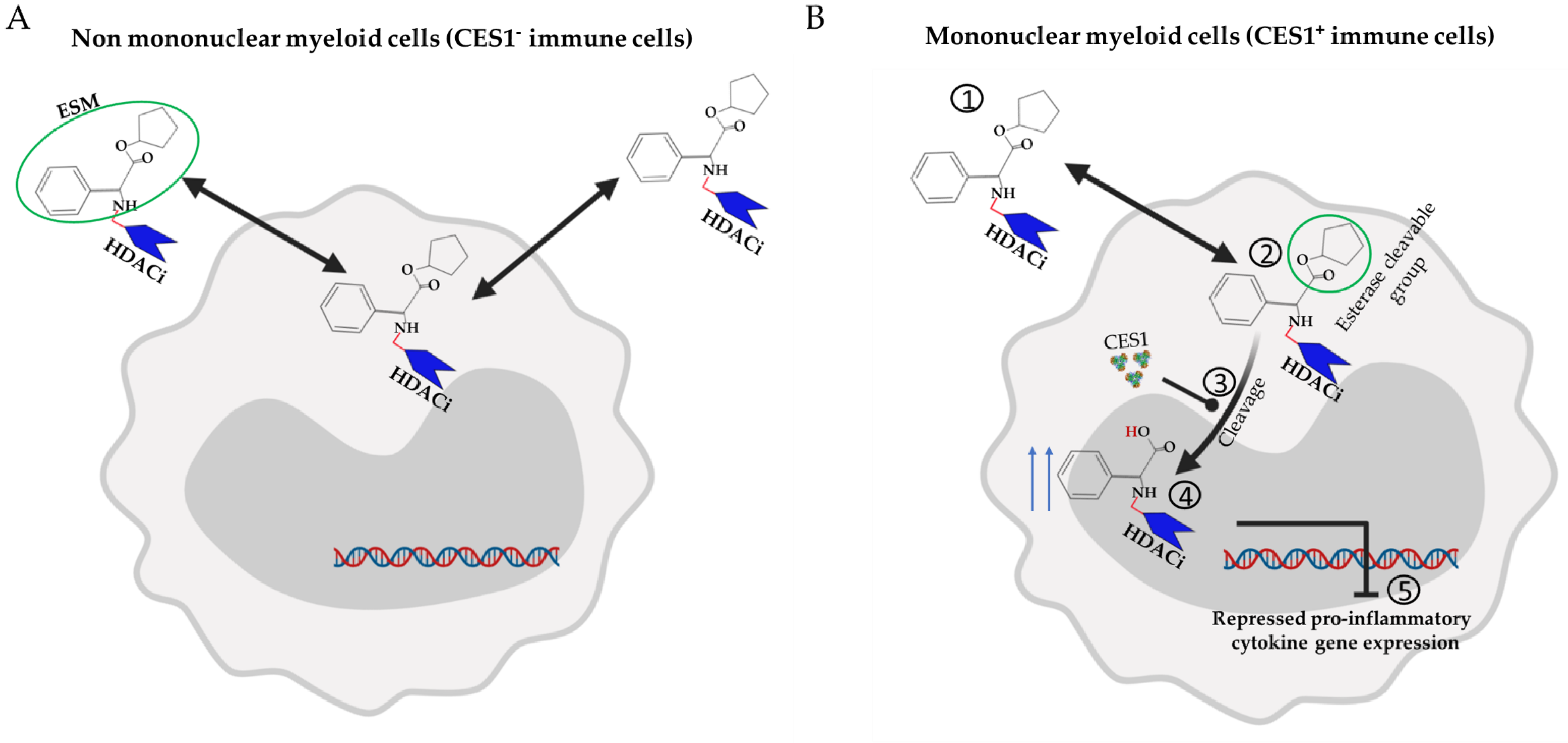
3.3. Cell-Specific Targeted Drug Delivery of Pan-HDACi
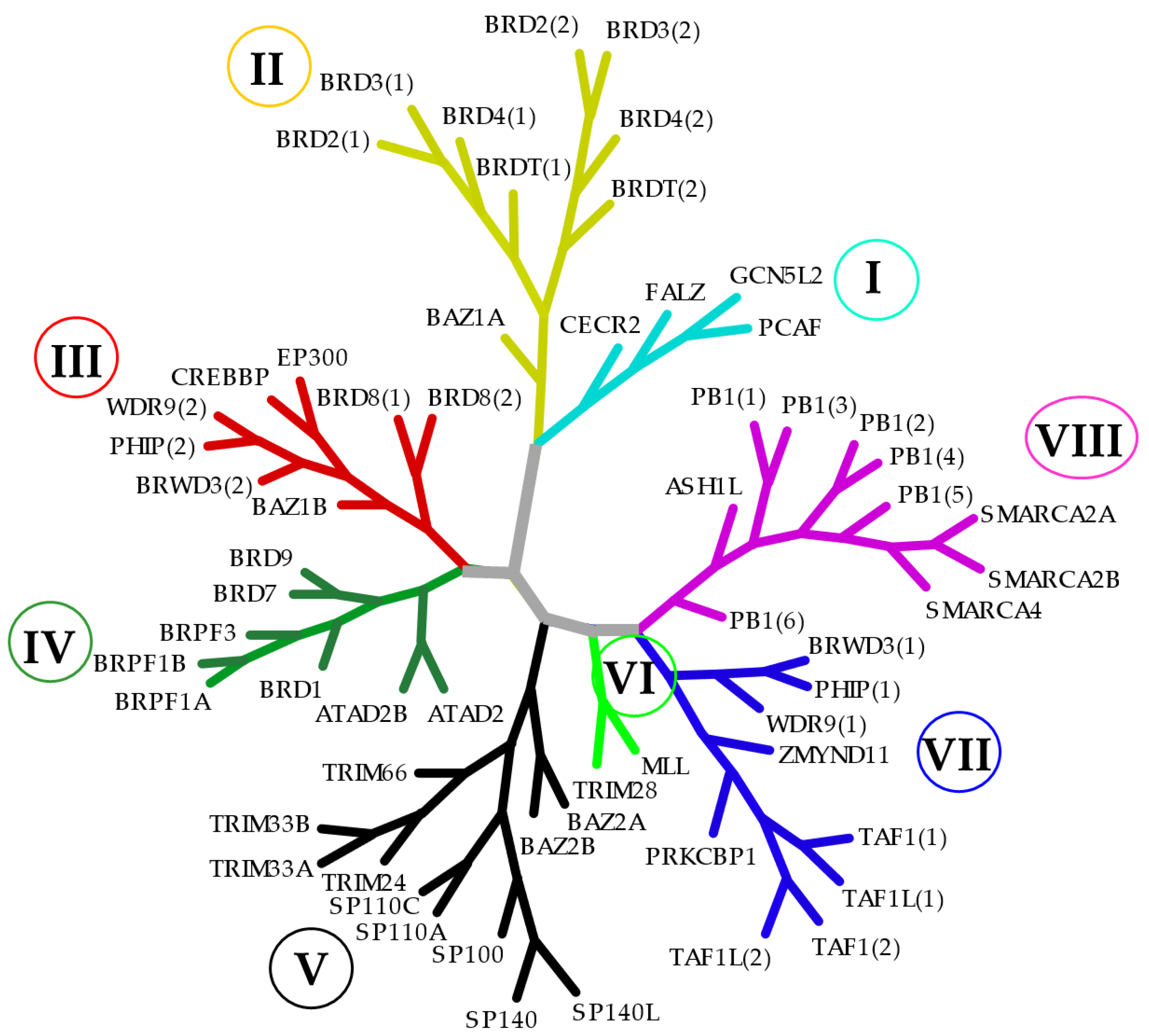
4. Advances in Selective Targeting of BCPs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitors | Targets | Degree of Selectivityin Cell Free Assay | Preclinical Models of Inflammatory and Autoimmune Diseases | Key Findings |
|---|---|---|---|---|
| Entinostat (MS-275) [39] | Class I HDAC | IC50s of 243, 453, and 248 nM for HDAC1, HDAC2, and HDAC3, respectively. | Collagen antibody-induced arthritis (mouse and rat) [46] | While pan-HDACi (SAHA) could not inhibit the onset of arthritis, Entinostat displayed strong antirheumatic activities. |
| Cigarette smoke-induced airway inflammation (mouse) [42] | While Entinostat attenuated inflammatory expression and neutrophil influx in the lungs, pan-HDACi (SAHA) was without effect. | |||
| Thioacetamide-induced hepatic inflammation (mouse) [44] | Entinostat but not class IIa and IIb HDACi suppressed chronic hepatic inflammation and fibrosis. | |||
| Cerulein-induced acute and chronic pancreatitis (mouse) [47] | Reduced infiltration of inflammatory immune cells. | |||
| Tacedinaline (CI994) [40] | Class I HDAC | IC50s of 0.9, 0.9, 1.2, and >20 μM for human HDAC 1, 2, 3, and 8, respectively | Titanium particle-induced calvarial osteolysis (mouse) [124] | Tacedinaline inhibited osteoclastogenesis through targeting NF-κB and the downstream c-Fos/NFATc1 signaling pathway. |
| TMP195 (TFMO 2) [54] | Class IIa HDAC | IC50s of 59, 60, 26, and 15 nM for HDAC4, HDAC5, HDAC7, and HDAC9, respectively. 100-fold selectivity over other HDACs (IC50s >10 µM) | Lipopolysaccharide-induced acute kidney injury (mouse) [56] | TMP195 inhibited multiple proinflammatory cytokines/chemokines and reduced the accumulation of inflammatory immune cells in the injured kidney. |
| RGFP966 [125] | HDAC3 | IC50 of 0.08 μM inhibits HDAC3 > 200-fold selectivity over other HDACs | Diabetic models (mouse) [72,73,74] | RGFP966 prevented diabetes-associated liver damage, -cerebral ischemia and –cardiomyopathy. |
| Osteoarthritis model (rat) [70] | RGFP966 inhibited the expression of inflammatory markers via modulating HDAC3/NF-kB pathway. | |||
| MI192 [68] | HDAC3 | IC50s of 16 and 30 nM, for HDAC2 and HDAC3, respectively. | Ex vivo-stimulated human peripheral blood mononuclear cells (PBMCs) of RA patients [68] | Unlike pan-HDACi, MI192 inhibited inflammatory response in PBMC of RA patients but not in PBMCs of healthy control. |
| Santacruzamate A (CAY10683) [105] | HDAC2 | IC50 of 119 pM for HDAC2 and with >3600-fold selectivity over other HDACs. | LPS-induced neuro-inflammation (mouse) [106] | Santacruzamate A suppressed neuro-inflammatory responses and TLR4/NF-κB signaling pathways. |
| Tubastatin A [79] | HDAC6 | IC50 of 15 nM for HDAC6. It is selective against all the other HDACs (1000-fold) except HDAC8 (57-fold). | Orthotopic lung transplantation model (mouse) [95] | Tubastatin A downregulated Th17 cell function and suppressed acute lung allograft rejection.Notably, this effect was observed only with HDAC6 inhibition but not with HDAC1i-, HDAC3i-, HDAC4i-, and HDAC8i-treated mice. |
| Collagen-induced arthritis (mice) [96] | Tubastatin A reduced IL-6 in paw tissues of arthritic mice. | |||
| ACY-738 [80] | HDAC6 | IC50 of 1.7 nM for HDAC6 and 60- to 1500-fold selectivity over class I HDACs | Model of systemic lupus erythematosus (SLE) (mouse) [94] | ACY-738 modulated both B cell and T cell differentiation, restored the aberrant B cell development and enhanced the frequency of splenic Tregs. |
| Experimental autoimmune encephalomyelitis model (mouse) [87] | ACY-738 delayed disease onset and reduced disease severity. | |||
| BML-281 (CAY10603) [76] | HDAC6 | IC50s of 2 pM; for HDAC6. BML-281 also inhibits HDAC1, HDAC2, HDAC3, HDAC8, and HDAC10, with IC50s of 271, 252, 0.42, and 90.7 nM. | DSS-induced colitis (mouse) [83] | CD19+ B cell influx into inflamed colon was reduced in mice treated with BML-281. |
| LPS-induced acute lung injury (mouse) [89] | BML-281 blocks inflammatory signaling and caspase-1 activation. | |||
| LTB2 [82] | HDAC6 | IC50 value of 3.9 nM | DSS-induced colitis (mouse) [82] | LTB2 prevented DSS-induced colitis. |
| Ricolinostat (ACY-1215) [77] | HDAC6 | IC50 of 5 nM for HDAC6. Ricolinostat also inhibits HDAC1, HDAC2, and HDAC3 with IC50s of 58, 48, and 51 nM, respectively. | Contact hypersensitivity (CHS) and experimental graft-versus-host disease (GVHD)-like disease (mouse) [91] | Ricolinostat prevented the development of CHS and GVHD-like disease by modulating CD8 T cell activation and functions; abrogated the induction of effector T cells from naive CD8 T cells |
| CKD-506 [78] | HDAC6 | IC50 of around 5 nM. IC50 values for HDAC1, HDAC2, HDAC7, and HDAC8 were in the range of 2000–5000 nM. | DSS- and adoptive T cell transfer-induced colitis (mouse) [81]. | CKD-506 ameliorated weight loss, disease activity, and histopathologic score and downregulated proinflammatory cytokines production. |
| Model of SLE (mouse) [78] | CKD-506 modulate both B cell and T cell differentiation, restoring the aberrant B cell development and enhancing frequency of splenic Tregs | |||
| Adjuvant-induced arthritis (mouse) [84] | Suppresses monocytes/macrophages inflammatory responses, improves Treg function, and ameliorates arthritis severity. | |||
| WK2-16 [102] | HDAC8 | - | Sepsis and LPS-induced neuro-inflammation (mouse) [63,102] | WK2-16 was able to reduce IL-6, TNF-α and MPP8. |
| PCI-34051 [103] | HDAC8 | IC50 of 10 nM for HDAC8 and with 200-fold selectivity over HDAC1 and 6 and more than 1000-fold selectivity over HDAC2, 3, and 10. | Ovalbumin-induced asthma (mice) [104] | PCI-34051 alleviated airway inflammation and disrupted HDAC8 interaction with Galectin-3, a protein involved in inflammation and pathogenesis of asthma. |
| ESM-HDAC528 [32] | Pan-HDAC | Selective accumulation of pan-HDACi in CES1+ cells. | Arthritis model [32] | ESM-HDAC528 achieved clinical improvement at lower dose of 1 mg/kg compared with 100 mg/kg of conventional pan-HDACi (SAHA) |
| DSS-induced colitis [116] | ESM-HDACi impaired monocytes differentiation in the inflamed tissue, and this was translated into modest improved colitis | |||
