
New Insights Regarding Diagnosis and Medication for Schizophrenia Based on Neuronal Synapse–Microglia Interaction

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Microglia–Synapse Interaction
3. Microglia–Synapse Interaction in Schizophrenia
4. Potential Diagnosis/Medication for Schizophrenia Based on Microglia–Synapse Interaction
4.1. Perspective for the Diagnosis of Schizophrenia
4.2. Perspective for Medication Drug Therapy in Schizophrenia
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Janoutova, J.; Janackova, P.; Sery, O.; Zeman, T.; Ambroz, P.; Kovalova, M.; Varechova, K.; Hosak, L.; Jirik, V.; Janout, V. Epidemiology and risk factors of schizophrenia. Neuroendocrinol. Lett. 2016, 37, 1–8. [Google Scholar] [PubMed]
- Besteher, B.; Brambilla, P.; Nenadic, I. Twin studies of brain structure and cognition in schizophrenia. Neurosci. Biobehav. Rev. 2020, 109, 103–113. [Google Scholar] [CrossRef] [PubMed]
- Torrey, E.F.; Yolken, R.H. Schizophrenia as a pseudogenetic disease: A call for more gene-environmental studies. Psychiatry Res. 2019, 278, 146–150. [Google Scholar] [CrossRef] [PubMed]
- Devor, A.; Andreassen, O.A.; Wang, Y.; Maki-Marttunen, T.; Smeland, O.B.; Fan, C.C.; Schork, A.J.; Holland, D.; Thompson, W.K.; Witoelar, A.; et al. Genetic evidence for role of integration of fast and slow neurotransmission in schizophrenia. Mol. Psychiatry 2017, 22, 792–801. [Google Scholar] [CrossRef]
- Howes, O.D.; McCutcheon, R.; Owen, M.J.; Murray, R.M. The Role of Genes, Stress, and Dopamine in the Development of Schizophrenia. Biol. Psychiatry 2017, 81, 9–20. [Google Scholar] [CrossRef] [Green Version]
- Hanif, F.; Amir, Q.U.; Washdev, W.; Bilwani, F.; Simjee, S.U.; Haque, Z. A Novel Variant in Dopamine Receptor Type 2 Gene is Associated with Schizophrenia. Arch. Med. Res. 2020, 52, 348–353. [Google Scholar] [CrossRef]
- Bassett, A.S.; Marshall, C.R.; Lionel, A.C.; Chow, E.W.C.; Scherer, S.W. Copy number variations and risk for schizophrenia in 22q11.2 deletion syndrome. Hum. Mol. Genet. 2008, 17, 4045–4053. [Google Scholar] [CrossRef] [Green Version]
- Wang, H.Y.; Liu, Y.; Yan, J.W.; Hu, X.L.; Zhu, D.M.; Xu, X.T.; Li, X.S. Gene polymorphisms of DISC1 is associated with schizophrenia: Evidence from a meta-analysis. Prog. Neuropsychopharmacol. Biol. Psychiatry 2018, 81, 64–73. [Google Scholar] [CrossRef]
- Inoue, K.; Ando, N.; Suzuki, E.; Hayashi, H.; Tsuji, D.; Itoh, K. Genotype distributions and allele frequencies of possible major depressive disorder-associated single nucleotide polymorphisms, cyclic adenosine monophosphate response element binding protein 1 rs4675690 and Piccolo rs2522833, in a Japanese population. Biol. Pharm. Bull. 2012, 35, 265–268. [Google Scholar] [CrossRef] [Green Version]
- Cen, X.; Nitta, A.; Ibi, D.; Zhao, Y.; Niwa, M.; Taguchi, K.; Hamada, M.; Ito, Y.; Ito, Y.; Wang, L.; et al. Identification of Piccolo as a regulator of behavioral plasticity and dopamine transporter internalization. Mol. Psychiatry 2008, 13, 451–463. [Google Scholar] [CrossRef] [Green Version]
- Seo, S.; Takayama, K.; Uno, K.; Ohi, K.; Hashimoto, R.; Nishizawa, D.; Ikeda, K.; Ozaki, N.; Nabeshima, T.; Miyamoto, Y.; et al. Functional Analysis of Deep Intronic SNP rs13438494 in Intron 24 of PCLO Gene. PLoS ONE 2013, 8, e76960. [Google Scholar] [CrossRef] [Green Version]
- Farrell, M.S.; Werge, T.; Sklar, P.; Owen, M.J.; Ophoff, R.A.; O’Donovan, M.C.; Corvin, A.; Cichon, S.; Sullivan, P.F. Evaluating historical candidate genes for schizophrenia. Mol. Psychiatry 2015, 20, 555–562. [Google Scholar] [CrossRef] [Green Version]
- Schizophrenia Working Group of the Psychiatric Genomics Consortium. Biological insights from 108 schizophrenia-associated genetic loci. Nature 2014, 511, 421–427. [Google Scholar] [CrossRef] [Green Version]
- International Schizophrenia Consortium. Common polygenic variation contributes to risk of schizophrenia and bipolar disorder. Nature 2009, 460, 748–752. [Google Scholar] [CrossRef]
- Shi, J.X.; Levinson, D.F.; Duan, J.B.; Sanders, A.R.; Zheng, Y.L.; Pe’er, I.; Dudbridge, F.; Holmans, P.A.; Whittemore, A.S.; Mowry, B.J.; et al. Common variants on chromosome 6p22.1 are associated with schizophrenia. Nature 2009, 460, 753–757. [Google Scholar] [CrossRef] [PubMed]
- Stefansson, H.; Ophoff, R.A.; Steinberg, S.; Andreassen, O.A.; Cichon, S.; Rujescu, D.; Werge, T.; Pietilainen, O.P.H.; Mors, O.; Mortensen, P.B.; et al. Common variants conferring risk of schizophrenia. Nature 2009, 460, 744–747. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mokhtari, R.; Lachman, H.M. The Major Histocompatibility Complex (MHC) in Schizophrenia: A Review. J. Clin. Cell. Immunol. 2016, 7, 479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Radewicz, K.; Garey, L.J.; Gentleman, S.M.; Reynolds, R. Increase in HLA-DR immunoreactive microglia in frontal and temporal cortex of chronic schizophrenics. J. Neuropathol. Exp. Neurol. 2000, 59, 137–150. [Google Scholar] [CrossRef] [Green Version]
- Steiner, J.; Bielau, H.; Brisch, R.; Danos, P.; Ullrich, O.; Mawrin, C.; Bernstein, H.G.; Bogerts, B. Immunological aspects in the neurobiology of suicide: Elevated microglial density in schizophrenia and depression is associated with suicide. J. Psychiatr. Res. 2008, 42, 151–157. [Google Scholar] [CrossRef]
