
Biomarkers for Homologous Recombination Deficiency in Cancer

Abstract
:1. Introduction
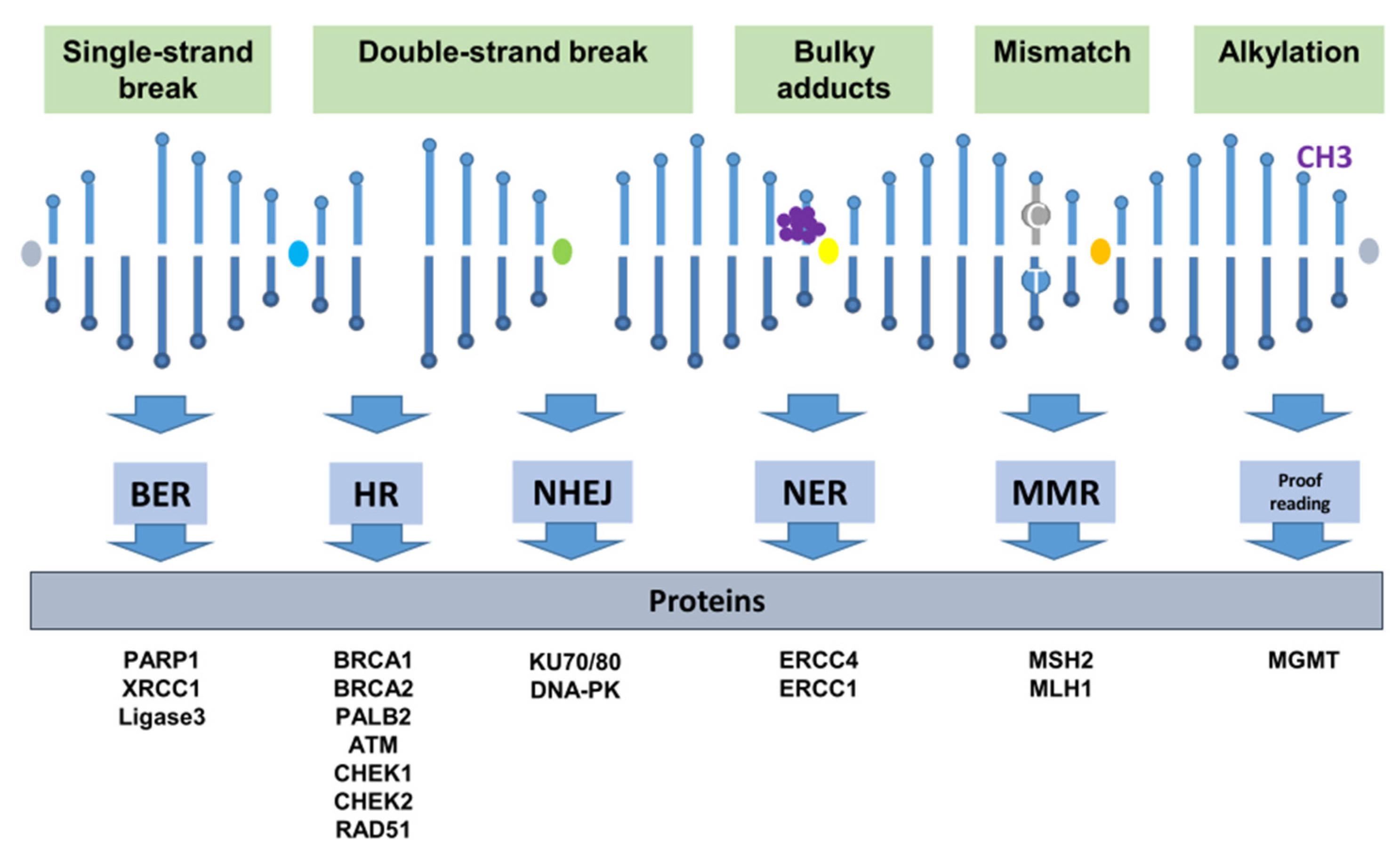
2. DNA Repair Mechanisms
2.1. Single-Strand Lesion Repair
2.2. Double-Strand Break Repair
2.2.1. Homologous Recombination Repair
2.2.2. Non-Homologous End Joining