| ZL0420 and ZL0454 [126,127] | BRD4 | For ZL0420, an IC50 of 27 nM against BRD4 BD1 and 32 nM against BRD4 BD2, and for ZL0454, an IC50 of 49 and 32 nM for BD1 and BD2. | TLR3-mediated acute airway inflammation (mice) [126] | The infiltration of neutrophils into the airway fluids and cytokine expression in the lung tissue were more effectively blocked by BRD4 inhibitors than pan-I-BET; (+)-JQ1 or RVX-208 |
| GSK761 [4] | SP140 | IC50 value of 77.79 ± 8.27 nM | CD14+ macrophages isolated from Crohn’s disease colonic tissues [4] | GSK761 reduced the spontaneous secretion of proinflammatory cytokines by macrophages |
| I-BRD9 [128,129] | BRD9 | pIC50 of 7.3 for BRD9, with pIC50 of 5.3 against BRD4. | Nur77 knockout-induced obesity (mice) [130] | Combining I-BRD9 with calcipotriol regulated the gut microbiota and improved intestinal mucosal barrier function. |
| GSK046 (iBET-BD2) [131] | BD2 of BET proteins | IC50s of 264 nM (BRD2 BD2), 98 nM (BRD3 BD2), 49 nM (BRD4 BD2), and 214 nM (BRDT BD2). | Models of RA and psoriasis (mouse) [131] | Immunomodulatory effects. |
| GSK778 (iBET-BD1) [131] | BD1 of BET proteins | IC50s of 75 nM (BRD2 BD1), 41 nM (BRD3 BD1), 41 nM (BRD4 BD1), and 143 nM (BRDT BD1), respectively | Cancer model (mouse) [131] | GSK778 phenocopied the effects of pan-BET inhibitors in cancer models. |
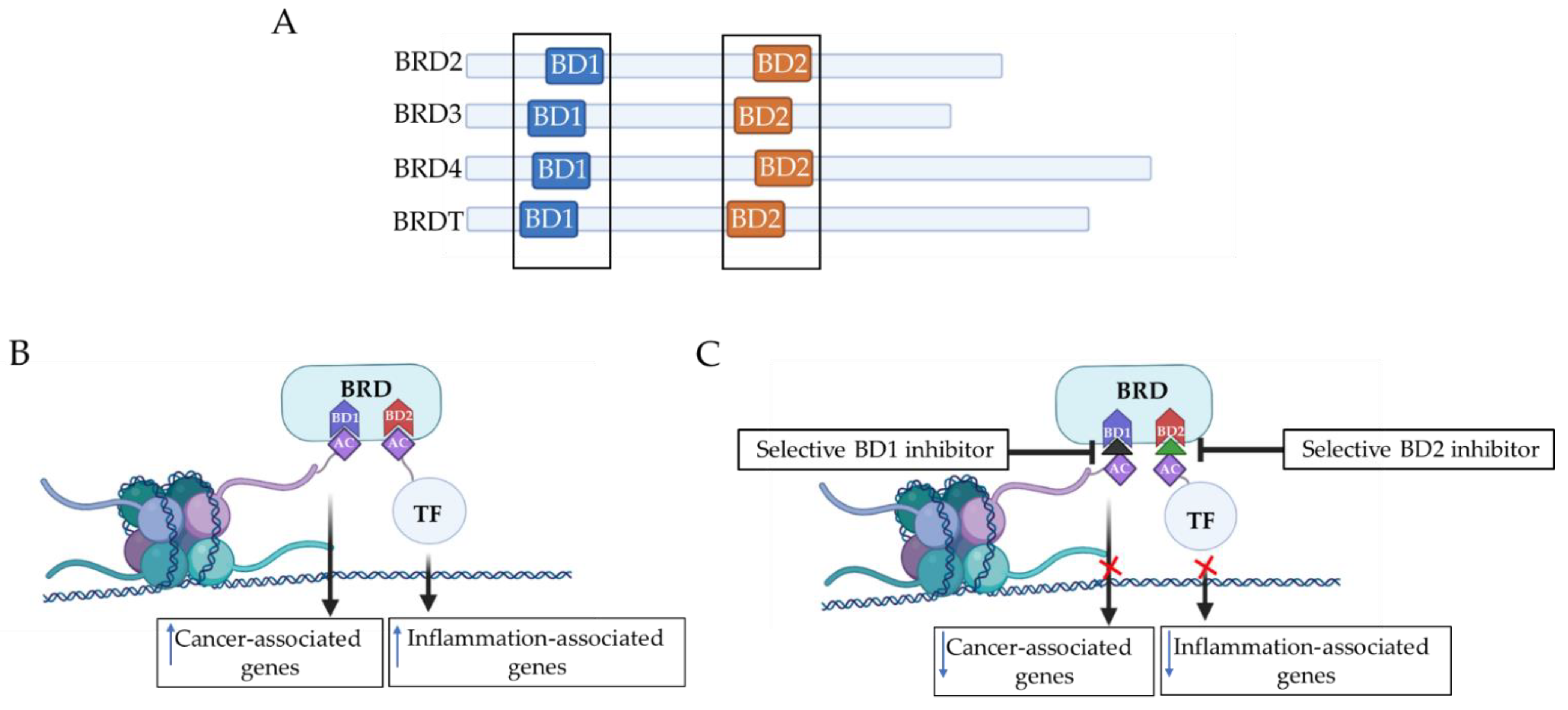
4.1. Domain-Selective Targeting (BD1 or BD2 Targeting)
4.2. Selective Targeting of Single BCP
4.2.1. BRD4 Inhibitors
4.2.2. SP140 Inhibitor
4.2.3. BRD9 Inhibitors
4.2.4. CREB Inhibitor
4.2.5. BRPF Inhibitors
4.3. Cell-Specific Targeted Drug Delivery of I-BET
5. Conclusions/Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
References
- Kondilis-Mangum, H.D.; Wade, P.A. Epigenetics and the adaptive immune response. Mol. Asp. Med. 2013, 34, 813–825. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lang, K.S.; Burow, A.; Kurrer, M.; Lang, P.A.; Recher, M. The role of the innate immune response in autoimmune disease. J. Autoimmun. 2007, 29, 206–212. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Li, S.; Subramaniam, S.; Shyy, J.Y.; Chien, S. Epigenetic Regulation: A New Frontier for Biomedical Engineers. Annu. Rev. Biomed. Eng. 2017, 19, 195–219. [Google Scholar] [CrossRef] [PubMed]
- Ghiboub, M.; Koster, J.; Craggs, P.D.; Li Yim, A.Y.F.; Shillings, A.; Hutchinson, S.; Bingham, R.P.; Gatfield, K.; Hageman, I.L.; Yao, G.; et al. Modulation of macrophage inflammatory function through selective inhibition of the epigenetic reader protein SP140. bioRxiv 2020. [Google Scholar] [CrossRef]
- Shanmugam, M.K.; Sethi, G. Role of epigenetics in inflammation-associated diseases. Subcell Biochem. 2013, 61, 627–657. [Google Scholar]
- Holoch, D.; Moazed, D. RNA-mediated epigenetic regulation of gene expression. Nat. Rev. Genet. 2015, 16, 71–84. [Google Scholar] [CrossRef]
- Zeng, Y.; Chen, T. DNA Methylation Reprogramming during Mammalian Development. Genes 2019, 10, 257. [Google Scholar] [CrossRef] [Green Version]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Araki, Y.; Mimura, T. The Histone Modification Code in the Pathogenesis of Autoimmune Diseases. Mediat. Inflamm. 2017, 2017, 1–12. [Google Scholar] [CrossRef]
- Hull, E.E.; Montgomery, M.R.; Leyva, K.J. HDAC Inhibitors as Epigenetic Regulators of the Immune System: Impacts on Cancer Therapy and Inflammatory Diseases. BioMed Res. Int. 2016, 2016, 1–15. [Google Scholar] [CrossRef] [Green Version]
- Morgado-Pascual, J.L.; Rayego-Mateos, S.; Tejedor, L.; Suarez-Alvarez, B.; Ruiz-Ortega, M. Bromodomain and Extraterminal Proteins as Novel Epigenetic Targets for Renal Diseases. Front. Pharm. 2019, 10, 1315. [Google Scholar] [CrossRef]
- Seto, E.; Yoshida, M. Erasers of Histone Acetylation: The Histone Deacetylase Enzymes. Cold Spring Harb. Perspect. Biol. 2014, 6, a018713. [Google Scholar] [CrossRef] [Green Version]
- Ferri, E.; Petosa, C.; McKenna, C.E. Bromodomains: Structure, function and pharmacology of inhibition. Biochem. Pharm. 2016, 106, 1–18. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Knapp, S. Targeting bromodomains: Epigenetic readers of lysine acetylation. Nat. Rev. Drug Discov. 2014, 13, 337–356. [Google Scholar] [CrossRef]
- Muller, S.; Filippakopoulos, P.; Knapp, S. Bromodomains as therapeutic targets. Expert Rev. Mol. Med. 2011, 13, e29. [Google Scholar] [CrossRef] [Green Version]
- Hancock, W.W.; Akimova, T.; Beier, U.H.; Liu, Y.; Wang, L. HDAC inhibitor therapy in autoimmunity and transplantation. Ann. Rheum. Dis. 2012, 71 (Suppl. 2), i46–i54. [Google Scholar] [CrossRef]
- Singh, A.K.; Bishayee, A.; Pandey, A.K. Targeting Histone Deacetylases with Natural and Synthetic Agents: An Emerging Anticancer Strategy. Nutrients 2018, 10, 731. [Google Scholar] [CrossRef] [Green Version]
- Glauben, R.; Batra, A.; Fedke, I.; Zeitz, M.; Lehr, H.A.; Leoni, F.; Mascagni, P.; Fantuzzi, G.; Dinarello, C.A.; Siegmund, B. Histone Hyperacetylation Is Associated with Amelioration of Experimental Colitis in Mice. J. Immunol. 2006, 176, 5015–5022. [Google Scholar] [CrossRef] [Green Version]
- Nadeem, A.; Al-Harbi, N.O.; Al-Harbi, M.M.; El-Sherbeeny, A.M.; Ahmad, S.F.; Siddiqui, N.; Ansari, M.A.; Zoheir, K.M.; Attia, S.M.; Al-Hosaini, K.A.; et al. Imiquimod-induced psoriasis-like skin inflammation is suppressed by BET bromodomain inhibitor in mice through RORC/IL-17A pathway modulation. Pharm. Res. 2015, 99, 248–257. [Google Scholar] [CrossRef]
- Rudman, M.D.; Choi, J.S.; Lee, H.E.; Tan, S.K.; Ayad, N.G.; Lee, J.K. Bromodomain and extraterminal domain-containing protein inhibition attenuates acute inflammation after spinal cord injury. Exp. Neurol. 2018, 309, 181–192. [Google Scholar] [CrossRef]
- Copsel, S.N.; Lightbourn, C.O.; Barreras, H.; Lohse, I.; Wolf, D.; Bader, C.S.; Manov, J.; Kale, B.J.; Shah, D.; Brothers, S.P.; et al. BET Bromodomain Inhibitors Which Permit Treg Function Enable a Combinatorial Strategy to Suppress GVHD in Pre-clinical Allogeneic HSCT. Front. Immunol. 2019, 9, 3104. [Google Scholar] [CrossRef] [PubMed]
- Friedrich, M.; Gerbeth, L.; Gerling, M.; Rosenthal, R.; Steiger, K.; Weidinger, C.; Keye, J.; Wu, H.; Schmidt, F.; Weichert, W.; et al. HDAC inhibitors promote intestinal epithelial regeneration via autocrine TGFβ1 signalling in inflammation. Mucosal Immunol. 2019, 12, 656–667. [Google Scholar] [CrossRef] [PubMed]