- Goldsmith, D.R.; Rapaport, M.H.; Miller, B.J. A meta-analysis of blood cytokine network alterations in psychiatric patients: Comparisons between schizophrenia, bipolar disorder and depression. Mol. Psychiatry 2016, 21, 1696–1709. [Google Scholar] [CrossRef] [PubMed]
- Orlovska-Waast, S.; Kohler-Forsberg, O.; Brix, S.W.; Nordentoft, M.; Kondziella, D.; Krogh, J.; Benros, M.E. Cerebrospinal fluid markers of inflammation and infections in schizophrenia and affective disorders: A systematic review and meta-analysis. Mol. Psychiatry 2019, 24, 869–887. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloomfield, P.S.; Selvaraj, S.; Veronese, M.; Rizzo, G.; Bertoldo, A.; Owen, D.R.; Bloomfield, M.A.P.; Bonoldi, I.; Kalk, N.; Turkheimer, F.; et al. Microglial Activity in People at Ultra High Risk of Psychosis and in Schizophrenia: An [(11)C]PBR28 PET Brain Imaging Study. Am. J. Psychiatry 2016, 173, 44–52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Van Berckel, B.N.; Bossong, M.G.; Boellaard, R.; Kloet, R.; Schuitemaker, A.; Caspers, E.; Luurtsema, G.; Windhorst, A.D.; Cahn, W.; Lammertsma, A.A.; et al. Microglia activation in recent-onset schizophrenia: A quantitative (R)-[11C]PK11195 positron emission tomography study. Biol. Psychiatry 2008, 64, 820–822. [Google Scholar] [CrossRef] [PubMed]
- Almulla, A.F.; Al-Rawi, K.F.; Maes, M.; Al-Hakeim, H.K. In schizophrenia, immune-inflammatory pathways are strongly associated with depressive and anxiety symptoms, which are part of a latent trait which comprises neurocognitive impairments and schizophrenia symptoms. J. Affect. Disord. 2021, 287, 316–326. [Google Scholar] [CrossRef]
- Stuss, D.T.; Kaplan, E.F.; Benson, D.F.; Weir, W.S.; Steven, C.; Sarazin, F.F. Evidence for the involvement of orbitofrontal cortex in memory functions: An interference effect. J. Comp. Physiol. Psychol. 1982, 96, 913–925. [Google Scholar] [CrossRef]
- Müller, H.F. Prefrontal cortex dysfunction as a common factor in psychosis. Acta Psychiatr. Scand. 1985, 71, 431–440. [Google Scholar] [CrossRef]
- Walton, E.; Hibar, D.P.; van Erp, T.G.M.; Potkin, S.G.; Roiz-Santianez, R.; Crespo-Facorro, B.; Suarez-Pinilla, P.; van Haren, N.E.M.; de Zwarte, S.M.C.; Kahn, R.S.; et al. Prefrontal cortical thinning links to negative symptoms in schizophrenia via the ENIGMA consortium. Psychol. Med. 2018, 48, 82–94. [Google Scholar] [CrossRef]
- Kuo, S.S.; Pogue-Geile, M.F. Variation in fourteen brain structure volumes in schizophrenia: A comprehensive meta-analysis of 246 studies. Neurosci. Biobehav. Rev. 2019, 98, 85–94. [Google Scholar] [CrossRef]
- Buchsbaum, M.S.; Nuechterlein, K.H.; Haier, R.J.; Wu, J.; Sicotte, N.; Hazlett, E.; Asarnow, R.; Potkin, S.; Guich, S. Glucose metabolic rate in normals and schizophrenics during the Continuous Performance Test assessed by positron emission tomography. Br. J. Psychiatry 1990, 156, 216–227. [Google Scholar] [CrossRef] [PubMed]
- Hommer, D.W.; Clem, T.; Litman, R.; Pickar, D. Maladaptive anticipatory saccades in schizophrenia. Biol. Psychiatry 1991, 30, 779–794. [Google Scholar] [CrossRef]
- Franke, P.; Maier, W.; Hain, C.; Klingler, T. Wisconsin Card Sorting Test: An indicator of vulnerability to schizophrenia? Schizophr. Res. 1992, 6, 243–249. [Google Scholar] [CrossRef]
- GoldmanRakic, P.S.; Selemon, L.D. Functional and anatomical aspects of prefrontal pathology in schizophrenia. Schizophr. Bull. 1997, 23, 437–458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ragland, J.D.; Laird, A.R.; Ranganath, C.; Blumenfeld, R.S.; Gonzales, S.M.; Glahn, D.C. Prefrontal activation deficits during episodic memory in schizophrenia. Am. J. Psychiatry 2009, 166, 863–874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yan, C.; Yang, T.; Yu, Q.J.; Jin, Z.; Cheung, E.F.C.; Liu, X.; Chan, R.C.K. Rostral medial prefrontal dysfunctions and consummatory pleasure in schizophrenia: A meta-analysis of functional imaging studies. Psychiatry Res. 2015, 231, 187–196. [Google Scholar] [CrossRef]
- Feinberg, I. Schizophrenia: Caused by a fault in programmed synaptic elimination during adolescence? J. Psychiatr. Res. 1982, 17, 319–334. [Google Scholar] [CrossRef]
- Selemon, L.D.; Zecevic, N. Schizophrenia: A tale of two critical periods for prefrontal cortical development. Transl. Psychiatry 2015, 5, e623. [Google Scholar] [CrossRef]
- Fillman, S.G.; Cloonan, N.; Catts, V.S.; Miller, L.C.; Wong, J.; McCrossin, T.; Cairns, M.; Weickert, C.S. Increased inflammatory markers identified in the dorsolateral prefrontal cortex of individuals with schizophrenia. Mol. Psychiatry 2013, 18, 206–214. [Google Scholar] [CrossRef] [Green Version]
- Paolicelli, R.C.; Bolasco, G.; Pagani, F.; Maggi, L.; Scianni, M.; Panzanelli, P.; Giustetto, M.; Ferreira, T.A.; Guiducci, E.; Dumas, L.; et al. Synaptic pruning by microglia is necessary for normal brain development. Science 2011, 333, 1456–1458. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kettenmann, H.; Kirchhoff, F.; Verkhratsky, A. Microglia: New roles for the synaptic stripper. Neuron 2013, 77, 10–18. [Google Scholar] [CrossRef] [Green Version]
- Wilton, D.K.; Dissing-Olesen, L.; Stevens, B. Neuron-Glia Signaling in Synapse Elimination. Annu. Rev. Neurosci. 2019, 42, 107–127. [Google Scholar] [CrossRef]
