
A Proteomic Atlas of Lineage and Cancer-Polarized Expression Modules in Myeloid Cells Modeling Immunosuppressive Tumor-Infiltrating Subsets

 , , , , ,
, , , , ,  , ,
, ,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cells and Mice
2.2. In Vitro Differentiation and Purification of Myeloid-Derived Suppressor Cells, Tumor-Associated Macrophages and Non-Polarized Macrophages
2.3. Cell Staining and Flow Cytometry
2.4. Western Blotting
2.5. Mass Spectrometry-Based Quantitative (Shotgun) Proteomics and Bioinformatics Analyses
2.6. Proteomic Data Analysis
2.7. Statistical Analyses
3. Results
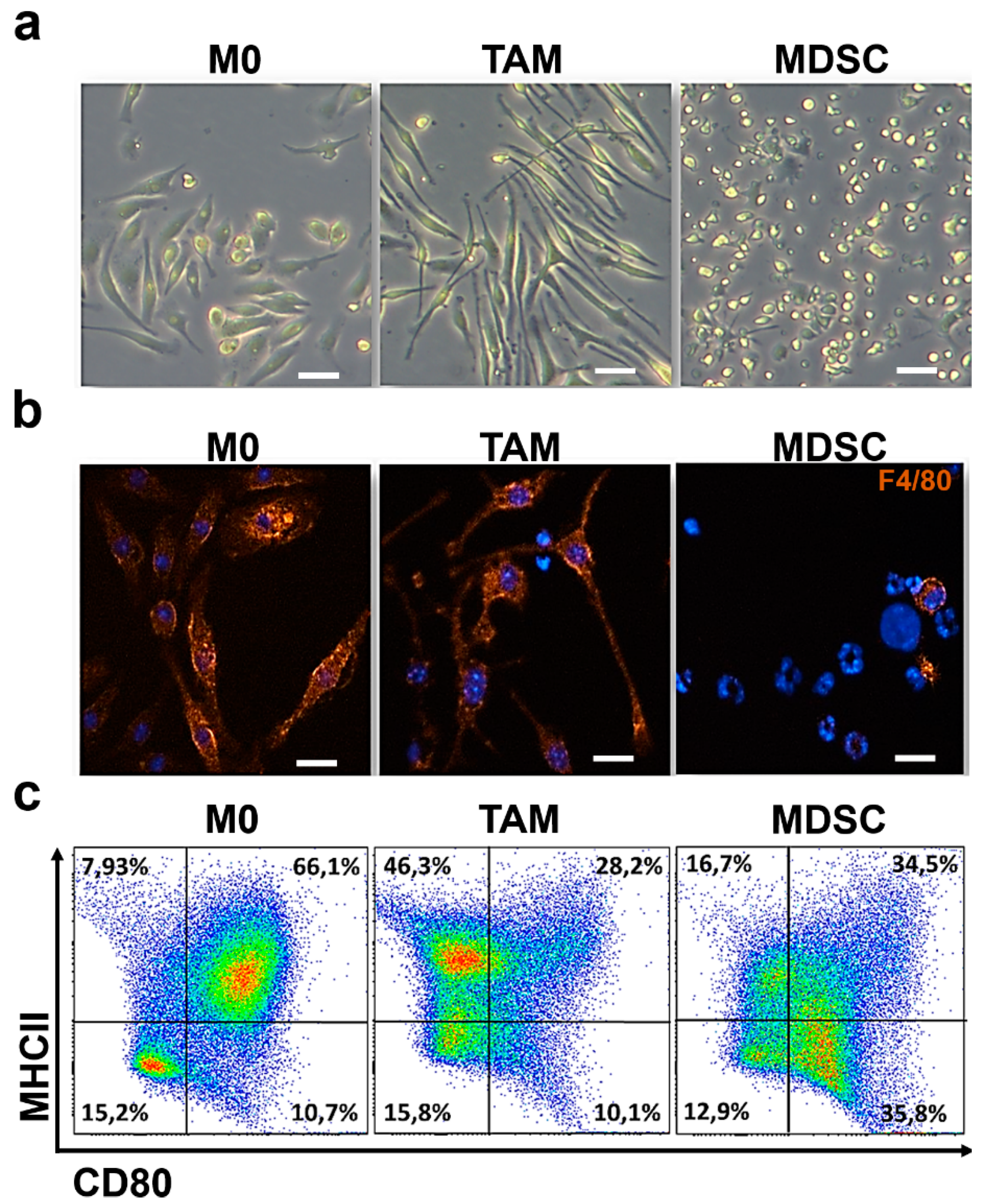
3.1. Ex Vivo Differentiation of Myeloid Cells Modeling Regulatory Subpopulations
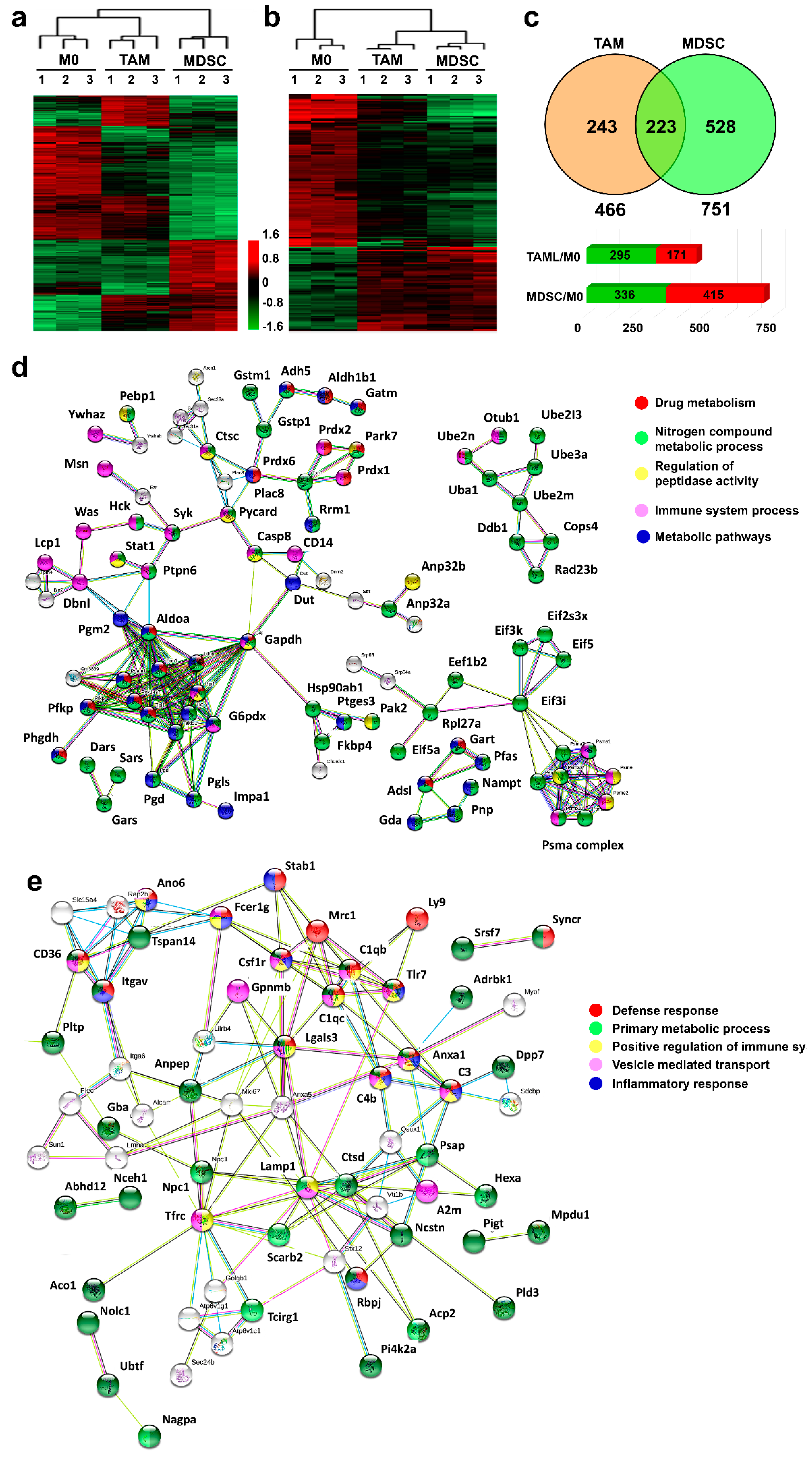
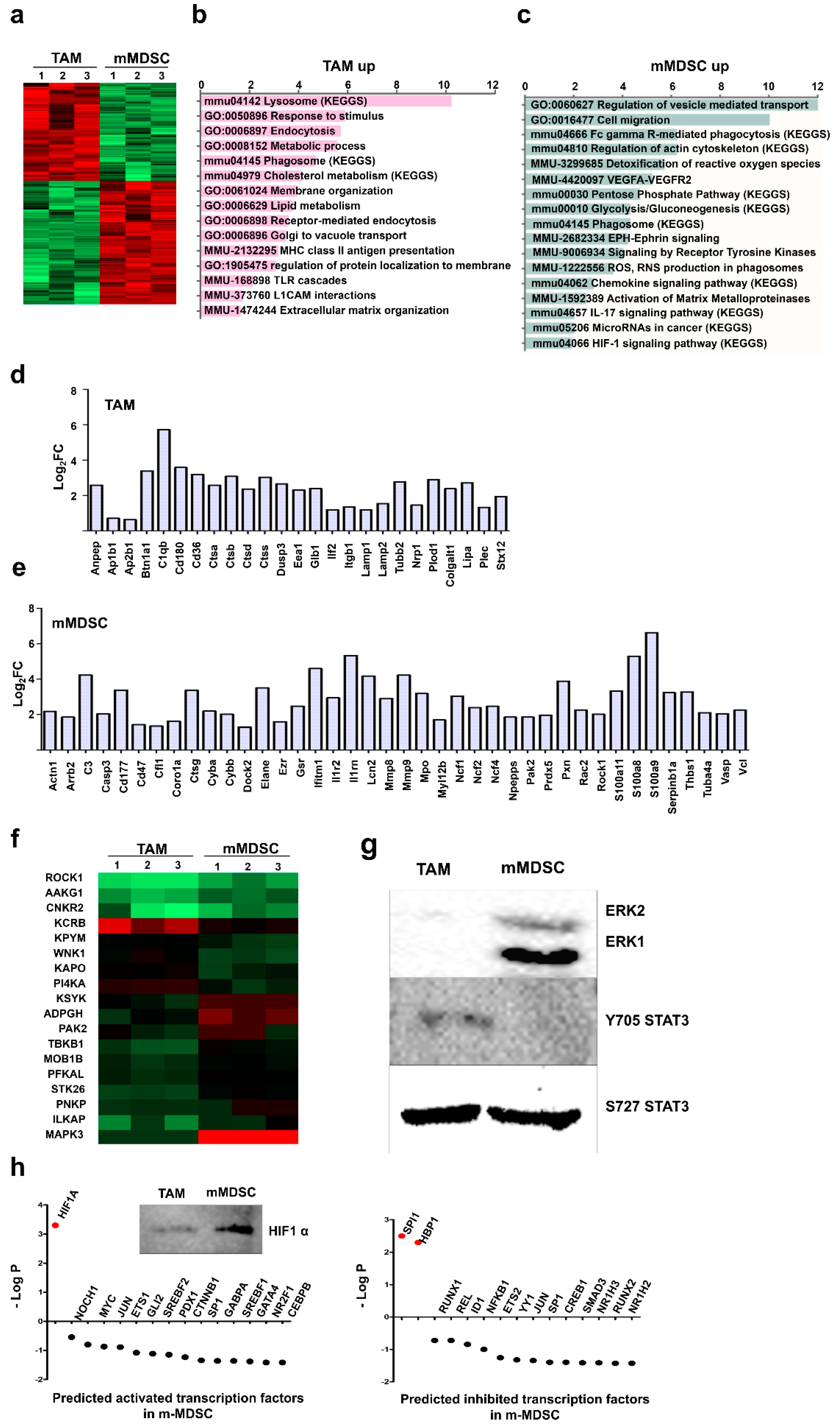
3.2. Proteome Profiles of Ex Vivo-Differentiated Myeloid Subsets Separate Lineage-Regulated from Tumor-Polarized Interactome Modules
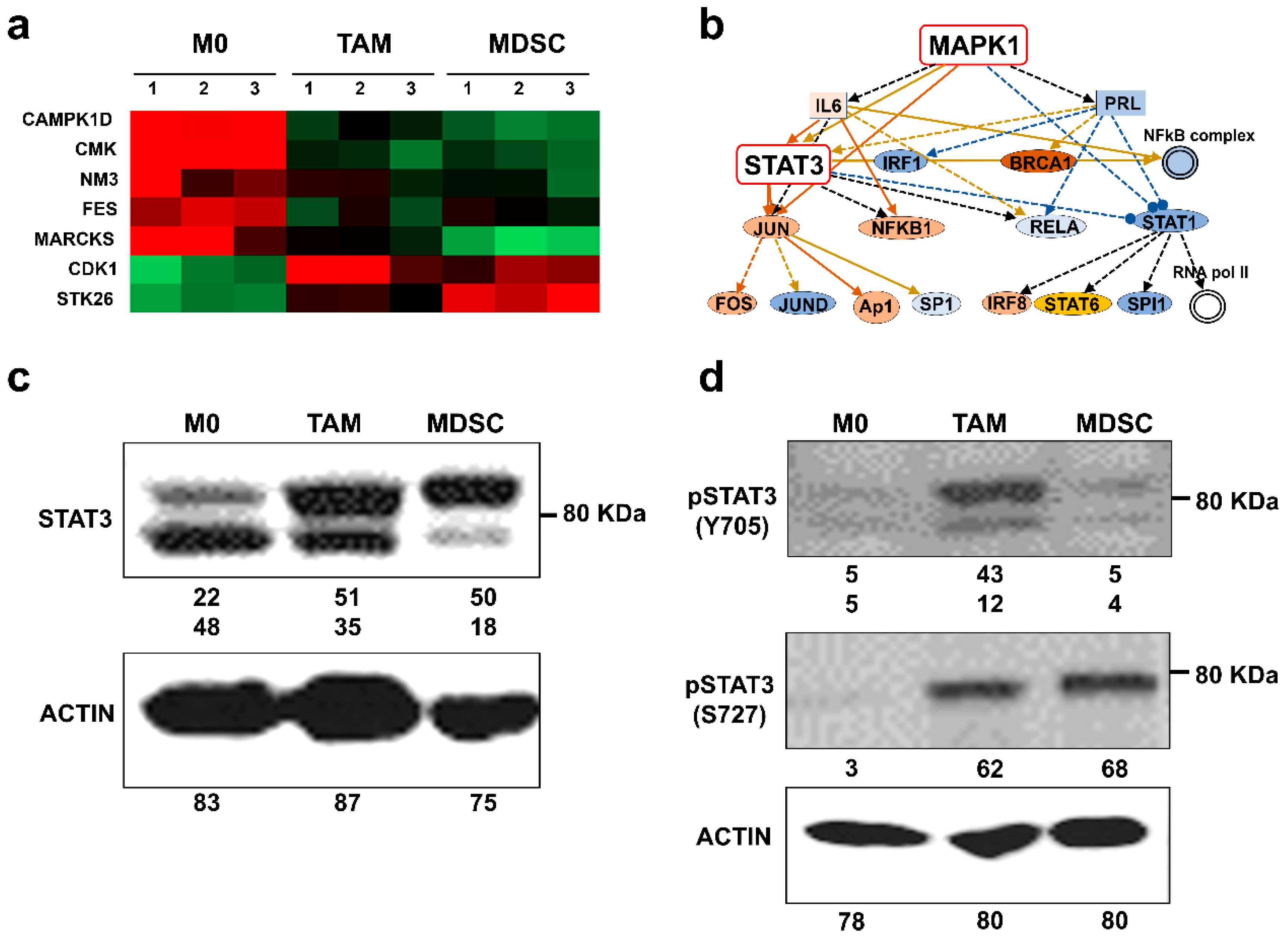
3.3. Kinase Profiles Associated to Cancer-Polarized Immunosuppressive Myeloid Cells
3.4. Unique Protein Expression Profiles Discriminate Ex Vivo-Differentiated Monocytic MDSCs from TAMs
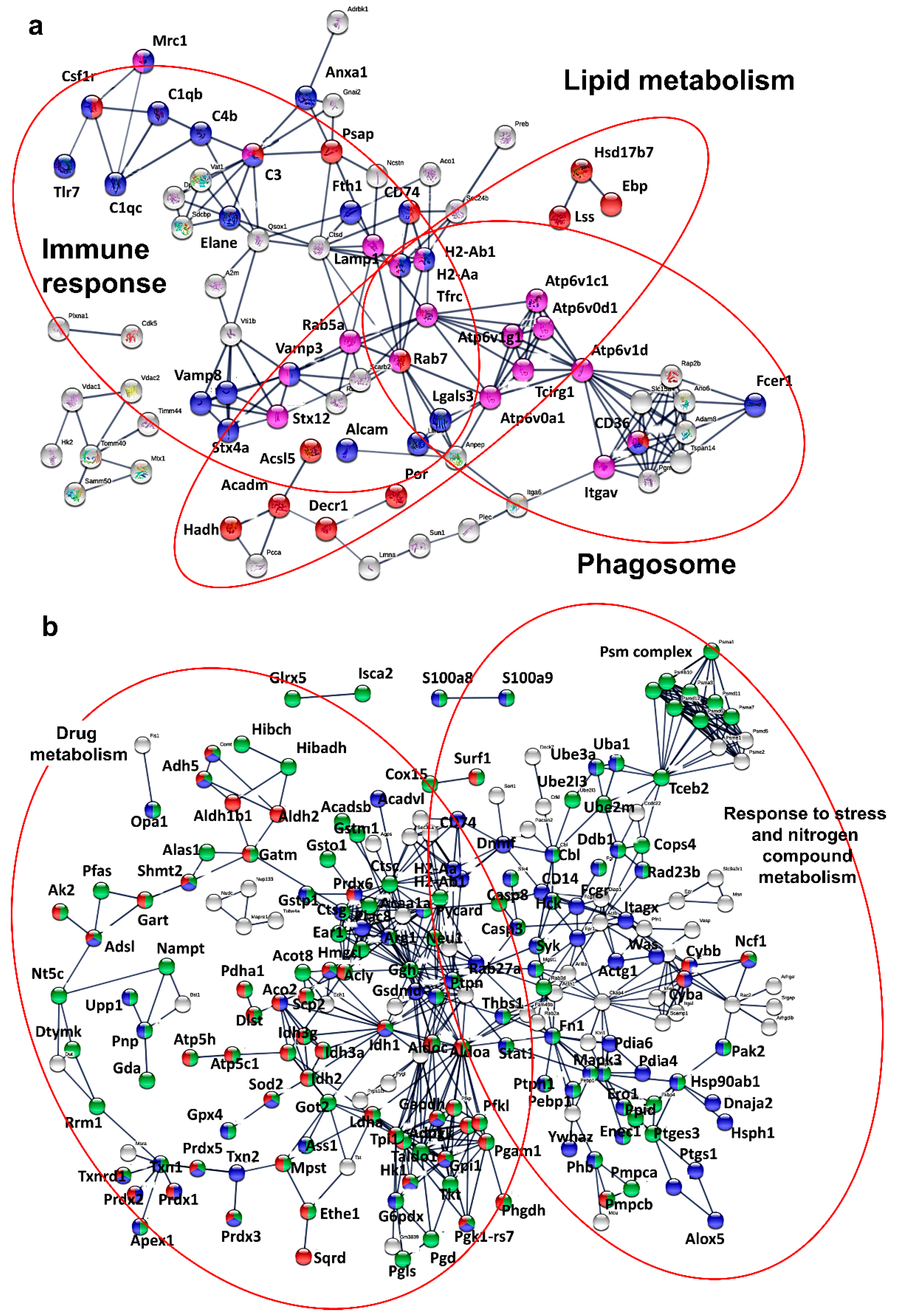
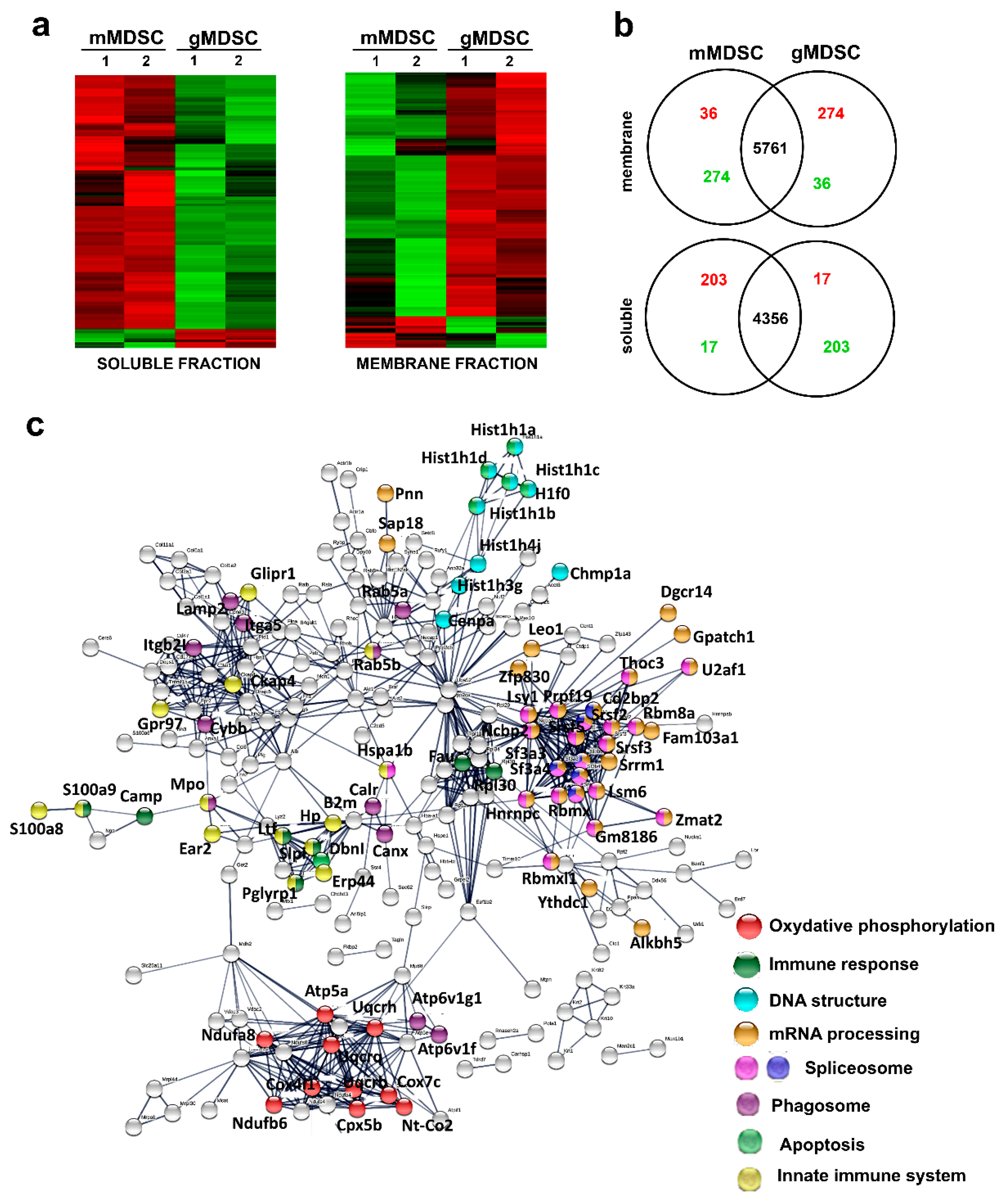
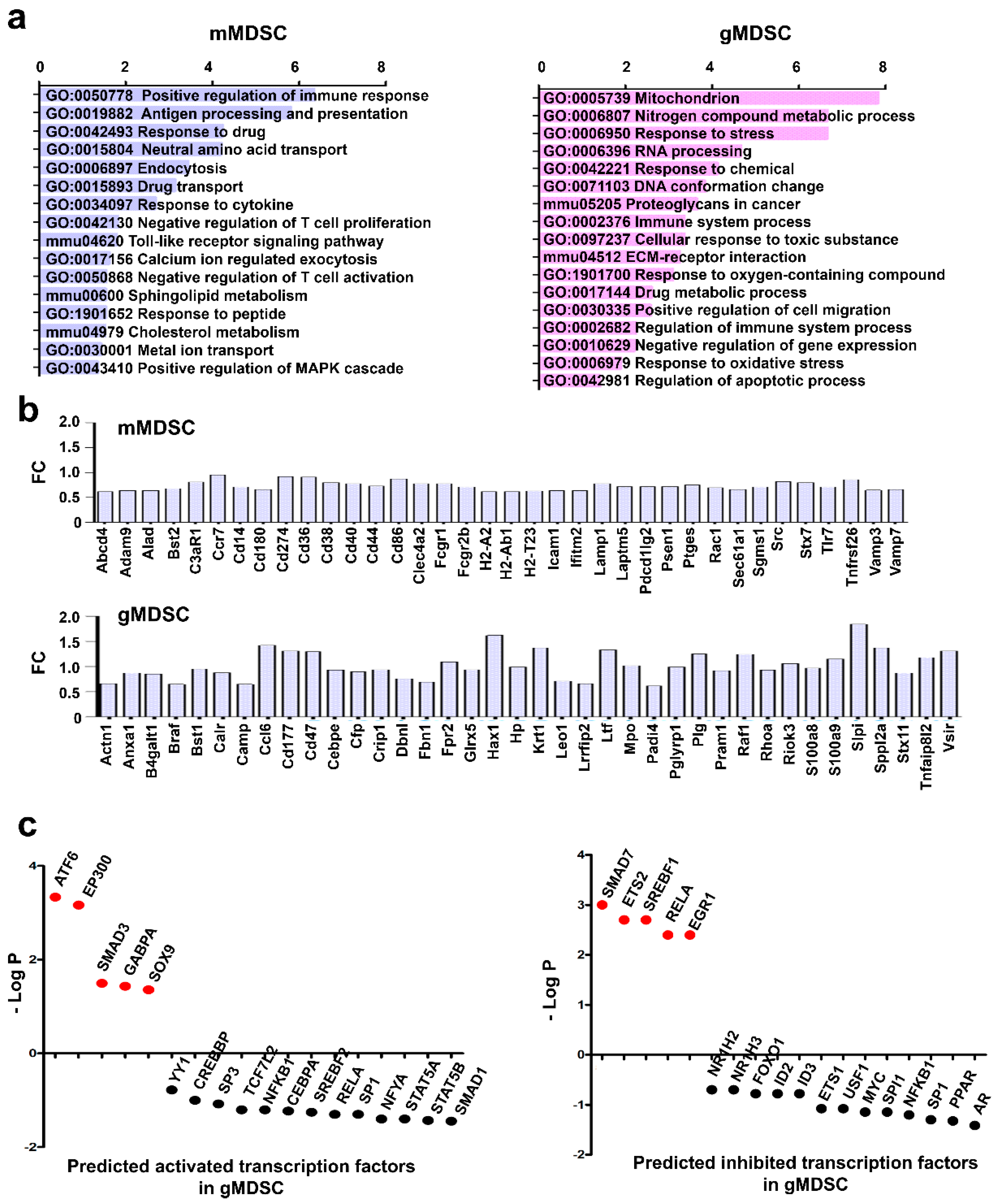
3.5. Key Nuclear Differential Pathways and Major Metabolic Routes Separate Monocytic from Granulocytic MDSCs
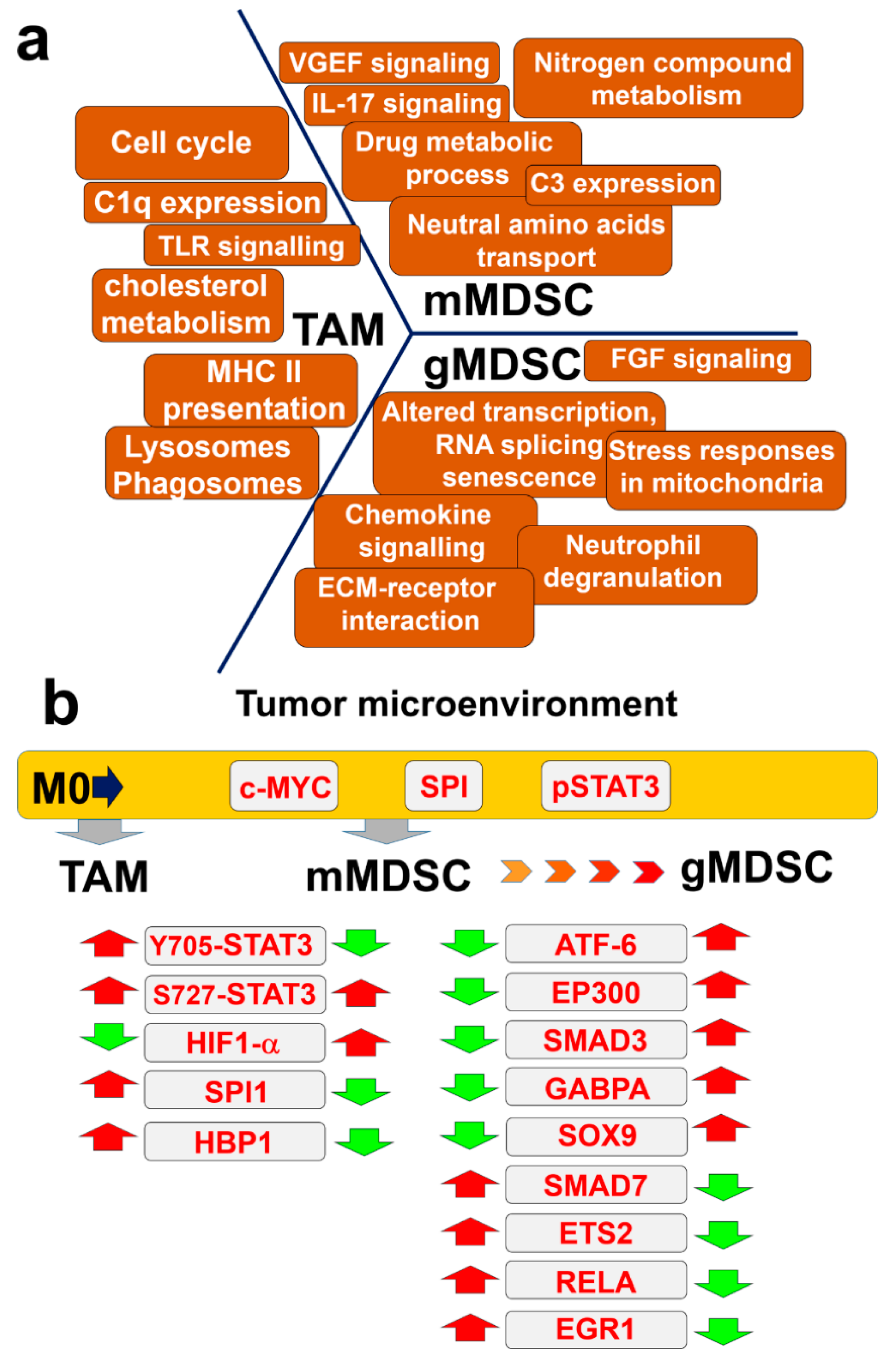
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Conflicts of Interest
References
- Chang, C.H.; Qiu, J.; O’Sullivan, D.; Buck, M.D.; Noguchi, T.; Curtis, J.D.; Chen, Q.; Gindin, M.; Gubin, M.M.; van der Windt, G.J.; et al. Metabolic Competition in the Tumor Microenvironment Is a Driver of Cancer Progression. Cell 2015, 162, 1229–1241. [Google Scholar] [CrossRef] [Green Version]
- Bronte, V.; Brandau, S.; Chen, S.H.; Colombo, M.P.; Frey, A.B.; Greten, T.F.; Mandruzzato, S.; Murray, P.J.; Ochoa, A.; Ostrand-Rosenberg, S.; et al. Recommendations for myeloid-derived suppressor cell nomenclature and characterization standards. Nat. Commun. 2016, 7, 12150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibanez-Vea, M.; Huang, H.; Martinez de Morentin, X.; Perez, E.; Gato, M.; Zuazo, M.; Arasanz, H.; Fernandez-Irigoyen, J.; Santamaria, E.; Fernandez-Hinojal, G.; et al. Characterization of Macrophage Endogenous S-Nitrosoproteome Using a Cysteine-Specific Phosphonate Adaptable Tag in Combination with TiO2 Chromatography. J. Proteome Res. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arce, F.; Breckpot, K.; Stephenson, H.; Karwacz, K.; Ehrenstein, M.R.; Collins, M.; Escors, D. Selective ERK activation differentiates mouse and human tolerogenic dendritic cells, expands antigen-specific regulatory T cells, and suppresses experimental inflammatory arthritis. Arthritis Rheum. 2011, 63, 84–95. [Google Scholar] [CrossRef]
- Liechtenstein, T.; Perez-Janices, N.; Gato, M.; Caliendo, F.; Kochan, G.; Blanco-Luquin, I.; Van der Jeught, K.; Arce, F.; Guerrero-Setas, D.; Fernandez-Irigoyen, J.; et al. A highly efficient tumor-infiltrating MDSC differentiation system for discovery of anti-neoplastic targets, which circumvents the need for tumor establishment in mice. Oncotarget 2014, 5, 7843–7857. [Google Scholar] [CrossRef] [Green Version]
- Veglia, F.; Perego, M.; Gabrilovich, D. Myeloid-derived suppressor cells coming of age. Nat. Immunol. 2018, 19, 108–119. [Google Scholar] [CrossRef]
- Bocanegra, A.; Fernandez-Hinojal, G.; Zuazo-Ibarra, M.; Arasanz, H.; Garcia-Granda, M.J.; Hernandez, C.; Ibanez, M.; Hernandez-Marin, B.; Martinez-Aguillo, M.; Lecumberri, M.J.; et al. PD-L1 Expression in Systemic Immune Cell Populations as a Potential Predictive Biomarker of Responses to PD-L1/PD-1 Blockade Therapy in Lung Cancer. Int. J. Mol. Sci. 2019, 20, 1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ibanez-Vea, M.; Zuazo, M.; Gato, M.; Arasanz, H.; Fernandez-Hinojal, G.; Escors, D.; Kochan, G. Myeloid-Derived Suppressor Cells in the Tumor Microenvironment: Current Knowledge and Future Perspectives. Arch. Immunol. Ther. Exp. 2017. [Google Scholar] [CrossRef] [PubMed]
- Friedman, A.D. C/EBPalpha in normal and malignant myelopoiesis. Trans. Int. J. Hematol. 2015, 101, 330–341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corzo, C.A.; Condamine, T.; Lu, L.; Cotter, M.J.; Youn, J.I.; Cheng, P.; Cho, H.I.; Celis, E.; Quiceno, D.G.; Padhya, T.; et al. HIF-1alpha regulates function and differentiation of myeloid-derived suppressor cells in the tumor microenvironment. J. Exp. Med. 2010, 207, 2439–2453. [Google Scholar] [CrossRef]