- Cui, S.-N.; Chen, Z.-Y.; Yang, X.-B.; Chen, L.; Yang, Y.-Y.; Pan, S.-W.; Wang, Y.-X.; Xu, J.-Q.; Zhou, T.; Xiao, H.-R.; et al. Trichostatin A modulates the macrophage phenotype by enhancing autophagy to reduce inflammation during polymicrobial sepsis. Int. Immunopharmacol. 2019, 77, 105973. [Google Scholar] [CrossRef]
- Ali, M.N.; Choijookhuu, N.; Takagi, H.; Srisowanna, N.; Huynh, M.N.N.; Yamaguchi, Y.; Oo, P.S.; Kyaw, M.T.H.; Sato, K.; Yamaguchi, R.; et al. The HDAC Inhibitor, SAHA, Prevents Colonic Inflammation by Suppressing Pro-inflammatory Cytokines and Chemokines in DSS-induced Colitis. Acta Histochem. Cytochem. 2018, 51, 33–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jahagirdar, R.; Attwell, S.; Marusic, S.; Bendele, A.; Shenoy, N.; McLure, K.G.; Gilham, D.; Norek, K.; Hansen, H.C.; Yu, R.; et al. RVX-297, a BET Bromodomain Inhibitor, Has Therapeutic Effects in Preclinical Models of Acute Inflammation and Autoimmune Disease. Mol. Pharm. 2017, 92, 694–706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schilderink, R.; Bell, M.; Reginato, E.; Patten, C.; Rioja, I.; Hilbers, F.W.; Kabala, P.A.; Reedquist, K.A.; Tough, D.F.; Tak, P.P.; et al. BET bromodomain inhibition reduces maturation and enhances tolerogenic properties of human and mouse dendritic cells. Mol. Immunol. 2016, 79, 66–76. [Google Scholar] [CrossRef] [PubMed]
- Yan, L.; Wang, F.; Xiaoxue, C.; Wang, J.; Zhao, Y.; Li, Y.; He, B. Zinc-dependent Deacetylase (HDAC) Inhibitors with Different Zinc Binding Groups. Curr. Top. Med. Chem. 2019, 19, 223–241. [Google Scholar]
- Subramanian, S.; Bates, S.E.; Wright, J.J.; Espinoza-Delgado, I.; Piekarz, R.L. Clinical Toxicities of Histone Deacetylase Inhibitors. Pharmaceuticals 2010, 3, 2751–2767. [Google Scholar] [CrossRef] [Green Version]
- van Veggel, M.; Westerman, E.; Hamberg, P. Clinical Pharmacokinetics and Pharmacodynamics of Panobinostat. Clin. Pharm. 2018, 57, 21–29. [Google Scholar] [CrossRef]
- Pervaiz, M.; Mishra, P.; Günther, S. Bromodomain Drug Discovery—The Past, the Present, and the Future. Chem. Rec. 2018, 18, 1808–1817. [Google Scholar] [CrossRef]
- Andrieu, G.; Belkina, A.C.; Denis, G.V. Clinical trials for BET inhibitors run ahead of the science. Drug Discov. Today Technol. 2016, 19, 45–50. [Google Scholar] [CrossRef] [Green Version]
- Needham, L.A.; Davidson, A.H.; Bawden, L.J.; Belfield, A.; Bone, E.A.; Brotherton, D.H.; Bryant, S.; Charlton, M.H.; Clark, V.L.; Davies, S.J.; et al. Drug Targeting to Monocytes and Macrophages Using Esterase-Sensitive Chemical Motifs. J. Pharm. Exp. 2011, 339, 132–142. [Google Scholar] [CrossRef] [Green Version]
- Ghiboub, M.; Zhao, J.; Yim, A.Y.F.L.; Schilderink, R.; Verseijden, C.; van Hamersveld, P.H.P.; Duarte, J.M.; Hakvoort, T.B.M.; Admiraal, I.; Harker, N.R.; et al. HDAC3 Mediates the Inflammatory Response and LPS Tolerance in Human Monocytes and Macrophages. Front. Immunol. 2020, 11, 550769. [Google Scholar] [CrossRef]
- Park, S.-Y.; Kim, J.-S. A short guide to histone deacetylases including recent progress on class II enzymes. Exp. Mol. Med. 2020, 52, 204–212. [Google Scholar] [CrossRef]
- Marié, I.J.; Chang, H.-M.; Levy, D.E. HDAC stimulates gene expression through BRD4 availability in response to IFN and in interferonopathies. J. Exp. Med. 2018, 215, 3194–3212. [Google Scholar] [CrossRef]
- Grabiec, A.M.; Korchynskyi, O.; Tak, P.P.; Reedquist, K.A. Histone deacetylase inhibitors suppress rheumatoid arthritis fibroblast-like synoviocyte and macrophage IL-6 production by accelerating mRNA decay. Ann. Rheum. Dis. 2011, 71, 424–431. [Google Scholar] [CrossRef]
- Tang, J.; Yan, H.; Zhuang, S. Histone deacetylases as targets for treatment of multiple diseases. Clin. Sci. 2013, 124, 651–662. [Google Scholar] [CrossRef] [Green Version]
- Cao, F.; Zwinderman, M.R.; Van Merkerk, R.; Ettema, P.E.; Quax, W.J.; Dekker, F.J. Inhibitory selectivity among class I HDACs has a major impact on inflammatory gene expression in macrophages. Eur. J. Med. Chem. 2019, 177, 457–466. [Google Scholar] [CrossRef]
- Saito, A.; Yamashita, T.; Mariko, Y.; Nosaka, Y.; Tsuchiya, K.; Ando, T.; Suzuki, T.; Tsuruo, T.; Nakanishi, O. A synthetic inhibitor of histone deacetylase, MS-27-275, with marked in vivo antitumor activity against human tumors. Proc. Natl. Acad. Sci. USA 1999, 96, 4592–4597. [Google Scholar] [CrossRef] [Green Version]
- Moradei, O.M.; Mallais, T.C.; Frechette, S.; Paquin, I.; Tessier, P.E.; Leit, S.M.; Fournel, M.; Bonfils, C.; Trachy-Bourget, M.-C.; Liu, J.; et al. Novel Aminophenyl Benzamide-Type Histone Deacetylase Inhibitors with Enhanced Potency and Selectivity. J. Med. Chem. 2007, 50, 5543–5546. [Google Scholar] [CrossRef]
- Cantley, M.D.; Fairlie, D.P.; Bartold, P.M.; Marino, V.; Gupta, P.K.; Haynes, D.R. Inhibiting histone deacetylase 1 suppresses both inflammation and bone loss in arthritis. Rheumatology 2015, 54, 1713–1723. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leus, N.G.J.; Bosch, T.V.D.; van der Wouden, P.E.; Krist, K.; Ourailidou, M.E.; Eleftheriadis, N.; Kistemaker, L.E.M.; Bos, S.; Gjaltema, R.A.F.; Mekonnen, S.A.; et al. HDAC1-3 inhibitor MS-275 enhances IL10 expression in RAW264.7 macrophages and reduces cigarette smoke-induced airway inflammation in mice. Sci. Rep. 2017, 7, 45047. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ryu, Y.; Kee, H.J.; Sun, S.; Seok, Y.M.; Choi, S.Y.; Kim, G.R.; Kee, S.-J.; Pflieger, M.; Kurz, T.; Kim, H.-S.; et al. Class I histone deacetylase inhibitor MS-275 attenuates vasoconstriction and inflammation in angiotensin II-induced hypertension. PLoS ONE 2019, 14, e0213186. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, Z.; Fitzsimmons, R.L.; Reid, R.C.; Ramnath, D.; Clouston, A.; Gupta, P.K.; Irvine, K.M.; Powell, E.E.; Schroder, K.; Stow, J.L.; et al. Inhibitors of class I histone deacetylases attenuate thioacetamide-induced liver fibrosis in mice by suppressing hepatic type 2 inflammation. Br. J. Pharm. 2019, 176, 3775–3790. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Zhang, W.; Jiao, F.; Li, X.; Zhang, H.; Wang, L.; Gong, Z. The Nephroprotective Effect of MS-275 on Lipopolysaccharide (LPS)-Induced Acute Kidney Injury by Inhibiting Reactive Oxygen Species (ROS)-Oxidative Stress and Endoplasmic Reticulum Stress. Med. Sci. Monit. 2018, 24, 2620–2630. [Google Scholar] [CrossRef] [PubMed]