- Bright, F.; Werry, E.L.; Dobson-Stone, C.; Piguet, O.; Ittner, L.M.; Halliday, G.M.; Hodges, J.R.; Kiernan, M.C.; Loy, C.T.; Kassiou, M.; et al. Neuroinflammation in frontotemporal dementia. Nat. Rev. Neurol. 2019, 15, 540–555. [Google Scholar] [CrossRef] [PubMed]
- Akiyama, H.; Barger, S.; Barnum, S.; Bradt, B.; Bauer, J.; Cole, G.M.; Cooper, N.R.; Eikelenboom, P.; Emmerling, M.; Fiebich, B.L.; et al. Inflammation and Alzheimer’s disease. Neurobiol. Aging 2000, 21, 383–421. [Google Scholar] [CrossRef]
- Izuo, N.; Kasahara, C.; Murakami, K.; Kume, T.; Maeda, M.; Irie, K.; Yokote, K.; Shimizu, T. Toxic Conformer of Aβ42 with a Turn at 22–23 is a Novel Therapeutic Target for Alzheimer’s Disease. Sci. Rep. 2017, 7, 11811. [Google Scholar] [CrossRef] [PubMed]
- Dubbelaar, M.L.; Kracht, L.; Eggen, B.J.L.; Boddeke, E.W.G.M. The Kaleidoscope of Microglial Phenotypes. Front. Immunol. 2018, 9, 1753. [Google Scholar] [CrossRef]
- Davalos, D.; Grutzendler, J.; Yang, G.; Kim, J.V.; Zuo, Y.; Jung, S.; Littman, D.R.; Dustin, M.L.; Gan, W.B. ATP mediates rapid microglial response to local brain injury in vivo. Nat. Neurosci. 2005, 8, 752–758. [Google Scholar] [CrossRef] [PubMed]
- Nimmerjahn, A.; Kirchhoff, F.; Helmchen, F. Resting microglial cells are highly dynamic surveillants of brain parenchyma in vivo. Science 2005, 308, 1314–1318. [Google Scholar] [CrossRef] [Green Version]
- Tremblay, M.E.; Lowery, R.L.; Majewska, A.K. Microglial interactions with synapses are modulated by visual experience. PLoS Biol. 2010, 8, e1000527. [Google Scholar] [CrossRef] [Green Version]
- Wake, H.; Moorhouse, A.J.; Jinno, S.; Kohsaka, S.; Nabekura, J. Resting microglia directly monitor the functional state of synapses in vivo and determine the fate of ischemic terminals. J. Neurosci. 2009, 29, 3974–3980. [Google Scholar] [CrossRef] [Green Version]
- Milinkeviciute, G.; Henningfield, C.M.; Muniakz, M.A.; Chokr, S.M.; Green, K.N.; Cramer, K.S. Microglia Regulate Pruning of Specialized Synapses in the Auditory Brainstem. Front. Neural. Circuits 2019, 13, 55. [Google Scholar] [CrossRef]
- Ma, X.K.; Chen, K.; Cui, Y.H.; Huang, G.N.; Nehme, A.; Zhang, L.; Li, H.D.; Wei, J.; Liong, K.; Liu, Q.; et al. Depletion of microglia in developing cortical circuits reveals its critical role in glutamatergic synapse development, functional connectivity, and critical period plasticity. J. Neurosci. Res. 2020, 98, 1968–1986. [Google Scholar] [CrossRef]
- Brown, G.C.; Neher, J.J. Eaten alive! Cell death by primary phagocytosis: ‘phagoptosis’. Trends Biochem. Sci. 2012, 37, 325–332. [Google Scholar] [CrossRef] [PubMed]
- Vilalta, A.; Brown, G.C. Neurophagy, the phagocytosis of live neurons and synapses by glia, contributes to brain development and disease. FEBS J. 2018, 285, 3566–3575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harrison, J.K.; Jiang, Y.; Chen, S.Z.; Xia, Y.Y.; Maciejewski, D.; McNamara, R.K.; Streit, W.J.; Salafranca, M.N.; Adhikari, S.; Thompson, D.A.; et al. Role for neuronally derived fractalkine in mediating interactions between neurons and CX3CR1-expressing microglia. Proc. Natl. Acad. Sci. USA 1998, 95, 10896–10901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jung, S.; Aliberti, J.; Graemmel, P.; Sunshine, M.J.; Kreutzberg, G.W.; Sher, A.; Littman, D.R. Analysis of fractalkine receptor CX(3)CR1 function by targeted deletion and green fluorescent protein reporter gene insertion. Mol. Cell. Biol. 2000, 20, 4106–4114. [Google Scholar] [CrossRef] [Green Version]
- Gunner, G.; Cheadle, L.; Johnson, K.M.; Ayata, P.; Badimon, A.; Mondo, E.; Nagy, M.A.; Liu, L.W.; Bemiller, S.M.; Kim, K.W.; et al. Sensory lesioning induces microglial synapse elimination via ADAM10 and fractalkine signaling. Nat. Neurosci. 2019, 22, 1075–1088. [Google Scholar] [CrossRef]
- Chekeni, F.B.; Elliott, M.R.; Sandilos, J.K.; Walk, S.F.; Kinchen, J.M.; Lazarowski, E.R.; Armstrong, A.J.; Penuela, S.; Laird, D.W.; Salvesen, G.S.; et al. Pannexin 1 channels mediate ’find-me’ signal release and membrane permeability during apoptosis. Nature 2010, 467, 863–867. [Google Scholar] [CrossRef] [Green Version]
- Haynes, S.E.; Hollopeter, G.; Yang, G.; Kurpius, D.; Dailey, M.E.; Gan, W.B.; Julius, D. The P2Y12 receptor regulates microglial activation by extracellular nucleotides. Nat. Neurosci. 2006, 9, 1512–1519. [Google Scholar] [CrossRef]
- Maeda, M.; Tsuda, M.; Tozaki-Saitoh, H.; Inoue, K.; Kiyama, H. Nerve injury-activated microglia engulf myelinated axons in a P2Y12 signaling-dependent manner in the dorsal horn. Glia 2010, 58, 1838–1846. [Google Scholar] [CrossRef]
- Sipe, G.O.; Lowery, R.L.; Tremblay, M.E.; Kelly, E.A.; Lamantia, C.E.; Majewska, A.K. Microglial P2Y12 is necessary for synaptic plasticity in mouse visual cortex. Nat. Commun. 2016, 7, 10905. [Google Scholar] [CrossRef]
- Cserep, C.; Posfai, B.; Lenart, N.; Fekete, R.; Laszlo, Z.I.; Lele, Z.; Orsolits, B.; Molnar, G.; Heindl, S.; Schwarcz, A.D.; et al. Microglia monitor and protect neuronal function through specialized somatic purinergic junctions. Science 2020, 367, 528–537. [Google Scholar] [CrossRef]
- Arandjelovic, S.; Ravichandran, K.S. Phagocytosis of apoptotic cells in homeostasis. Nat. Immunol. 2015, 16, 907–917. [Google Scholar] [CrossRef] [Green Version]