- Sica, A.; Schioppa, T.; Mantovani, A.; Allavena, P. Tumour-associated macrophages are a distinct M2 polarised population promoting tumour progression: Potential targets of anti-cancer therapy. Eur. J. Cancer 2006, 42, 717–727. [Google Scholar] [CrossRef] [PubMed]
- Tcyganov, E.; Mastio, J.; Chen, E.; Gabrilovich, D.I. Plasticity of myeloid-derived suppressor cells in cancer. Curr. Opin. Immunol. 2018, 51, 76–82. [Google Scholar] [CrossRef]
- Kumar, V.; Cheng, P.; Condamine, T.; Mony, S.; Languino, L.R.; McCaffrey, J.C.; Hockstein, N.; Guarino, M.; Masters, G.; Penman, E.; et al. CD45 Phosphatase Inhibits STAT3 Transcription Factor Activity in Myeloid Cells and Promotes Tumor-Associated Macrophage Differentiation. Immunity 2016, 44, 303–315. [Google Scholar] [CrossRef] [Green Version]
- Escors, D.; Lopes, L.; Lin, R.; Hiscott, J.; Akira, S.; Davis, R.J.; Collins, M.K. Targeting dendritic cell signalling to regulate the response to immunisation. Blood 2008, 111, 3050–3061. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gato-Canas, M.; Martinez de Morentin, X.; Blanco-Luquin, I.; Fernandez-Irigoyen, J.; Zudaire, I.; Liechtenstein, T.; Arasanz, H.; Lozano, T.; Casares, N.; Chaikuad, A.; et al. A core of kinase-regulated interactomes defines the neoplastic MDSC lineage. Oncotarget 2015, 6, 27160–27175. [Google Scholar] [CrossRef] [PubMed]
- Gato-Canas, M.; Zuazo, M.; Arasanz, H.; Ibanez-Vea, M.; Lorenzo, L.; Fernandez-Hinojal, G.; Vera, R.; Smerdou, C.; Martisova, E.; Arozarena, I.; et al. PDL1 Signals through Conserved Sequence Motifs to Overcome Interferon-Mediated Cytotoxicity. Cell Rep. 2017, 20, 1818–1829. [Google Scholar] [CrossRef] [Green Version]
- Zuazo, M.; Arasanz, H.; Fernandez-Hinojal, G.; Garcia-Granda, M.J.; Gato, M.; Bocanegra, A.; Martinez, M.; Hernandez, B.; Teijeira, L.; Morilla, I.; et al. Functional systemic CD4 immunity is required for clinical responses to PD-L1/PD-1 blockade therapy. EMBO Mol. Med. 2019, 11, e10293. [Google Scholar] [CrossRef]
- Shevchenko, A.; Tomas, H.; Havlis, J.; Olsen, J.V.; Mann, M. In-gel digestion for mass spectrometric characterization of proteins and proteomes. Nat. Protoc. 2006, 1, 2856–2860. [Google Scholar] [CrossRef]