- Lin, H.-S.; Hu, C.-Y.; Chan, H.-Y.; Liew, Y.-Y.; Huang, H.-P.; Lepescheux, L.; Bastianelli, E.; Baron, R.; Rawadi, G.; Clément-Lacroix, P. Anti-rheumatic activities of histone deacetylase (HDAC) inhibitors in vivo in collagen-induced arthritis in rodents. Br. J. Pharm. 2007, 150, 862–872. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bombardo, M.; Saponara, E.; Malagola, E.; Chen, R.; Seleznik, G.M.; Haumaitre, C.; Quilichini, E.; Zabel, A.; Reding, T.; Graf, R.; et al. Class I histone deacetylase inhibition improves pancreatitis outcome by limiting leukocyte recruitment and acinar-to-ductal metaplasia. Br. J. Pharm. 2017, 174, 3865–3880. [Google Scholar] [CrossRef] [Green Version]
- Walewski, J.; Paszkiewicz-Kozik, E.; Borsaru, G.; Hellmann, A.; Janikova, A.; Warszewska, A.; Mais, A.; Ammendola, A.; Herz, T.; Krauss, B.; et al. Resminostat in patients with relapsed or refractory Hodgkin lymphoma: Results of the phase II SAPHIRE study. Leuk. Lymphoma 2019, 60, 675–684. [Google Scholar] [CrossRef]
- Kitazono, S.; Fujiwara, Y.; Nakamichi, S.; Mizugaki, H.; Nokihara, H.; Yamamoto, N.; Yamada, Y.; Inukai, E.; Nakamura, O.; Tamura, T. A phase I study of resminostat in Japanese patients with advanced solid tumors. Cancer Chemother. Pharm. 2015, 75, 1155–1161. [Google Scholar] [CrossRef]
- Boumber, Y.; Younes, A.; Garcia-Manero, G. Mocetinostat (MGCD0103): A review of an isotype-specific histone deacetylase inhibitor. Expert Opin. Investig. Drugs 2011, 20, 823–829. [Google Scholar] [CrossRef] [Green Version]
- Knipstein, J.; Gore, L. Entinostat for treatment of solid tumors and hematologic malignancies. Expert Opin. Investig. Drugs 2011, 20, 1455–1467. [Google Scholar] [CrossRef]
- Ruiz, R.; Raez, E.L.; Rolfo, C. Entinostat (SNDX-275) for the treatment of non-small cell lung cancer. Expert Opin. Investig. Drugs 2015, 24, 1101–1109. [Google Scholar] [CrossRef]
- Giannopoulou, A.F.; Velentzas, A.D.; Konstantakou, E.G.; Avgeris, M.; Katarachia, S.A.; Papandreou, N.C.; Kalavros, N.I.; Mpakou, V.E.; Iconomidou, V.; Anastasiadou, E.; et al. Revisiting Histone Deacetylases in Human Tumorigenesis: The Paradigm of Urothelial Bladder Cancer. Int. J. Mol. Sci. 2019, 20, 1291. [Google Scholar] [CrossRef] [Green Version]
- Lobera, M.; Madauss, K.P.; Pohlhaus, D.T.; Wright, Q.G.; Trocha, M.; Schmidt, D.R.; Baloglu, E.; Trump, R.P.; Head, M.S.; Hofmann, G.A.; et al. Selective class IIa histone deacetylase inhibition via a nonchelating zinc-binding group. Nat. Chem. Biol. 2013, 9, 319–325. [Google Scholar] [CrossRef]
- Guerriero, J.L.; Sotayo, A.; Ponichtera, H.E.; Castrillon, J.A.; Pourzia, A.L.; Schad, S.; Johnson, S.F.; Carrasco, R.D.; Lazo, S.; Bronson, R.T.; et al. Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nat. Cell Biol. 2017, 543, 428–432. [Google Scholar] [CrossRef]
- Zhang, W.; Guan, Y.; Bayliss, G.P.; Zhuang, S. Class IIa HDAC inhibitor TMP195 alleviates lipopolysaccharide-induced acute kidney injury. Am. J. Physiol. Physiol. 2020, 319, F1015–F1026. [Google Scholar] [CrossRef]
- Yanginlar, C.; Logie, C. HDAC11 is a regulator of diverse immune functions. Biochim. Biophys. Acta 2018, 1861, 54–59. [Google Scholar] [CrossRef]
- Janczura, K.J.; Volmar, C.H.; Sartor, G.C.; Rao, S.J.; Ricciardi, N.R.; Lambert, G.; Brothers, S.P.; Wahlestedt, C. Inhibition of HDAC3 reverses Alzheimer’s disease-related pathologies in vitro and in the 3xTg-AD mouse model. Proc. Natl. Acad. Sci. USA 2018, 115, E11148–E11157. [Google Scholar] [CrossRef] [Green Version]
- Leus, N.G.; van der Wouden, P.E.; van den Bosch, T.; Hooghiemstra, W.T.; Ourailidou, M.E.; Kistemaker, L.E.; Bischoff, R.; Gosens, R.; Haisma, H.J.; Dekker, F.J. HDAC 3-selective inhibitor RGFP966 demonstrates anti-inflammatory properties in RAW 264.7 macrophages and mouse precision-cut lung slices by attenuating NF-κB p65 transcriptional activity. Biochem. Pharmacol. 2016, 108, 58–74. [Google Scholar] [CrossRef] [Green Version]
- Zhang, L.; Chen, Y.; Jiang, Q.; Song, W.; Zhang, L. Therapeutic potential of selective histone deacetylase 3 inhibition. Eur. J. Med. Chem. 2019, 162, 534–542. [Google Scholar] [CrossRef]
- Cao, F.; Zwinderman, M.R.; Dekker, F.J. The Process and Strategy for Developing Selective Histone Deacetylase 3 Inhibitors. Molecules 2018, 23, 551. [Google Scholar] [CrossRef] [Green Version]
- Huang, W.-J.; Wang, Y.-C.; Chao, S.-W.; Yang, C.-Y.; Chen, L.-C.; Lin, M.-H.; Hou, W.-C.; Chen, M.-Y.; Lee, T.-L.; Yang, P.; et al. Synthesis and biological evaluation of ortho-aryl N-hydroxycinnamides as potent histone deacetylase (HDAC) 8 isoform-selective inhibitors. ChemMedChem 2012, 7, 1815–1824. [Google Scholar] [CrossRef]
- Jan, J.-S.; Chou, Y.-C.; Cheng, Y.-W.; Chen, C.-K.; Huang, W.-J.; Hsiao, G. The Novel HDAC8 Inhibitor WK2-16 Attenuates Lipopolysaccharide-Activated Matrix Metalloproteinase-9 Expression in Human Monocytic Cells and Improves Hypercytokinemia in vivo. Int. J. Mol. Sci. 2017, 18, 1394. [Google Scholar] [CrossRef] [Green Version]
- Ran, J.; Zhou, J. Targeted inhibition of histone deacetylase 6 in inflammatory diseases. Thorac. Cancer 2019, 10, 405–412. [Google Scholar] [CrossRef] [Green Version]
- Kutil, Z.; Mikešová, J.; Zessin, M.; Meleshin, M.; Nováková, Z.; Alquicer, G.; Kozikowski, A.; Sippl, W.; Bařinka, C.; Schutkowski, M. Continuous Activity Assay for HDAC11 Enabling Reevaluation of HDAC Inhibitors. ACS Omega 2019, 4, 19895–19904. [Google Scholar] [CrossRef] [Green Version]
- Solomon, J.M.; Pasupuleti, R.; Xu, L.; McDonagh, T.; Curtis, R.; DiStefano, P.S.; Huber, L.J. Inhibition of SIRT1 Catalytic Activity Increases p53 Acetylation but Does Not Alter Cell Survival following DNA Damage. Mol. Cell. Biol. 2006, 26, 28–38. [Google Scholar] [CrossRef] [Green Version]
- Chen, X.; Wales, P.; Quinti, L.; Zuo, F.; Moniot, S.; Hérisson, F.; Rauf, N.A.; Wang, H.; Silverman, R.B.; Ayata, C.; et al. The Sirtuin-2 Inhibitor AK7 Is Neuroprotective in Models of Parkinson’s Disease but Not Amyotrophic Lateral Sclerosis and Cerebral Ischemia. PLoS ONE 2015, 10, e0116919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gillespie, J.; Savic, S.; Wong, C.; Hempshall, A.; Inman, M.; Emery, P.; Grigg, R.; McDermott, M.F. Histone deacetylases are dysregulated in rheumatoid arthritis and a novel histone deacetylase 3-selective inhibitor reduces interleukin-6 production by peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Rheum. 2012, 64, 418–422. [Google Scholar] [CrossRef] [PubMed]