- Tufail, Y.; Cook, D.; Fourgeaud, L.; Powers, C.J.; Merten, K.; Clark, C.L.; Hoffman, E.; Ngo, A.; Sekiguchi, K.J.; O’Shea, C.C.; et al. Phosphatidylserine Exposure Controls Viral Innate Immune Responses by Microglia. Neuron 2017, 93, 574–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soulard, C.; Salsac, C.; Mouzat, K.; Hilaire, C.; Roussel, J.; Mezghrani, A.; Lumbroso, S.; Raoul, C.; Scamps, F. Spinal Motoneuron TMEM16F Acts at C-boutons to Modulate Motor Resistance and Contributes to ALS Pathogenesis. Cell Rep. 2020, 30, 2581–2593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filipello, F.; Morini, R.; Corradini, I.; Zerbi, V.; Canzi, A.; Michalski, B.; Erreni, M.; Markicevic, M.; Starvaggi-Cucuzza, C.; Otero, K.; et al. The Microglial Innate Immune Receptor TREM2 Is Required for Synapse Elimination and Normal Brain Connectivity. Immunity 2018, 48, 979–991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Chiou, B.; Gilman, C.K.; Luo, R.; Koshi, T.; Yu, D.K.; Oak, H.C.; Giera, S.; Johnson-Venkatesh, E.; Muthukumar, A.K.; et al. A splicing isoform of GPR56 mediates microglial synaptic refinement via phosphatidylserine binding. EMBO J. 2020, 39, e104136. [Google Scholar] [CrossRef] [PubMed]
- Biatynicki-Birula, R. The 100th anniversary of Wassermann-Neisser-Bruck reaction. Clin. Dermatol. 2008, 26, 79–88. [Google Scholar] [CrossRef]
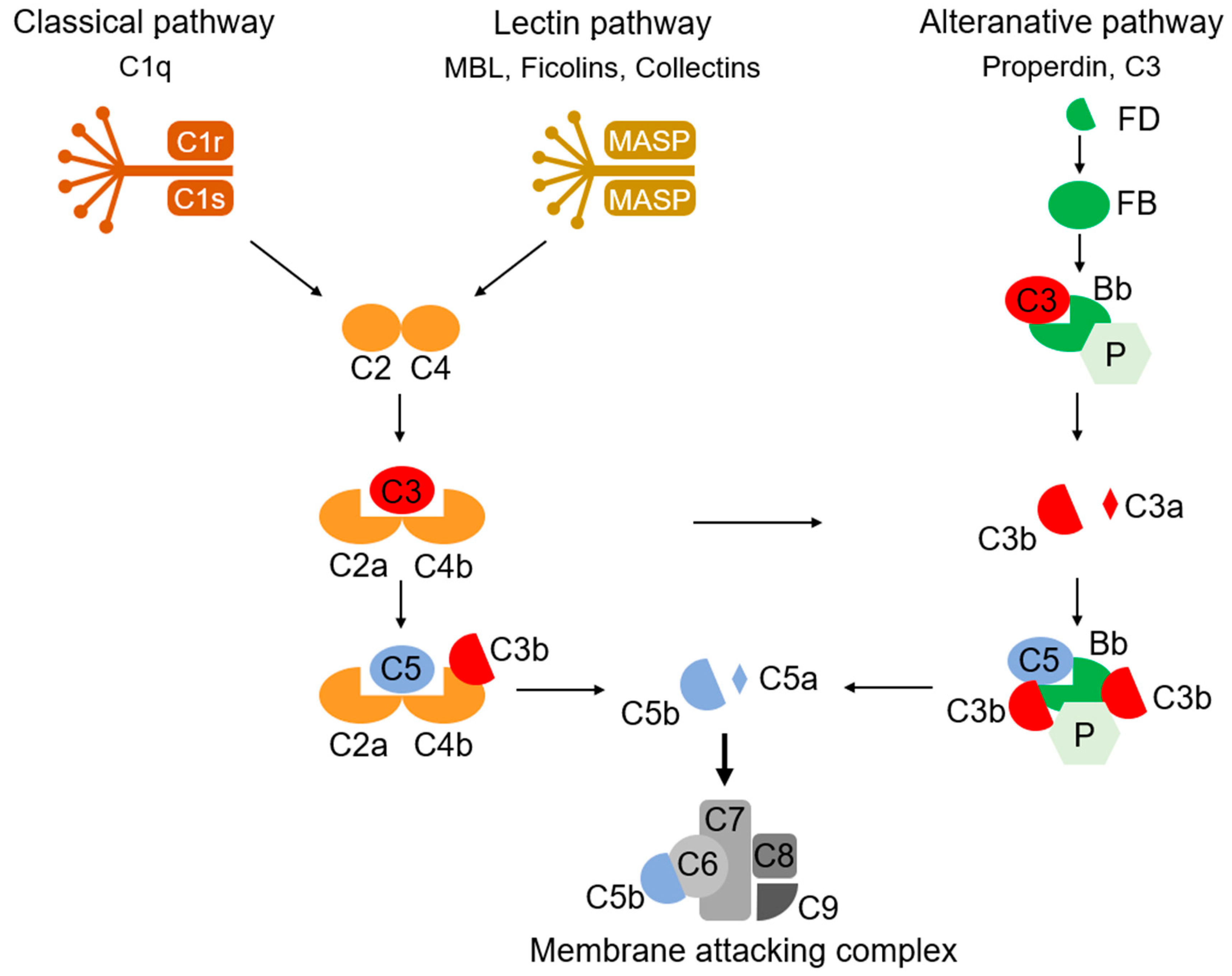
- Carroll, M.C. The role of complement and complement receptors in induction and regulation of immunity. Annu. Rev. Immunol. 1998, 16, 545–568. [Google Scholar] [CrossRef]
- Dunkelberger, J.R.; Song, W.C. Complement and its role in innate and adaptive immune responses. Cell Res. 2010, 20, 34–50. [Google Scholar] [CrossRef] [Green Version]
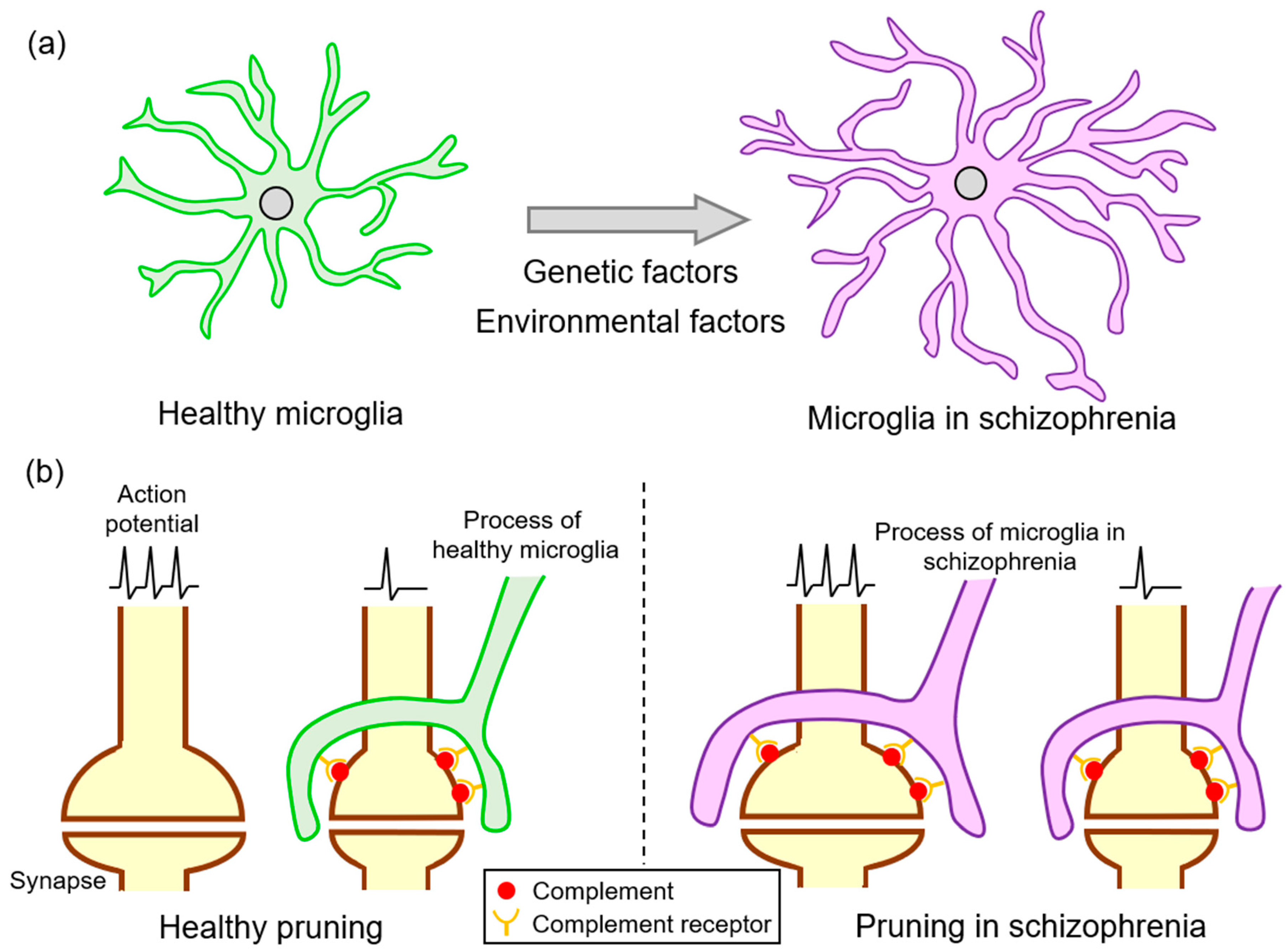
- Stevens, B.; Allen, N.J.; Vazquez, L.E.; Howell, G.R.; Christopherson, K.S.; Nouri, N.; Micheva, K.D.; Mehalow, A.K.; Huberman, A.D.; Stafford, B.; et al. The classical complement cascade mediates CNS synapse elimination. Cell 2007, 131, 1164–1178. [Google Scholar] [CrossRef] [Green Version]
- Schartz, N.D.; Tenner, A.J. The good, the bad, and the opportunities of the complement system in neurodegenerative disease. J. Neuroinflammation 2020, 17, 354. [Google Scholar] [CrossRef]
- Shi, Q.Q.; Colodner, K.J.; Matousek, S.B.; Merry, K.; Hong, S.Y.; Kenison, J.E.; Frost, J.L.; Le, K.X.; Li, S.M.; Dodart, J.C.; et al. Complement C3-Deficient Mice Fail to Display Age-Related Hippocampal Decline. J. Neurosci. 2015, 35, 13029–13042. [Google Scholar] [CrossRef] [PubMed]
- Anderson, S.R.; Zhang, J.M.; Steele, M.R.; Romero, C.O.; Kautzman, A.G.; Schafer, D.P.; Vetter, M.L. Complement Targets Newborn Retinal Ganglion Cells for Phagocytic Elimination by Microglia. J. Neurosci. 2019, 39, 2025–2040. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gyorffy, B.A.; Kun, J.; Torok, G.; Bulyaki, E.; Borhegyi, Z.; Gulyassy, P.; Kis, V.; Szocsics, P.; Micsonai, A.; Matko, J. Local apoptotic-like mechanisms underlie complement-mediated synaptic pruning. Proc. Natl. Acad. Sci. USA 2018, 115, 6303–6308. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sekar, A.; Bialas, A.R.; de Rivera, H.; Davis, A.; Hammond, T.R.; Kamitaki, N.; Tooley, K.; Presumey, J.; Baum, M.; Van Doren, V.; et al. Schizophrenia risk from complex variation of complement component 4. Nature 2016, 530, 177–183. [Google Scholar] [CrossRef] [Green Version]
- Nomura, K.; Vilalta, A.; Allendorf, D.H.; Hornik, T.C.; Brown, G.C. Activated Microglia Desialylate and Phagocytose Cells via Neuraminidase, Galectin-3, and Mer Tyrosine Kinase. J. Immunol. 2017, 198, 4792–4801. [Google Scholar] [CrossRef] [Green Version]
- Angata, T.; Kerr, S.C.; Greaves, D.R.; Varki, N.M.; Crocker, P.R.; Varki, A. Cloning and characterization of human Siglec-11—A recently evolved signaling molecule that can interact with SHP-1 and SHP-2 and is expressed by tissue macrophages, including brain microglia. J. Biol. Chem. 2002, 277, 24466–24474. [Google Scholar] [CrossRef] [Green Version]
- Puigdellivol, M.; Allendorf, D.H.; Brown, G.C. Sialylation and Galectin-3 in Microglia-Mediated Neuroinflammation and Neurodegeneration. Front. Cell. Neurosci. 2020, 14, 162. [Google Scholar] [CrossRef]
- Klaus, C.; Hansen, J.N.; Ginolhac, A.; Gerard, D.; Gnanapragassam, V.S.; Horstkorte, R.; Rossdam, C.; Buettner, F.F.R.; Sauter, T.; Sinkkonen, L.; et al. Reduced sialylation triggers homeostatic synapse and neuronal loss in middle-aged mice. Neurobiol. Aging 2020, 88, 91–107. [Google Scholar] [CrossRef]
- Lehrman, E.K.; Wilton, D.K.; Litvina, E.Y.; Welsh, C.A.; Chang, S.T.; Frouin, A.; Walker, A.J.; Heller, M.D.; Umemori, H.; Chen, C.F.; et al. CD47 Protects Synapses from Excess Microglia-Mediated Pruning during Development. Neuron 2018, 100, 120–134. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.D.; Coulthard, L.G.; Woodruff, T.M. Complement dysregulation in the central nervous system during development and disease. Semin. Immunol. 2019, 45, 101340. [Google Scholar] [CrossRef]