- Lachen-Montes, M.; Gonzalez-Morales, A.; Zelaya, M.V.; Perez-Valderrama, E.; Ausin, K.; Ferrer, I.; Fernandez-Irigoyen, J.; Santamaria, E. Olfactory bulb neuroproteomics reveals a chronological perturbation of survival routes and a disruption of prohibitin complex during Alzheimer’s disease progression. Sci. Rep. 2017, 7, 9115. [Google Scholar] [CrossRef] [PubMed]
- Tyanova, S.; Temu, T.; Sinitcyn, P.; Carlson, A.; Hein, M.Y.; Geiger, T.; Mann, M.; Cox, J. The Perseus computational platform for comprehensive analysis of (prote)omics data. Nat. Methods 2016, 13, 731–740. [Google Scholar] [CrossRef]
- Ibáñez-Vea, M.; Kempf, S.J.; Larsen, M.R. Characterization of the Phosphoproteome and Sialoproteome in Brain Tissues by Mass Spectrometry. In Current Proteomic Approaches Applied to Brain Function; Humana Press: New York, NY, USA, 2017. [Google Scholar] [CrossRef]
- Engholm-Keller, K.; Birck, P.; Storling, J.; Pociot, F.; Mandrup-Poulsen, T.; Larsen, M.R. TiSH—A robust and sensitive global phosphoproteomics strategy employing a combination of TiO2, SIMAC, and HILIC. J. Proteom. 2012, 75, 5749–5761. [Google Scholar] [CrossRef]
- Liechtenstein, T.; Perez-Janices, N.; Blanco-Luquin, I.; Schwarze, J.; Dufait, I.; Lanna, A.; De Ridder, M.; Guerrero-Setas, D.; Breckpot, K.; Escors, D. Anti-melanoma vaccines engineered to simultaneously modulate cytokine priming and silence PD-L1 characterized using ex vivo myeloid-derived suppressor cells as a readout of therapeutic efficacy. Oncoimmunology 2014, 3, e29178. [Google Scholar] [CrossRef] [Green Version]
- Karwacz, K.; Bricogne, C.; Macdonald, D.; Arce, F.; Bennett, C.L.; Collins, M.; Escors, D. PD-L1 co-stimulation contributes to ligand-induced T cell receptor down-modulation on CD8(+) T cells. EMBO Mol. Med. 2011, 3, 581–592. [Google Scholar] [CrossRef]
- Gato, M.; Blanco-Luquin, I.; Zudaire, M.; de Morentin, X.M.; Perez-Valderrama, E.; Zabaleta, A.; Kochan, G.; Escors, D.; Fernandez-Irigoyen, J.; Santamaria, E. Drafting the proteome landscape of myeloid-derived suppressor cells. Proteomics 2016, 16, 367–378. [Google Scholar] [CrossRef] [PubMed]
- Dufait, I.; Pardo, J.; Escors, D.; De Vlaeminck, Y.; Jiang, H.; Keyaerts, M.; De Ridder, M.; Breckpot, K. Perforin and Granzyme B Expressed by Murine Myeloid-Derived Suppressor Cells: A Study on Their Role in Outgrowth of Cancer Cells. Cancers 2019, 11, 808. [Google Scholar] [CrossRef] [Green Version]