- Lei, L.; Bai, G.; Wang, X.; Liu, S.; Xia, J.; Wu, S.; Huan, Y.; Shen, Z. Histone deacetylase 3-selective inhibitor RGFP966 ameliorates impaired glucose tolerance through β-cell protection. Toxicol. Appl. Pharm. 2020, 406, 115189. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Ji, L.; Yang, Y.; Wei, Y.; Zhang, X.; Gang, Y.; Lu, J.; Bai, L. The Therapeutic Effects of Treadmill Exercise on Osteoarthritis in Rats by Inhibiting the HDAC3/NF-KappaB Pathway in vivo and in vitro. Front. Physiol. 2019, 10, 1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, W.; Sun, X.; Ba, G.; Tang, R.; Lin, H. RGFP966, a selective HDAC3 inhibitor, ameliorates allergic and inflammatory responses in an OVA-induced allergic rhinitis mouse model. Int. Immunopharmacol. 2021, 93, 107400. [Google Scholar] [CrossRef]
- Zhang, J.; Xu, Z.; Gu, J.; Jiang, S.; Liu, Q.; Zheng, Y.; Freedman, J.H.; Sun, J.; Cai, L. HDAC3 inhibition in diabetic mice may activate Nrf2 preventing diabetes-induced liver damage and FGF21 synthesis and secretion leading to aortic protection. Am. J. Physiol. Metab. 2018, 315, E150–E162. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.-J.; Zhao, Q.-C.; Xia, M.-X.; Chen, J.; Chen, Y.-T.; Cao, X.; Liu, Y.; Yuan, Z.-Q.; Wang, X.-Y.; Xu, Y. The HDAC3 inhibitor RGFP966 ameliorated ischemic brain damage by downregulating the AIM2 inflammasome. FASEB J. 2019, 34, 648–662. [Google Scholar] [CrossRef] [Green Version]
- Xu, Z.; Tong, Q.; Zhang, Z.; Wang, S.; Zheng, Y.; Liu, Q.; Qian, L.-B.; Chen, S.-Y.; Sun, J.; Cai, L. Inhibition of HDAC3 prevents diabetic cardiomyopathy in OVE26 mice via epigenetic regulation of DUSP5-ERK1/2 pathway. Clin. Sci. 2017, 131, 1841–1857. [Google Scholar] [CrossRef] [Green Version]
- Angiolilli, C.; Kabala, P.A.; Grabiec, A.M.; van Baarsen, I.M.; Ferguson, B.S.; García, S.; Fernandez, B.M.; McKinsey, T.A.; Tak, P.P.; Fossati, G.; et al. Histone deacetylase 3 regulates the inflammatory gene expression programme of rheumatoid arthritis fibroblast-like synoviocytes. Ann. Rheum. Dis. 2017, 76, 277–285. [Google Scholar] [CrossRef]
- Kozikowski, A.P.; Tapadar, S.; Luchini, D.N.; Kim, K.H.; Billadeau, D.D. Use of the Nitrile Oxide Cycloaddition (NOC) Reaction for Molecular Probe Generation: A New Class of Enzyme Selective Histone Deacetylase Inhibitors (HDACIs) Showing Picomolar Activity at HDAC6. J. Med. Chem. 2008, 51, 4370–4373. [Google Scholar] [CrossRef] [Green Version]
- Santo, L.; Hideshima, T.; Kung, A.L.; Tseng, J.-C.; Tamang, D.; Yang, M.; Jarpe, M.; van Duzer, J.H.; Mazitschek, R.; Ogier, W.C.; et al. Preclinical activity, pharmacodynamic, and pharmacokinetic properties of a selective HDAC6 inhibitor, ACY-1215, in combination with bortezomib in multiple myeloma. Blood 2012, 119, 2579–2589. [Google Scholar] [CrossRef]
- Choi, E.W.; Song, J.W.; Ha, N.; Choi, Y.I.; Kim, S. CKD-506, a novel HDAC6-selective inhibitor, improves renal outcomes and survival in a mouse model of systemic lupus erythematosus. Sci. Rep. 2018, 8, 17297. [Google Scholar] [CrossRef]
- Butler, K.V.; Kalin, J.; Brochier, C.; Vistoli, G.; Langley, B.; Kozikowski, A.P. Rational Design and Simple Chemistry Yield a Superior, Neuroprotective HDAC6 Inhibitor, Tubastatin, A. J. Am. Chem. Soc. 2010, 132, 10842–10846. [Google Scholar] [CrossRef] [Green Version]
- Jochems, J.; Boulden, J.; Lee, B.G.; Blendy, J.A.; Jarpe, M.; Mazitschek, R.; van Duzer, J.H.; Jones, S.; Berton, O. Antidepressant-Like Properties of Novel HDAC6-Selective Inhibitors with Improved Brain Bioavailability. Neuropsychopharmacology 2014, 39, 389–400. [Google Scholar] [CrossRef]
- Lee, J.W.; Lee, S.-M.; Chun, J.; Im, J.P.; Seo, S.-K.; Ha, N.; Choi, Y.I.; Kim, J.S. Novel Histone Deacetylase 6 Inhibitor CKD-506 Inhibits NF-κB Signaling in Intestinal Epithelial Cells and Macrophages and Ameliorates Acute and Chronic Murine Colitis. Inflamm. Bowel Dis. 2020, 26, 852–862. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Wang, R.; Xu, H.; Song, Y.; Qi, Y. A Highly Potent and Selective Histone Deacetylase 6 Inhibitor Prevents DSS-Induced Colitis in Mice. Biol. Pharm. Bull. 2017, 40, 936–940. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Do, A.; Reid, R.C.; Lohman, R.-J.; Sweet, M.J.; Fairlie, D.P.; Iyer, A. An HDAC6 Inhibitor Confers Protection and Selectively Inhibits B-Cell Infiltration in DSS-Induced Colitis in Mice. J. Pharm. Exp. 2017, 360, 140–151. [Google Scholar] [CrossRef] [PubMed]
- Park, J.K.; Jang, Y.J.; Oh, B.R.; Shin, J.; Bae, D.; Ha, N.; Choi, Y.I.; Youn, G.S.; Park, J.; Lee, E.Y.; et al. Therapeutic potential of CKD-506, a novel selective histone deacetylase 6 inhibitor, in a murine model of rheumatoid arthritis. Arthritis Res. 2020, 22, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Vishwakarma, S.; Iyer, L.R.; Muley, M.; Singh, P.K.; Shastry, A.; Saxena, A.; Kulathingal, J.; Vijaykanth, G.; Raghul, J.; Rajesh, N.; et al. Tubastatin, a selective histone deacetylase 6 inhibitor shows anti-inflammatory and anti-rheumatic effects. Int. Immunopharmacol. 2013, 16, 72–78. [Google Scholar] [CrossRef] [PubMed]
- Ren, J.; Panther, E.; Liao, X.; Grammer, A.C.; Lipsky, P.E.; Reilly, C.M. The Impact of Protein Acetylation/Deacetylation on Systemic Lupus Erythematosus. Int. J. Mol. Sci. 2018, 19, 4007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopresti, P. The Selective HDAC6 Inhibitor ACY-738 Impacts Memory and Disease Regulation in an Animal Model of Multiple Sclerosis. Front. Neurol. 2019, 10. [Google Scholar] [CrossRef] [Green Version]
- Ren, Y.; Su, X.; Kong, L.; Li, M.; Zhao, X.; Yu, N.; Kang, J. Therapeutic effects of histone deacetylase inhibitors in a murine asthma model. Inflamm. Res. 2016, 65, 995–1008. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Zhou, X.; Shetty, S.; Hou, G.; Wang, Q.; Fu, J. HDAC6 inhibition blocks inflammatory signaling and caspase-1 activation in LPS-induced acute lung injury. Toxicol. Appl. Pharm. 2019, 370, 178–183. [Google Scholar] [CrossRef]
- Ellis, J.D.; Neil, D.A.; Inston, N.G.; Jenkinson, E.; Drayson, M.T.; Hampson, P.; Shuttleworth, S.J.; Ready, A.R.; Cobbold, M. Inhibition of Histone Deacetylase 6 Reveals a Potent Immunosuppressant Effect in Models of Transplantation. Transplantation 2016, 100, 1667–1674. [Google Scholar] [CrossRef]