- Cooper, J.D.; Ozcan, S.; Gardner, R.M.; Rustogi, N.; Wicks, S.; van Rees, G.F.; Leweke, F.M.; Dalman, C.; Karlsson, H.; Bahn, S. Schizophrenia-risk and urban birth are associated with proteomic changes in neonatal dried blood spots. Transl. Psychiatry 2017, 7, 1290. [Google Scholar] [CrossRef] [Green Version]
- Gallego, J.A.; Blanco, E.A.; Morell, C.; Lencz, T.; Malhotra, A.K. Complement component C4 levels in the cerebrospinal fluid and plasma of patients with schizophrenia. Neuropsychopharmacology 2020, 46. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.C.; Zhou, N.; Liu, R.; Rao, W.W.; Yang, M.J.; Cao, B.N.; Kang, G.J.; Kang, Q.; Zhu, X.J.; Li, R.X.; et al. Association Between Polymorphisms of the Complement 3 Gene and Schizophrenia in a Han Chinese Population. Cell. Physiol. Biochem. 2018, 46, 2480–2486. [Google Scholar] [CrossRef]
- Crider, A.; Feng, T.; Pandya, C.D.; Davis, T.; Nair, A.; Ahmed, A.O.; Baban, B.; Turecki, G.; Pillai, A. Complement component 3a receptor deficiency attenuates chronic stress-induced monocyte infiltration and depressive-like behavior. Brain Behav. Immun. 2018, 70, 246–256. [Google Scholar] [CrossRef]
- Ishii, T.; Hattori, K.; Miyakawa, T.; Watanabe, K.; Hidese, S.; Sasayama, D.; Ota, M.; Teraishi, T.; Hori, H.; Yoshida, S.; et al. Increased cerebrospinal fluid complement C5 levels in major depressive disorder and schizophrenia. Biochem. Biophys. Res. Commun. 2018, 497, 683–688. [Google Scholar] [CrossRef]
- Comer, A.L.; Jinadasa, T.; Sriram, B.; Phadke, R.A.; Kretsge, L.N.; Nguyen, T.P.H.; Antognetti, G.; Gilbert, J.P.; Lee, J.; Newmark, E.R.; et al. Increased expression of schizophrenia-associated gene C4 leads to hypoconnectivity of prefrontal cortex and reduced social interaction. PLoS Biol. 2020, 18, e3000604. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, M.; Yalcin, E.; Presumey, J.; Aw, E.; Ma, M.H.; Whelan, C.W.; Stevens, B.; McCarroll, S.A.; Carroll, M.C. Overexpression of schizophrenia susceptibility factor human complement C4A promotes excessive synaptic loss and behavioral changes in mice. Nat. Neurosci. 2021, 24, 214–224. [Google Scholar] [CrossRef]
- Sellgren, C.M.; Gracias, J.; Watmuff, B.; Biag, J.D.; Thanos, J.M.; Whittredge, P.B.; Fu, T.; Worringer, K.; Brown, H.E.; Wang, J.; et al. Increased synapse elimination by microglia in schizophrenia patient-derived models of synaptic pruning. Nat. Neurosci. 2019, 22, 374–385. [Google Scholar] [CrossRef] [PubMed]
- Davies, C.; Segre, G.; Estrade, A.; Radua, J.; De Micheli, A.; Provenzani, U.; Oliver, D.; de Pablo, G.S.; Ramella-Cravaro, V.; Besozzi, M.; et al. Prenatal and perinatal risk and protective factors for psychosis: A systematic review and meta-analysis. Lancet Psychiatry 2020, 7, 399–410. [Google Scholar] [CrossRef]
- Zimmer, A.; Youngblood, A.; Adnane, A.; Miller, B.J.; Goldsmith, D.R. Prenatal exposure to viral infection and neuropsychiatric disorders in offspring: A review of the literature and recommendations for the COVID-19 pandemic. Brain Behav. Immun. 2021, 91, 756–770. [Google Scholar] [CrossRef]
- Glynn, M.W.; Elmer, B.M.; Garay, P.A.; Liu, X.B.; Needleman, L.A.; El-Sabeawy, F.; McAllister, A.K. MHCI negatively regulates synapse density during the establishment of cortical connections. Nat. Neurosci. 2011, 14, 442–451. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Elmer, B.M.; Estes, M.L.; Barrow, S.L.; McAllister, A.K. MHCI requires MEF2 transcription factors to negatively regulate synapse density during development and in disease. J. Neurosci. 2013, 33, 13791–13804. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Severance, E.G.; Gressitt, K.L.; Buka, S.L.; Cannon, T.D.; Yolken, R.H. Maternal complement C1q and increased odds for psychosis in adult offspring. Schizophr. Res. 2014, 159, 14–19. [Google Scholar] [CrossRef] [Green Version]
- Flegr, J. Effects of toxoplasma on human behavior. Schizophr. Bull. 2007, 33, 757–760. [Google Scholar] [CrossRef] [Green Version]
- Kannan, G.; Pletnikov, M.V. Toxoplasma Gondii and Cognitive Deficits in Schizophrenia: An Animal Model Perspective. Schizophr. Bull. 2012, 38, 1155–1161. [Google Scholar] [CrossRef] [PubMed]
- Xiao, J.C.; Li, Y.; Gressitt, K.L.; He, H.; Kannan, G.; Schultz, T.L.; Svezhova, N.; Carruthers, V.B.; Pletnikov, M.V.; Yolken, R.H.; et al. Cerebral complement C1q activation in chronic Toxoplasma infection. Brain Behav. Immun. 2016, 58, 52–56. [Google Scholar] [CrossRef] [Green Version]
- Li, Y.; Severance, E.G.; Viscidi, R.P.; Yolken, R.H.; Xiao, J. Persistent Toxoplasma Infection of the Brain Induced Neurodegeneration Associated with Activation of Complement and Microglia. Infect. Immun. 2019, 87, e00139-19. [Google Scholar] [CrossRef] [Green Version]
- Xin, Y.R.; Jiang, J.X.; Hu, Y.; Pan, J.P.; Mi, X.N.; Gao, Q.; Xiao, F.; Zhang, W.; Luo, H.M. The Immune System Drives Synapse Loss During Lipopolysaccharide-Induced Learning and Memory Impairment in Mice. Front. Aging Neurosci. 2019, 11, 279. [Google Scholar] [CrossRef]
- Li, S.M.; Li, B.; Zhang, L.; Zhang, G.F.; Sun, J.; Ji, M.H.; Yang, J.J. A complement-microglial axis driving inhibitory synapse related protein loss might contribute to systemic inflammation-induced cognitive impairment. Int. Immunopharmacol. 2020, 87, 106814. [Google Scholar] [CrossRef]
- Sato, C.; Kitajima, K. Disialic, oligosialic and polysialic acids: Distribution, functions and related disease. J. Biochem. 2013, 154, 115–136. [Google Scholar] [CrossRef] [Green Version]