- Dufait, I.; Schwarze, J.K.; Liechtenstein, T.; Leonard, W.; Jiang, H.; Law, K.; Verovski, V.; Escors, D.; De Ridder, M.; Breckpot, K. Ex vivo generation of myeloid-derived suppressor cells that model the tumor immunosuppressive environment in colorectal cancer. Oncotarget 2015, 6, 12369–12382. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Combes, F.; Mc Cafferty, S.; Meyer, E.; Sanders, N.N. Off-Target and Tumor-Specific Accumulation of Monocytes, Macrophages and Myeloid-Derived Suppressor Cells after Systemic Injection. Neoplasia 2018, 20, 848–856. [Google Scholar] [CrossRef] [PubMed]
- Merino, M.; Lozano, T.; Casares, N.; Lana, H.; Troconiz, I.F.; Ten Hagen, T.L.M.; Kochan, G.; Berraondo, P.; Zalba, S.; Garrido, M.J. Dual activity of PD-L1 targeted Doxorubicin immunoliposomes promoted an enhanced efficacy of the antitumor immune response in melanoma murine model. J. Nanobiotechnol. 2021, 19, 102. [Google Scholar] [CrossRef] [PubMed]
- Sica, A.; Strauss, L. Energy metabolism drives myeloid-derived suppressor cell differentiation and functions in pathology. J. Leukoc. Biol. 2017, 102, 325–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aliper, A.M.; Frieden-Korovkina, V.P.; Buzdin, A.; Roumiantsev, S.A.; Zhavoronkov, A. Interactome analysis of myeloid-derived suppressor cells in murine models of colon and breast cancer. Oncotarget 2014, 5, 11345–11353. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, G.; Yan, H.; Ye, S.; Tong, C.; Ying, Q.L. STAT3 phosphorylation at tyrosine 705 and serine 727 differentially regulates mouse ESC fates. Stem Cells 2014, 32, 1149–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brodsky, I.E. JAK-ing into M1/M2 Polarization SteErs Salmonella-Containing Macrophages Away from Immune Attack to Promote Bacterial Persistence. Cell Host Microbe 2020, 27, 3–5. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Mouton, A.J.; Lindsey, M.L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 2018, 191, 15–28. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giurisato, E.; Xu, Q.; Lonardi, S.; Telfer, B.; Russo, I.; Pearson, A.; Finegan, K.G.; Wang, W.; Wang, J.; Gray, N.S.; et al. Myeloid ERK5 deficiency suppresses tumor growth by blocking protumor macrophage polarization via STAT3 inhibition. Proc. Natl. Acad. Sci. USA 2018, 115, E2801–E2810. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.C.; Everts, B.; Ivanova, Y.; O’Sullivan, D.; Nascimento, M.; Smith, A.M.; Beatty, W.; Love-Gregory, L.; Lam, W.Y.; O’Neill, C.M.; et al. Cell-intrinsic lysosomal lipolysis is essential for alternative activation of macrophages. Nat. Immunol. 2014, 15, 846–855. [Google Scholar] [CrossRef] [Green Version]
- Genini, D.; Brambilla, L.; Laurini, E.; Merulla, J.; Civenni, G.; Pandit, S.; D’Antuono, R.; Perez, L.; Levy, D.E.; Pricl, S.; et al. Mitochondrial dysfunction induced by a SH2 domain-targeting STAT3 inhibitor leads to metabolic synthetic lethality in cancer cells. Proc. Natl. Acad. Sci. USA 2017, 114, E4924–E4933. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, F.; Sonveaux, P.; Rabbani, Z.N.; Liu, S.; Yan, B.; Huang, Q.; Vujaskovic, Z.; Dewhirst, M.W.; Li, C.Y. Regulation of HIF-1alpha stability through S-nitrosylation. Mol. Cell 2007, 26, 63–74. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Liu, J.; Shao, J.; Qin, Y.; Ji, Q.; Zhang, X.; Du, J. Cathepsin S-mediated autophagic flux in tumor-associated macrophages accelerate tumor development by promoting M2 polarization. Mol. Cancer 2014, 13, 43. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ewald, S.E.; Engel, A.; Lee, J.; Wang, M.; Bogyo, M.; Barton, G.M. Nucleic acid recognition by Toll-like receptors is coupled to stepwise processing by cathepsins and asparagine endopeptidase. J. Exp. Med. 2011, 208, 643–651. [Google Scholar] [CrossRef] [PubMed]
- Matsumoto, F.; Saitoh, S.; Fukui, R.; Kobayashi, T.; Tanimura, N.; Konno, K.; Kusumoto, Y.; Akashi-Takamura, S.; Miyake, K. Cathepsins are required for Toll-like receptor 9 responses. Biochem. Biophys. Res. Commun. 2008, 367, 693–699. [Google Scholar] [CrossRef] [PubMed]
- Garcia-Cattaneo, A.; Gobert, F.X.; Muller, M.; Toscano, F.; Flores, M.; Lescure, A.; Del Nery, E.; Benaroch, P. Cleavage of Toll-like receptor 3 by cathepsins B and H is essential for signaling. Proc. Natl. Acad. Sci. USA 2012, 109, 9053–9058. [Google Scholar] [CrossRef] [Green Version]