- Tsuji, G.; Okiyama, N.; Villarroel, V.A.; Katz, S.I. Histone deacetylase 6 inhibition impairs effector CD8 T-cell functions during skin inflammation. J. Allergy Clin. Immunol. 2015, 135, 1228–1239. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Zhao, T.; Liu, B.; Halaweish, I.; Mazitschek, R.; Duan, X.; Alam, H.B. Inhibition of histone deacetylase 6 improves long-term survival in a lethal septic model. J. Trauma Acute Care Surg. 2015, 78, 378–385. [Google Scholar] [CrossRef] [Green Version]
- Zhang, W.-B.; Zhang, H.-Y.; Jiao, F.-Z.; Wang, L.-W.; Zhang, H.; Gong, Z.-J. Histone deacetylase 6 inhibitor ACY-1215 protects against experimental acute liver failure by regulating the TLR4-MAPK/NF-κB pathway. Biomed. Pharmacother. 2018, 97, 818–824. [Google Scholar] [CrossRef]
- Regna, N.L.; Vieson, M.D.; Luo, X.M.; Chafin, C.B.; Puthiyaveetil, A.G.; Hammond, S.E.; Caudell, D.L.; Jarpe, M.B.; Reilly, C.M. Specific HDAC6 inhibition by ACY-738 reduces SLE pathogenesis in NZB/W mice. Clin. Immunol. 2016, 162, 58–73. [Google Scholar] [CrossRef] [Green Version]
- Zhou, W.; Yang, J.; Saren, G.; Zhao, H.; Cao, K.; Fu, S.; Pan, X.; Zhang, H.; Wang, A.; Chen, X. HDAC6-specific inhibitor suppresses Th17 cell function via the HIF-1α pathway in acute lung allograft rejection in mice. Theranostics 2020, 10, 6790–6805. [Google Scholar] [CrossRef]
- Lee, J.; Hong, E.C.; Jeong, H.; Hwang, J.W.; Kim, H.; Bae, E.-K.; Ahn, J.K.; Choi, Y.-L.; Han, J.; Cha, H.-S.; et al. A novel histone deacetylase 6-selective inhibitor suppresses synovial inflammation and joint destruction in a collagen antibody-induced arthritis mouse model. Int. J. Rheum. Dis. 2015, 18, 514–523. [Google Scholar] [CrossRef]
- Yee, A.J.; Bensinger, W.I.; Supko, J.G.; Voorhees, P.M.; Berdeja, J.G.; Richardson, P.G.; Libby, E.N.; Wallace, E.E.; Birrer, E.N.; Burke, J.N.; et al. Ricolinostat plus lenalidomide, and dexamethasone in relapsed or refractory multiple myeloma: A multicentre phase 1b trial. Lancet Oncol. 2016, 17, 1569–1578. [Google Scholar] [CrossRef]
- Vogl, D.T.; Raje, N.; Jagannath, S.; Richardson, P.; Hari, P.; Orlowski, R.; Supko, J.G.; Tamang, D.; Yang, M.; Jones, S.S.; et al. Ricolinostat, the First Selective Histone Deacetylase 6 Inhibitor, in Combination with Bortezomib and Dexamethasone for Relapsed or Refractory Multiple Myeloma. Clin. Cancer Res. 2017, 23, 3307–3315. [Google Scholar] [CrossRef] [Green Version]
- Shultz, M.D.; Cao, X.; Chen, C.H.; Cho, Y.S.; Davis, N.R.; Eckman, J.; Fan, J.; Fekete, A.; Firestone, B.; Flynn, J.; et al. Optimization of the in Vitro Cardiac Safety of Hydroxamate-Based Histone Deacetylase Inhibitors. J. Med. Chem. 2011, 54, 4752–4772. [Google Scholar] [CrossRef]
- Kopljar, I.; Gallacher, D.J.; De Bondt, A.; Cougnaud, L.; Vlaminckx, E.; Wyngaert, I.V.D.; Lu, H.R. Functional and Transcriptional Characterization of Histone Deacetylase Inhibitor-Mediated Cardiac Adverse Effects in Human Induced Pluripotent Stem Cell-Derived Cardiomyocytes. Stem Cells Transl. Med. 2016, 5, 602–612. [Google Scholar] [CrossRef] [Green Version]
- Li, P.; Ninomiya, H.; Kurata, Y.; Kato, M.; Miake, J.; Yamamoto, Y.; Igawa, O.; Nakai, A.; Higaki, K.; Toyoda, F.; et al. Reciprocal Control of hERG Stability by Hsp70 and Hsc70 With Implication for Restoration of LQT2 Mutant Stability. Circ. Res. 2011, 108, 458–468. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, F.-L.; Yen, J.-L.; Kuo, Y.-C.; Kang, J.-J.; Cheng, Y.-W.; Huang, W.-J.; Hsiao, G. HADC8 Inhibitor WK2-16 Therapeutically Targets Lipopolysaccharide-Induced Mouse Model of Neuroinflammation and Microglial Activation. Int. J. Mol. Sci. 2019, 20, 410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balasubramanian, S.; Ramos, J.; Luo, W.; Sirisawad, M.; Verner, E.; Buggy, J.J. A novel histone deacetylase 8 (HDAC8)-specific inhibitor PCI-34051 induces apoptosis in T-cell lymphomas. Leukemia 2008, 22, 1026–1034. [Google Scholar] [CrossRef] [PubMed]
- Zuberi, R.I.; Hsu, D.K.; Kalayci, O.; Chen, H.-Y.; Sheldon, H.K.; Yu, L.; Apgar, J.R.; Kawakami, T.; Lilly, C.M.; Liu, F.-T. Critical Role for Galectin-3 in Airway Inflammation and Bronchial Hyperresponsiveness in a Murine Model of Asthma. Am. J. Pathol. 2004, 165, 2045–2053. [Google Scholar] [CrossRef] [Green Version]
- Pavlik, C.M.; Wong, C.Y.B.; Ononye, S.; Lopez, D.D.; Engene, N.; McPhail, K.L.; Gerwick, W.H.; Balunas, M.J. Santacruzamate A, a Potent and Selective Histone Deacetylase Inhibitor from the Panamanian Marine Cyanobacterium cf. Symploca sp. J. Nat. Prod. 2013, 76, 2026–2033. [Google Scholar] [CrossRef] [Green Version]
- Jiao, F.-Z.; Wang, Y.; Zhang, H.-Y.; Zhang, W.-B.; Wang, L.-W.; Gong, Z.-J. Histone Deacetylase 2 Inhibitor CAY10683 Alleviates Lipopolysaccharide Induced Neuroinflammation through Attenuating TLR4/NF-κB Signaling Pathway. Neurochem. Res. 2018, 43, 1161–1170. [Google Scholar] [CrossRef]
- Poralla, L.; Stroh, T.; Erben, U.; Sittig, M.; Liebig, S.; Siegmund, B.; Glauben, R. Histone deacetylase 5 regulates the inflammatory response of macrophages. J. Cell. Mol. Med. 2015, 19, 2162–2171. [Google Scholar] [CrossRef]
- Liao, W.; Sun, J.; Liu, W.; Li, W.; Jia, J.; Ou, F.; Su, K.; Zheng, Y.; Zhang, Z.; Sun, Y. HDAC10 upregulation contributes to interleukin 1β-mediated inflammatory activation of synovium-derived mesenchymal stem cells in temporomandibular joint. J. Cell. Physiol. 2019, 234, 12646–12662. [Google Scholar] [CrossRef]
- Son, S.I.; Cao, J.; Zhu, C.-L.; Miller, S.P.; Lin, H. Activity-Guided Design of HDAC11-Specific Inhibitors. ACS Chem. Biol. 2019, 14, 1393–1397. [Google Scholar] [CrossRef]
- Oishi, Y.; Manabe, I. Macrophages in inflammation, repair and regeneration. Int. Immunol. 2018, 30, 511–528. [Google Scholar] [CrossRef]
- Koh, T.J.; DiPietro, L.A. Inflammation and wound healing: The role of the macrophage. Expert Rev. Mol. Med. 2011, 13, e23. [Google Scholar] [CrossRef] [Green Version]
- Lian, J.; Nelson, R.; Lehner, R. Carboxylesterases in lipid metabolism: From mouse to human. Protein Cell 2018, 9, 178–195. [Google Scholar] [CrossRef]
- Satoh, T.; Hemmerlein, B.; Zschunke, F.; Radzun, H.J. In situ Detection of Human Monocyte/Macrophage Serine Esterase-1 mRNA Expression in Human Tissues. Pathobiology 1999, 67, 158–162. [Google Scholar] [CrossRef]
- Imai, T. Human Carboxylesterase Isozymes: Catalytic Properties and Rational Drug Design. Drug Metab. Pharm. 2006, 21, 173–185. [Google Scholar] [CrossRef]