- Barbeau, D.; Liang, J.; Robitalille, Y.; Quirion, R.; Srivastava, L. Decreased expression of the embryonic form of the neural cell adhesion molecule in schizophrenic brains. Proc. Natl. Acad. Sci. USA 1995, 92, 2785–2789. [Google Scholar] [CrossRef] [Green Version]
- Gilabert-Juan, J.; Varea, E.; Guirado, R.; Blasco-Ibanez, J.M.; Crespo, C.; Nacher, J. Alterations in the expression of PSA-NCAM and synaptic proteins in the dorsolateral prefrontal cortex of psychiatric disorder patients. Neurosci. Lett. 2012, 530, 97–102. [Google Scholar] [CrossRef] [PubMed]
- Piras, F.; Schiff, M.; Chiapponi, C.; Bossu, P.; Muhlenhoff, M.; Caltagirone, C.; Gerardy-Schahn, R.; Hildebrandt, H.; Spalletta, G. Brain structure, cognition and negative symptoms in schizophrenia are associated with serum levels of polysialic acid-modified NCAM. Transl. Psychiatry 2015, 5, e658. [Google Scholar] [CrossRef]
- Arai, M.; Yamada, K.; Toyota, T.; Obata, N.; Haga, S.; Yoshida, Y.; Nakamura, K.; Minabe, Y.; Ujike, H.; Sora, I.; et al. Association between polymorphisms in the promoter region of the sialyltransferase 8B (SIAT8B) gene and schizophrenia. Biol. Psychiatry 2006, 59, 652–659. [Google Scholar] [CrossRef]
- Tao, R.; Li, C.; Zheng, Y.L.; Qin, W.; Zhang, J.; Li, X.W.; Xu, Y.F.; Shi, Y.Y.; Feng, G.Y.; He, L. Positive association between SIAT8B and schizophrenia in the Chinese Han population. Schizophr. Res. 2007, 90, 108–114. [Google Scholar] [CrossRef] [PubMed]
- Gilabert-Juan, J.; Nacher, J.; Sanjuan, J.; Molto, M.D. Sex-specific association of the ST8SIAII gene with schizophrenia in a Spanish population. Psychiatry Res. 2013, 210, 1293–1295. [Google Scholar] [CrossRef]
- Angata, K.; Long, J.M.; Bukalo, O.; Lee, W.; Dityatev, A.; Wynshaw-Boris, A.; Schachner, M.; Fukuda, M.; Marth, J.D. Sialyltransferase ST8Sia-II assembles a subset of polysialic acid that directs hippocampal axonal targeting and promotes fear behavior. J. Biol. Chem. 2004, 279, 32603–32613. [Google Scholar] [CrossRef] [Green Version]
- Mori, Y.; Yoshino, Y.; Ochi, S.; Yamazaki, K.; Kawabe, K.; Abe, M.; Kitano, T.; Ozaki, Y.; Yoshida, T.; Numata, S.; et al. TREM2 mRNA Expression in Leukocytes Is Increased in Alzheimer’s Disease and Schizophrenia. PLoS ONE 2015, 10, e0136835. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, Y.; Kawabe, K.; Yamazaki, K.; Watanabe, S.; Numata, S.; Mori, Y.; Yoshida, T.; Iga, J.; Ohmori, T.; Ueno, S.I. Elevated TREM2 mRNA expression in leukocytes in schizophrenia but not major depressive disorder. J. Neural Transm. 2016, 123, 637–641. [Google Scholar] [CrossRef] [PubMed]
- Yoshino, Y.; Ozaki, Y.; Yamazaki, K.; Sao, T.; Mori, Y.; Ochi, S.; Iga, J.; Ueno, S. DNA Methylation Changes in Intron 1 of Triggering Receptor Expressed on Myeloid Cell 2 in Japanese Schizophrenia Subjects. Front. Neurosci. 2017, 11, 275. [Google Scholar] [CrossRef] [Green Version]
- Onwordi, E.C.; Halff, E.F.; Whitehurst, T.; Mansur, A.; Cotel, M.C.; Wells, L.; Creeney, H.; Bonsall, D.; Rogdaki, M.; Shatalina, E.; et al. Synaptic density marker SV2A is reduced in schizophrenia patients and unaffected by antipsychotics in rats. Nat. Commun. 2020, 11, 246. [Google Scholar] [CrossRef] [PubMed]
- Germann, M.; Brederoo, S.G.; Sommer, I.E.C. Abnormal synaptic pruning during adolescence underlying the development of psychotic disorders. Curr. Opin. Psychiatry 2021, 34, 222–227. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.L.; Hu, K.; Shao, T.; Hou, L.; Zhang, S.J.; Ye, W.J.; Josephson, L.; Meyer, J.H.; Zhang, M.R.; Vasdev, N.; et al. Recent developments on PET radiotracers for TSPO and their applications in neuroimaging. Acta Pharm. Sin. B 2021, 11, 373–393. [Google Scholar] [CrossRef] [PubMed]
- Marques, T.R.; Ashok, A.H.; Pillinger, T.; Veronese, M.; Turkheimer, F.E.; Dazzan, P.; Sommer, I.E.C.; Howes, O.D. Neuroinflammation in schizophrenia: Meta-analysis of in vivo microglial imaging studies. Psychol. Med. 2019, 49, 2186–2196. [Google Scholar] [CrossRef] [Green Version]
- Sneeboer, M.A.M.; van der Doef, T.; Litjens, M.; Psy, N.B.B.; Melief, J.; Hol, E.M.; Kahn, R.S.; de Witte, L.D. Microglial activation in schizophrenia: Is translocator 18 kDa protein (TSPO) the right marker? Schizophr. Res. 2020, 215, 167–172. [Google Scholar] [CrossRef]
- Narayanaswami, V.; Dahl, K.; Bernard-Gauthier, V.; Josephson, L.; Cumming, P.; Vasdev, N. Emerging PET Radiotracers and Targets for Imaging of Neuroinflammation in Neurodegenerative Diseases: Outlook Beyond TSPO. Mol. Imaging 2018, 17. [Google Scholar] [CrossRef] [Green Version]
- Karlstrom, S.; Nordvall, G.; Sohn, D.; Hettman, A.; Turek, D.; Ahlin, K.; Kers, A.; Claesson, M.; Slivo, C.; Lo-Alfredsson, Y.; et al. Substituted 7-amino-5-thio-thiazolo[4,5-d]pyrimidines as potent and selective antagonists of the fractalkine receptor (CX3CR1). J. Med. Chem. 2013, 56, 3177–3190. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Yan, X.; Zhao, T.; Xu, Q.; Peng, Q.; Hu, R.; Quan, S.; Zhou, Y.; Xing, G. Targeting C3a/C5a receptors inhibits human mesangial cell proliferation and alleviates immunoglobulin A nephropathy in mice. Clin. Exp. Immunol. 2017, 189, 60–70. [Google Scholar] [CrossRef] [Green Version]