- Gehad, A.E.; Lichtman, M.K.; Schmults, C.D.; Teague, J.E.; Calarese, A.W.; Jiang, Y.; Watanabe, R.; Clark, R.A. Nitric oxide-producing myeloid-derived suppressor cells inhibit vascular E-selectin expression in human squamous cell carcinomas. J. Investig. Dermatol. 2012, 132, 2642–2651. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roumenina, L.T.; Daugan, M.V.; Noe, R.; Petitprez, F.; Vano, Y.A.; Sanchez-Salas, R.; Becht, E.; Meilleroux, J.; Clec’h, B.L.; Giraldo, N.A.; et al. Tumor Cells Hijack Macrophage-Produced Complement C1q to Promote Tumor Growth. Cancer Immunol. Res. 2019, 7, 1091–1105. [Google Scholar] [CrossRef] [PubMed]
- Hsieh, C.C.; Chou, H.S.; Yang, H.R.; Lin, F.; Bhatt, S.; Qin, J.; Wang, L.; Fung, J.J.; Qian, S.; Lu, L. The role of complement component 3 (C3) in differentiation of myeloid-derived suppressor cells. Blood 2013, 121, 1760–1768. [Google Scholar] [CrossRef]
- Smith, I.A.; Knezevic, B.R.; Ammann, J.U.; Rhodes, D.A.; Aw, D.; Palmer, D.B.; Mather, I.H.; Trowsdale, J. BTN1A1, the mammary gland butyrophilin, and BTN2A2 are both inhibitors of T cell activation. J. Immunol. 2010, 184, 3514–3525. [Google Scholar] [CrossRef]
- Avasarala, S.; Wu, P.Y.; Khan, S.Q.; Yanlin, S.; Van Scoyk, M.; Bao, J.; Di Lorenzo, A.; David, O.; Bedford, M.T.; Gupta, V.; et al. PRMT6 Promotes Lung Tumor Progression via the Alternate Activation of Tumor-Associated Macrophages. Mol. Cancer Res. 2020, 18, 166–178. [Google Scholar] [CrossRef] [Green Version]
- Sinha, P.; Okoro, C.; Foell, D.; Freeze, H.H.; Ostrand-Rosenberg, S.; Srikrishna, G. Proinflammatory S100 proteins regulate the accumulation of myeloid-derived suppressor cells. J. Immunol. 2008, 181, 4666–4675. [Google Scholar] [CrossRef] [Green Version]
- Cheng, P.; Corzo, C.A.; Luetteke, N.; Yu, B.; Nagaraj, S.; Bui, M.M.; Ortiz, M.; Nacken, W.; Sorg, C.; Vogl, T.; et al. Inhibition of dendritic cell differentiation and accumulation of myeloid-derived suppressor cells in cancer is regulated by S100A9 protein. J. Exp. Med. 2008, 205, 2235–2249. [Google Scholar] [CrossRef] [PubMed]
- Denholtz, M.; Zhu, Y.; He, Z.; Lu, H.; Isoda, T.; Dohrmann, S.; Nizet, V.; Murre, C. Upon microbial challenge, human neutrophils undergo rapid changes in nuclear architecture and chromatin folding to orchestrate an immediate inflammatory gene program. Genes Dev. 2020, 34, 149–165. [Google Scholar] [CrossRef] [PubMed]