- Ossenkoppele, G.J.; Lowenberg, B.; Zachee, P.; Vey, N.; Breems, D.; van de Loosdrecht, A.A.; Davidson, A.H.; Wells, G.; Needham, L.; Bawden, L.; et al. A phase I first-in-human study with tefinostat-a monocyte/macrophage targeted histone deacetylase inhibitor-in patients with advanced haematological malignancies. Br. J. Haematol. 2013, 162, 191–201. [Google Scholar] [CrossRef]
- Elfiky, A.; Ghiboub, M.; van Hamersveld, H.P.; Welting, O.; van den Berg, I.; Rahman, S.; de Winther, M.; Becker, M.; Wildenberg, M.E.; Furze, R.; et al. Mo1104 Mononuclear Myeloid Cell Targeted Histone Deactylase (HDAC) Inhibitor Demonstrates Potent Activity in Monocytes and Impairs Colon Monocytes to Macrophage Differentation During Acute DSS Colitis. Gastroenterology 2020, 158, 789–790. [Google Scholar] [CrossRef]
- Luque-Martin, R.; van den Bossche, J.; Furze, R.C.; Neele, A.E.; van der Velden, S.; Gijbels, M.J.J.; van Roomen, C.P.P.A.; Bernard, S.G.; de Jonge, W.J.; Rioja, I.; et al. Targeting Histone Deacetylases in Myeloid Cells Inhibits Their Maturation and Inflammatory Function with Limited Effects on Atherosclerosis. Front. Pharm. 2019, 10, 1242. [Google Scholar] [CrossRef]
- Fan, X.; Zhang, H.; Cheng, Y.; Jiang, X.; Zhu, J.; Jin, T. Double Roles of Macrophages in Human Neuroimmune Diseases and Their Animal Models. Mediat. Inflamm. 2016, 2016, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.-C.; Zou, X.-B.; Chai, Y.-F.; Yao, Y.-M. Macrophage Polarization in Inflammatory Diseases. Int. J. Biol. Sci. 2014, 10, 520–529. [Google Scholar] [CrossRef]
- Fujisawa, T.; Filippakopoulos, P. Functions of bromodomain-containing proteins and their roles in homeostasis and cancer. Nat. Rev. Mol. Cell Biol. 2017, 18, 246–262. [Google Scholar] [CrossRef]
- Meslamani, J.; Smith, S.G.; Sanchez, R.; Zhou, M.-M. Structural features and inhibitors of bromodomains. Drug Discov. Today Technol. 2016, 19, 3–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.-I.; Robson, S.C.; Chung, C.-W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nat. Cell Biol. 2011, 478, 529–533. [Google Scholar] [CrossRef] [Green Version]
- Cochran, A.G.; Conery, A.R.; Sims, R.J. Bromodomains: A new target class for drug development. Nat. Rev. Drug Discov. 2019, 18, 609–628. [Google Scholar] [CrossRef] [PubMed]
- Guo, D.; Hong, D.; Wang, P.; Wang, J.; Chen, L.; Zhao, W.; Zhang, L.; Yao, C.; Chu, B.; Chen, S.; et al. Histone deacetylase inhibitor CI-994 inhibits osteoclastogenesis via suppressing NF-κB and the downstream c-Fos/NFATc1 signaling pathways. Eur. J. Pharmacol. 2019, 848, 96–104. [Google Scholar] [CrossRef]
- Malvaez, M.; McQuown, S.C.; Rogge, G.A.; Astarabadi, M.; Jacques, V.; Carreiro, S.; Rusche, J.R.; Wood, M.A. HDAC3-selective inhibitor enhances extinction of cocaine-seeking behavior in a persistent manner. Proc. Natl. Acad. Sci. USA 2013, 110, 2647–2652. [Google Scholar] [CrossRef] [Green Version]
- Liu, Z.; Tian, B.; Chen, H.; Wang, P.; Brasier, A.R.; Zhou, J. Discovery of potent and selective BRD4 inhibitors capable of blocking TLR3-induced acute airway inflammation. Eur. J. Med. Chem. 2018, 151, 450–461. [Google Scholar] [CrossRef]
- Tian, B.; Liu, Z.; Litvinov, J.; Maroto, R.; Jamaluddin, M.; Rytting, E.; Patrikeev, I.; Ochoa, L.; Vargas, G.; Motamedi, M.; et al. Efficacy of Novel Highly Specific Bromodomain-Containing Protein 4 Inhibitors in Innate Inflammation–Driven Airway Remodeling. Am. J. Respir. Cell Mol. Biol. 2019, 60, 68–83. [Google Scholar] [CrossRef]
- Theodoulou, N.H.; Bamborough, P.; Bannister, A.J.; Becher, I.; Bit, R.A.; Che, K.H.; Chung, C.-W.; Dittmann, A.; Drewes, G.; Drewry, D.H.; et al. Discovery of I-BRD9, a Selective Cell Active Chemical Probe for Bromodomain Containing Protein 9 Inhibition. J. Med. Chem. 2016, 59, 1425–1439. [Google Scholar] [CrossRef] [Green Version]
- Martin, L.J.; Koegl, M.; Bader, G.; Cockcroft, X.-L.; Fedorov, O.; Fiegen, D.; Gerstberger, T.; Hofmann, M.H.; Hohmann, A.F.; Kessler, D.; et al. Structure-Based Design of an in vivo Active Selective BRD9 Inhibitor. J. Med. Chem. 2016, 59, 4462–4475. [Google Scholar] [CrossRef]
- Lv, Q.; Yang, A.; Shi, W.; Chen, F.; Liu, Y.; Liu, Y.; Wang, D. Calcipotriol and iBRD9 reduce obesity in Nur77 knockout mice by regulating the gut microbiota, improving intestinal mucosal barrier function. Int. J. Obes. 2020, 44, 1052–1061. [Google Scholar] [CrossRef] [Green Version]
- Gilan, O.; Rioja, I.; Knezevic, K.; Bell, M.J.; Yeung, M.M.; Harker, N.R.; Lam, E.Y.N.; Chung, C.-W.; Bamborough, P.; Petretich, M.; et al. Selective targeting of BD1 and BD2 of the BET proteins in cancer and immunoinflammation. Science 2020, 368, 387–394. [Google Scholar] [CrossRef]
- Taniguchi, Y. The Bromodomain and Extra-Terminal Domain (BET) Family: Functional Anatomy of BET Paralogous Proteins. Int. J. Mol. Sci. 2016, 17, 1849. [Google Scholar] [CrossRef] [Green Version]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.-W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nat. Cell Biol. 2010, 468, 1119–1123. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, M.C.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef] [Green Version]
- Wyce, A.; Ganji, G.; Smitheman, K.N.; Chung, C.-W.; Korenchuk, S.; Bai, Y.; Barbash, O.; Le, B.; Craggs, P.D.; McCabe, M.T.; et al. BET Inhibition Silences Expression of MYCN and BCL2 and Induces Cytotoxicity in Neuroblastoma Tumor Models. PLoS ONE 2013, 8, e72967. [Google Scholar] [CrossRef] [Green Version]
- Chan, C.H.; Fang, C.; Qiao, Y.; Yarilina, A.; Prinjha, R.K.; Ivashkiv, L.B. BET bromodomain inhibition suppresses transcriptional responses to cytokine-Jak-STAT signaling in a gene-specific manner in human monocytes. Eur. J. Immunol. 2015, 45, 287–297. [Google Scholar] [CrossRef]
- Miller, T.C.R.; Simon, B.; Rybin, V.; Grötsch, H.; Curtet, S.; Khochbin, S.; Carlomagno, T.; Müller, C.W. A bromodomain–DNA interaction facilitates acetylation-dependent bivalent nucleosome recognition by the BET protein BRDT. Nat. Commun. 2016, 7, 13855. [Google Scholar] [CrossRef] [Green Version]
- Baud, M.G.J.; Lin-Shiao, E.; Cardote, T.; Tallant, C.; Pschibul, A.; Chan, K.-H.; Zengerle, M.; Garcia, J.R.; Kwan, T.T.-L.; Ferguson, F.M.; et al. A bump-and-hole approach to engineer controlled selectivity of BET bromodomain chemical probes. Science 2014, 346, 638–641. [Google Scholar] [CrossRef] [Green Version]
- Shi, J.; Wang, E.; Milazzo, J.P.; Wang, Z.; Kinney, J.B.; Vakoc, C.R. Discovery of cancer drug targets by CRISPR-Cas9 screening of protein domains. Nat. Biotechnol. 2015, 33, 661–667. [Google Scholar] [CrossRef] [Green Version]