- Laskaris, L.; Zalesky, A.; Weickert, C.S.; Di Biase, M.A.; Chana, G.; Baune, B.T.; Bousman, C.; Nelson, B.; McGorry, P.; Everall, I.; et al. Investigation of peripheral complement factors across stages of psychosis. Schizophr. Res. 2019, 204, 30–37. [Google Scholar] [CrossRef]
- Mongan, D.; Sabherwal, S.; Susai, S.R.; Focking, M.; Cannon, M.; Cotter, D.R. Peripheral complement proteins in schizophrenia: A systematic review and meta-analysis of serological studies. Schizophr. Res. 2020, 222, 58–72. [Google Scholar] [CrossRef]
- Zhang, T.; Tang, Y.; Yang, X.; Wang, X.; Ding, S.; Huang, K.; Liu, Y.; Lang, B. Expression of GSK3β, PICK1, NEFL, C4, NKCC1 and Synaptophysin in peripheral blood mononuclear cells of the first-episode schizophrenia patients. Asian J. Psychiatry 2021, 55, 102520. [Google Scholar] [CrossRef]
- Hill, A.F. Extracellular Vesicles and Neurodegenerative Diseases. J. Neurosci. 2019, 39, 9269–9273. [Google Scholar] [CrossRef]
- Vassileff, N.; Cheng, L.; Hill, A.F. Extracellular vesicles—Propagators of neuropathology and sources of potential biomarkers and therapeutics for neurodegenerative diseases. J. Cell Sci. 2020, 133, jcs243139. [Google Scholar] [CrossRef] [PubMed]
- Fiandaca, M.S.; Kapogiannis, D.; Mapstone, M.; Boxer, A.; Eitan, E.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Federoff, H.J.; Miller, B.L.; et al. Identification of preclinical Alzheimer’s disease by a profile of pathogenic proteins in neurally derived blood exosomes: A case-control study. Alzheimers Dement. 2015, 11, 600–607. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goetzl, E.J.; Boxer, A.; Schwartz, J.B.; Abner, E.L.; Petersen, R.C.; Miller, B.L.; Kapogiannis, D. Altered lysosomal proteins in neural-derived plasma exosomes in preclinical Alzheimer disease. Neurology 2015, 85, 40–47. [Google Scholar] [CrossRef] [Green Version]
- Goetzl, E.J.; Srihari, V.H.; Guloksuz, S.; Ferrara, M.; Tek, C.; Heninger, G.R. Neural cell-derived plasma exosome protein abnormalities implicate mitochondrial impairment in first episodes of psychosis. FASEB J. 2021, 35, e21339. [Google Scholar] [CrossRef] [PubMed]
- Goetzl, E.J.; Srihari, V.H.; Guloksuz, S.; Ferrara, M.; Tek, C.; Heninger, G.R. Decreased mitochondrial electron transport proteins and increased complement mediators in plasma neural-derived exosomes of early psychosis. Transl. Psychiatry 2020, 10, 361. [Google Scholar] [CrossRef]
- Kawata, K.; Mitsuhashi, M.; Aldret, R. A Preliminary Report on Brain-Derived Extracellular Vesicle as Novel Blood Biomarkers for Sport-Related Concussions. Front. Neurol. 2018, 9, 239. [Google Scholar] [CrossRef] [PubMed]
- Bennett, M.L.; Bennett, F.C.; Liddelow, S.A.; Ajami, B.; Zamanian, J.L.; Fernhoff, N.B.; Mulinyawe, S.B.; Bohlen, C.J.; Adil, A.; Tucker, A.; et al. New tools for studying microglia in the mouse and human CNS. Proc. Natl. Acad. Sci. USA 2016, 113, E1738–E1746. [Google Scholar] [CrossRef] [Green Version]
- Miyanohara, J.; Kakae, M.; Nagayasu, K.; Nakagawa, T.; Mori, Y.; Arai, K.; Shirakawa, H.; Kaneko, S. TRPM2 Channel Aggravates CNS Inflammation and Cognitive Impairment via Activation of Microglia in Chronic Cerebral Hypoperfusion. J. Neurosci. 2018, 38, 3520–3533. [Google Scholar] [CrossRef]
- Shin, D.A.; Kim, T.U.; Chang, M.C. Minocycline for Controlling Neuropathic Pain: A Systematic Narrative Review of Studies in Humans. J. Pain Res. 2021, 14, 139–145. [Google Scholar] [CrossRef]
- Mizoguchi, H.; Takuma, K.; Fukakusa, A.; Ito, Y.; Nakatani, A.; Ibi, D.; Kim, H.C.; Yamada, K. Improvement by minocycline of methamphetamine-induced impairment of recognition memory in mice. Psychopharmacology 2008, 196, 233–241. [Google Scholar] [CrossRef]
- Zhu, F.; Liu, Y.; Zhao, J.; Zheng, Y. Minocycline alleviates behavioral deficits and inhibits microglial activation induced by intrahippocampal administration of Granulocyte-Macrophage Colony-Stimulating Factor in adult rats. Neuroscience 2014, 266, 275–281. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.L.; Sullivan, K.M.; McEvoy, J.P.; McMahon, R.P.; Wehring, H.J.; Gold, J.M.; Liu, F.; Warfel, D.; Vyas, G.; Richardson, C.M.; et al. Adjunctive Minocycline in Clozapine-Treated Schizophrenia Patients With Persistent Symptoms. J. Clin. Psychopharmacol. 2015, 35, 374–381. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.L.; Zheng, H.B.; Wu, R.R.; Kosten, T.R.; Zhang, X.Y.; Zhao, J.P. The effect of minocycline on amelioration of cognitive deficits and pro-inflammatory cytokines levels in patients with schizophrenia. Schizophr. Res. 2019, 212, 92–98. [Google Scholar] [CrossRef]
- Dominic, M.R. Adverse Reactions Induced by Minocycline: A Review of Literature. Curr. Drug Saf. 2021, 16. [Google Scholar] [CrossRef] [PubMed]
- Fan, C.Q.; Long, Y.F.; Wang, L.Y.; Liu, X.H.; Liu, Z.C.; Lan, T.; Li, Y.; Yu, S.Y. N-Acetylcysteine Rescues Hippocampal Oxidative Stress-Induced Neuronal Injury via Suppression of p38/JNK Signaling in Depressed Rats. Front. Cell. Neurosci. 2020, 14, 554613. [Google Scholar] [CrossRef]
- Steullet, P.; Cabungcal, J.H.; Monin, A.; Dwir, D.; O’Donnell, P.; Cuenod, M.; Do, K.Q. Redox dysregulation, neuroinflammation, and NMDA receptor hypofunction: A "central hub" in schizophrenia pathophysiology? Schizophr. Res. 2016, 176, 41–51. [Google Scholar] [CrossRef] [Green Version]