- Veglia, F.; Hashimoto, A.; Dweep, H.; Sanseviero, E.; De Leo, A.; Tcyganov, E.; Kossenkov, A.; Mulligan, C.; Nam, B.; Masters, G.; et al. Analysis of classical neutrophils and polymorphonuclear myeloid-derived suppressor cells in cancer patients and tumor-bearing mice. J. Exp. Med. 2021, 218. [Google Scholar] [CrossRef]
- Youn, J.I.; Collazo, M.; Shalova, I.N.; Biswas, S.K.; Gabrilovich, D.I. Characterization of the nature of granulocytic myeloid-derived suppressor cells in tumor-bearing mice. J. Leukoc. Biol. 2012, 91, 167–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]








Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Blanco, E.; Ibañez-Vea, M.; Hernandez, C.; Drici, L.; Martínez de Morentin, X.; Gato, M.; Ausin, K.; Bocanegra, A.; Zuazo, M.; Chocarro, L.; et al. A Proteomic Atlas of Lineage and Cancer-Polarized Expression Modules in Myeloid Cells Modeling Immunosuppressive Tumor-Infiltrating Subsets. J. Pers. Med. 2021, 11, 542. https://doi.org/10.3390/jpm11060542
Blanco E, Ibañez-Vea M, Hernandez C, Drici L, Martínez de Morentin X, Gato M, Ausin K, Bocanegra A, Zuazo M, Chocarro L, et al. A Proteomic Atlas of Lineage and Cancer-Polarized Expression Modules in Myeloid Cells Modeling Immunosuppressive Tumor-Infiltrating Subsets. Journal of Personalized Medicine. 2021; 11(6):542. https://doi.org/10.3390/jpm11060542
Chicago/Turabian StyleBlanco, Ester, Maria Ibañez-Vea, Carlos Hernandez, Lylia Drici, Xabier Martínez de Morentin, Maria Gato, Karina Ausin, Ana Bocanegra, Miren Zuazo, Luisa Chocarro, and et al. 2021. "A Proteomic Atlas of Lineage and Cancer-Polarized Expression Modules in Myeloid Cells Modeling Immunosuppressive Tumor-Infiltrating Subsets" Journal of Personalized Medicine 11, no. 6: 542. https://doi.org/10.3390/jpm11060542
APA StyleBlanco, E., Ibañez-Vea, M., Hernandez, C., Drici, L., Martínez de Morentin, X., Gato, M., Ausin, K., Bocanegra, A., Zuazo, M., Chocarro, L., Arasanz, H., Fernandez-Hinojal, G., Fernandez-Irigoyen, J., Smerdou, C., Garnica, M., Echaide, M., Fernandez, L., Morente, P., Ramos-Castellanos, P., ... Kochan, G. (2021). A Proteomic Atlas of Lineage and Cancer-Polarized Expression Modules in Myeloid Cells Modeling Immunosuppressive Tumor-Infiltrating Subsets. Journal of Personalized Medicine, 11(6), 542. https://doi.org/10.3390/jpm11060542