- Faivre, E.J.; McDaniel, K.F.; Albert, D.H.; Mantena, S.R.; Plotnik, J.P.; Wilcox, D.; Zhang, L.; Bui, M.H.; Sheppard, G.S.; Wang, L.; et al. Selective inhibition of the BD2 bromodomain of BET proteins in prostate cancer. Nat. Cell Biol. 2020, 578, 306–310. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Knapp, S. Next-generation epigenetic inhibitors. Science 2020, 368, 367–368. [Google Scholar] [CrossRef] [PubMed]
- Zhang, G.; Plotnikov, A.N.; Rusinova, E.; Shen, T.; Morohashi, K.; Joshua, J.; Zeng, L.; Mujtaba, S.; Ohlmeyer, M.; Zhou, M.-M. Structure-Guided Design of Potent Diazobenzene Inhibitors for the BET Bromodomains. J. Med. Chem. 2013, 56, 9251–9264. [Google Scholar] [CrossRef] [PubMed]
- Nowak, R.P.; DeAngelo, S.L.; Buckley, D.; He, Z.; Donovan, K.A.; An, J.; Safaee, N.; Jedrychowski, M.P.; Ponthier, C.M.; Ishoey, M.; et al. Plasticity in binding confers selectivity in ligand-induced protein degradation. Nat. Chem. Biol. 2018, 14, 706–714. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Hu, Y.; Zhou, B.; Fernandez-Salas, E.; Yang, C.-Y.; Liu, L.; McEachern, D.; Przybranowski, S.; Wang, M.; Stuckey, J.; et al. Discovery of QCA570 as an Exceptionally Potent and Efficacious Proteolysis Targeting Chimera (PROTAC) Degrader of the Bromodomain and Extra-Terminal (BET) Proteins Capable of Inducing Complete and Durable Tumor Regression. J. Med. Chem. 2018, 61, 6685–6704. [Google Scholar] [CrossRef]
- Yim, A.Y.F.L.; Duijvis, N.W.; Zhao, J.; de Jonge, W.J.; D’Haens, G.R.A.M.; Mannens, M.M.A.M.; Mul, A.N.P.M.; Velde, A.A.T.; Henneman, P. Peripheral blood methylation profiling of female Crohn’s disease patients. Clin. Epigenet. 2016, 8, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Franke, A.; McGovern, D.P.B.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn’s disease susceptibility loci. Nat. Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef] [Green Version]
- Sima, X.; He, J.; Peng, J.; Xu, Y.; Zhang, F.; Deng, L. The genetic alteration spectrum of the SWI/SNF complex: The oncogenic roles of BRD9 and ACTL6A. PLoS ONE 2019, 14, e0222305. [Google Scholar] [CrossRef] [Green Version]
- Loo, C.-S.; Gatchalian, J.; Liang, Y.; Leblanc, M.; Xie, M.; Ho, J.; Venkatraghavan, B.; Hargreaves, D.C.; Zheng, Y. A Genome-wide CRISPR Screen Reveals a Role for the Non-canonical Nucleosome-Remodeling BAF Complex in Foxp3 Expression and Regulatory T Cell Function. Immunity 2020, 53, 143–157. [Google Scholar] [CrossRef]
- Wen, A.Y.; Sakamoto, K.M.; Miller, L.S. The Role of the Transcription Factor CREB in Immune Function. J. Immunol. 2010, 185, 6413–6419. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.; Xu, W.; Wang, H.; Cao, M.; Li, M.; Zhao, J.; Hu, Y.; Wang, Y.; Li, S.; Xie, Y.; et al. Inhibition of CREB-mediated ZO-1 and activation of NF-κB-induced IL-6 by colonic epithelial MCT4 destroys intestinal barrier function. Cell Prolif. 2019, 52, e12673. [Google Scholar] [CrossRef] [Green Version]
- Takeba, Y.; Suzuki, N.; Wakisaka, S.; Takeno, M.; Kaneko, A.; Asai, T.; Sakane, T. Involvement of cAMP responsive element binding protein (CREB) in the synovial cell hyperfunction in patients with rheumatoid arthritis. Clin. Exp. Rheumatol. 2000, 18, 47–55. [Google Scholar]
- Chekler, E.L.P.; Pellegrino, J.A.; Lanz, T.A.; Denny, R.A.; Flick, A.C.; Coe, J.; Langille, J.; Basak, A.; Liu, S.; Stock, I.A.; et al. Transcriptional Profiling of a Selective CREB Binding Protein Bromodomain Inhibitor Highlights Therapeutic Opportunities. Chem. Biol. 2015, 22, 1588–1596. [Google Scholar] [CrossRef]
- Best, J.L.; Amezcua, C.A.; Mayr, B.; Flechner, L.; Murawsky, C.M.; Emerson, B.; Zor, T.; Gardner, K.H.; Montminy, M. Identification of small-molecule antagonists that inhibit an activator: Coactivator interaction. Proc. Natl. Acad. Sci. USA 2004, 101, 17622–17627. [Google Scholar] [CrossRef] [Green Version]
- Igoe, N.; Bayle, E.D.; Tallant, C.; Fedorov, O.; Meier, J.C.; Savitsky, P.; Rogers, C.; Morias, Y.; Scholze, S.; Boyd, H.; et al. Design of a Chemical Probe for the Bromodomain and Plant Homeodomain Finger-Containing (BRPF) Family of Proteins. J. Med. Chem. 2017, 60, 6998–7011. [Google Scholar] [CrossRef]
- Meier, J.C.; Tallant, C.; Fedorov, O.; Witwicka, H.; Hwang, S.-Y.; Van Stiphout, R.G.; Lambert, J.-P.; Rogers, C.; Yapp, C.; Gerstenberger, B.S.; et al. Selective Targeting of Bromodomains of the Bromodomain-PHD Fingers Family Impairs Osteoclast Differentiation. ACS Chem. Biol. 2017, 12, 2619–2630. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brown, J.A.; Jones, K.L.; Prinjha, R.K.; Witherington, J. Covalent conjugates of bet inhibitors and alpha amino acid esters. U.S. Patent 15/559,518, 22 September 2016. [Google Scholar]
- Felice, C.; Lewis, A.; Armuzzi, A.; Lindsay, J.O.; Silver, A. Review article: Selective histone deacetylase isoforms as potential therapeutic targets in inflammatory bowel diseases. Aliment. Pharm. 2014, 41, 26–38. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghiboub, M.; Elfiky, A.M.I.; de Winther, M.P.J.; Harker, N.R.; Tough, D.F.; de Jonge, W.J. Selective Targeting of Epigenetic Readers and Histone Deacetylases in Autoimmune and Inflammatory Diseases: Recent Advances and Future Perspectives. J. Pers. Med. 2021, 11, 336. https://doi.org/10.3390/jpm11050336
Ghiboub M, Elfiky AMI, de Winther MPJ, Harker NR, Tough DF, de Jonge WJ. Selective Targeting of Epigenetic Readers and Histone Deacetylases in Autoimmune and Inflammatory Diseases: Recent Advances and Future Perspectives. Journal of Personalized Medicine. 2021; 11(5):336. https://doi.org/10.3390/jpm11050336
Chicago/Turabian StyleGhiboub, Mohammed, Ahmed M. I. Elfiky, Menno P. J. de Winther, Nicola R. Harker, David F. Tough, and Wouter J. de Jonge. 2021. "Selective Targeting of Epigenetic Readers and Histone Deacetylases in Autoimmune and Inflammatory Diseases: Recent Advances and Future Perspectives" Journal of Personalized Medicine 11, no. 5: 336. https://doi.org/10.3390/jpm11050336
APA StyleGhiboub, M., Elfiky, A. M. I., de Winther, M. P. J., Harker, N. R., Tough, D. F., & de Jonge, W. J. (2021). Selective Targeting of Epigenetic Readers and Histone Deacetylases in Autoimmune and Inflammatory Diseases: Recent Advances and Future Perspectives. Journal of Personalized Medicine, 11(5), 336. https://doi.org/10.3390/jpm11050336