- Smaga, I.; Frankowska, M.; Filip, M. N-acetylcysteine as a new prominent approach for treating psychiatric disorders. Br. J. Pharmacol. 2021. [Google Scholar] [CrossRef]
- Hillmen, P.; Hall, C.; Marsh, J.C.W.; Elebute, M.; Bombara, M.P.; Petro, B.E.; Cullen, M.J.; Richards, S.J.; Rollins, S.A.; Mojcik, C.F.; et al. Effect of eculizumab on hemolysis and transfusion requirements in patients with paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 2004, 350, 552–559. [Google Scholar] [CrossRef] [Green Version]
- Hillmen, P.; Young, N.S.; Schubert, J.; Brodsky, R.A.; Socie, G.; Muus, P.; Roth, A.; Szer, J.; Elebute, M.O.; Nakamura, R.; et al. The complement inhibitor eculizumab in paroxysmal nocturnal hemoglobinuria. N. Engl. J. Med. 2006, 355, 1233–1243. [Google Scholar] [CrossRef]
- Notaro, R.; Sica, M. C3-mediated extravascular hemolysis in PNH on eculizumab: Mechanism and clinical implications. Semin. Hematol. 2018, 55, 130–135. [Google Scholar] [CrossRef]
- De Castro, C.; Grossi, F.; Weitz, I.C.; Maciejewski, J.; Sharma, V.; Roman, E.; Brodsky, R.A.; Tan, L.; Casoli, C.D.; Mehdi, D.E.; et al. C. C3 inhibition with pegcetacoplan in subjects with paroxysmal nocturnal hemoglobinuria treated with eculizumab. Am. J. Hematol. 2020, 95, 1334–1343. [Google Scholar] [CrossRef]
- Tanaka, Y.; Takeuchi, T.; Umehara, H.; Nanki, T.; Yasuda, N.; Tago, F.; Kawakubo, M.; Kitahara, Y.; Hojo, S.; Kawano, T.; et al. Safety, pharmacokinetics, and efficacy of E6011, an antifractalkine monoclonal antibody, in a first-in-patient phase 1/2 study on rheumatoid arthritis. Mod. Rheumatol. 2018, 28, 58–65. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Hoshino-Negishi, K.; Kuboi, Y.; Tago, F.; Yasuda, N.; Imai, T. Emerging Role of Fractalkine in the Treatment of Rheumatic Diseases. Immunotargets Ther. 2020, 9, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Takeuchi, T.; Yamanaka, H.; Nanki, T.; Umehara, H.; Yasuda, N.; Tago, F.; Kitahara, Y.; Kawakubo, M.; Torii, K.; et al. Efficacy and Safety of E6011, an Anti-Fractalkine Monoclonal Antibody, in Patients With Active Rheumatoid Arthritis With Inadequate Response to Methotrexate: Results of a Randomized, Double-Blind, Placebo-Controlled Phase II Study. Arthritis Rheumatol. 2021, 73, 587–595. [Google Scholar] [CrossRef] [PubMed]
- Matsuoka, K.; Naganuma, M.; Hibi, T.; Tsubouchi, H.; Oketani, K.; Katsurabara, T.; Hojo, S.; Takenaka, O.; Kawano, T.; Imai, T.; et al. Phase 1 study on the safety and efficacy of E6011, antifractalkine antibody, in patients with Crohn’s disease. J. Gastroenterol. Hepatol. 2021. [Google Scholar] [CrossRef]
- Yates, D. Stop, don’t prune me! Nat. Rev. Neurosci. 2018, 19, 712–713. [Google Scholar] [CrossRef]
- Butler, C.A.; Popescu, A.; Kitchener, E.; Allendorf, D.H.; Puigdellívol, M.; Brown, G.C. Microglial phagocytosis of neurons in neurodegeneration, and its regulation. J. Neurochem. 2021. [Google Scholar] [CrossRef]
- Koshimizu, H.; Takao, K.; Matozaki, T.; Ohnishi, H.; Miyakawa, T. Comprehensive behavioral analysis of cluster of differentiation 47 knockout mice. PLoS ONE 2014, 9, e89584. [Google Scholar] [CrossRef] [Green Version]
- Ding, X.; Wang, J.; Huang, M.; Chen, Z.; Liu, J.; Zhang, Q.; Zhang, C.; Xiang, Y.; Zen, K.; Li, L. Loss of microglial SIRPα promotes synaptic pruning in preclinical models of neurodegeneration. Nat. Commun. 2021, 12, 2030. [Google Scholar] [CrossRef] [PubMed]
- Reissmann, S.; Filatova, M.P. New generation of cell-penetrating peptides: Functionality and potential clinical application. J. Pept. Sci. 2021, 27, e3300. [Google Scholar] [CrossRef]
- Tashima, T. Smart Strategies for Therapeutic Agent Delivery into Brain across the Blood-Brain Barrier Using Receptor-Mediated Transcytosis. Chem. Pharm. Bull. 2020, 68, 316–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mignani, S.; Shi, X.Y.; Karpus, A.; Majoral, J.P. Non-invasive intranasal administration route directly to the brain using dendrimer nanoplatforms: An opportunity to develop new CNS drugs. Eur. J. Med. Chem. 2021, 209, 112905. [Google Scholar] [CrossRef]
- Hallschmid, M. Intranasal Insulin for Alzheimer’s Disease. CNS Drugs 2021, 35, 21–37. [Google Scholar] [CrossRef] [PubMed]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Izuo, N.; Nitta, A. New Insights Regarding Diagnosis and Medication for Schizophrenia Based on Neuronal Synapse–Microglia Interaction. J. Pers. Med. 2021, 11, 371. https://doi.org/10.3390/jpm11050371
Izuo N, Nitta A. New Insights Regarding Diagnosis and Medication for Schizophrenia Based on Neuronal Synapse–Microglia Interaction. Journal of Personalized Medicine. 2021; 11(5):371. https://doi.org/10.3390/jpm11050371
Chicago/Turabian StyleIzuo, Naotaka, and Atsumi Nitta. 2021. "New Insights Regarding Diagnosis and Medication for Schizophrenia Based on Neuronal Synapse–Microglia Interaction" Journal of Personalized Medicine 11, no. 5: 371. https://doi.org/10.3390/jpm11050371
APA StyleIzuo, N., & Nitta, A. (2021). New Insights Regarding Diagnosis and Medication for Schizophrenia Based on Neuronal Synapse–Microglia Interaction. Journal of Personalized Medicine, 11(5), 371. https://doi.org/10.3390/jpm11050371