2.3. Deficiency of Homologous Recombination Repair and BRCAness
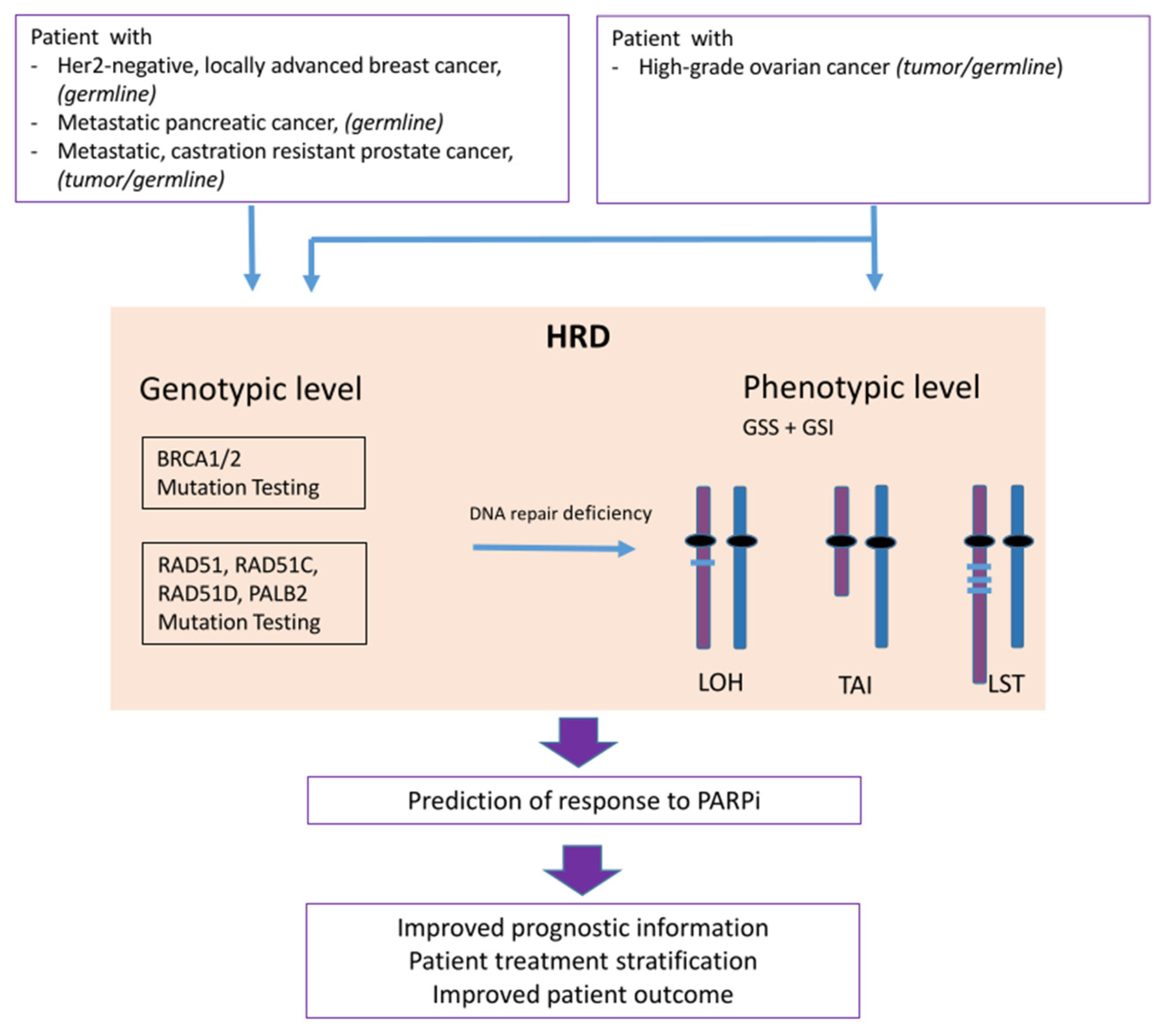
3. PARP Inhibition and Homologous Recombination Repair Deficiency
3.1. The Concept of Synthetic Lethality
3.2. Biomarkers for PARPi: Genes Involved in the Homologous Recombination Repair Pathway
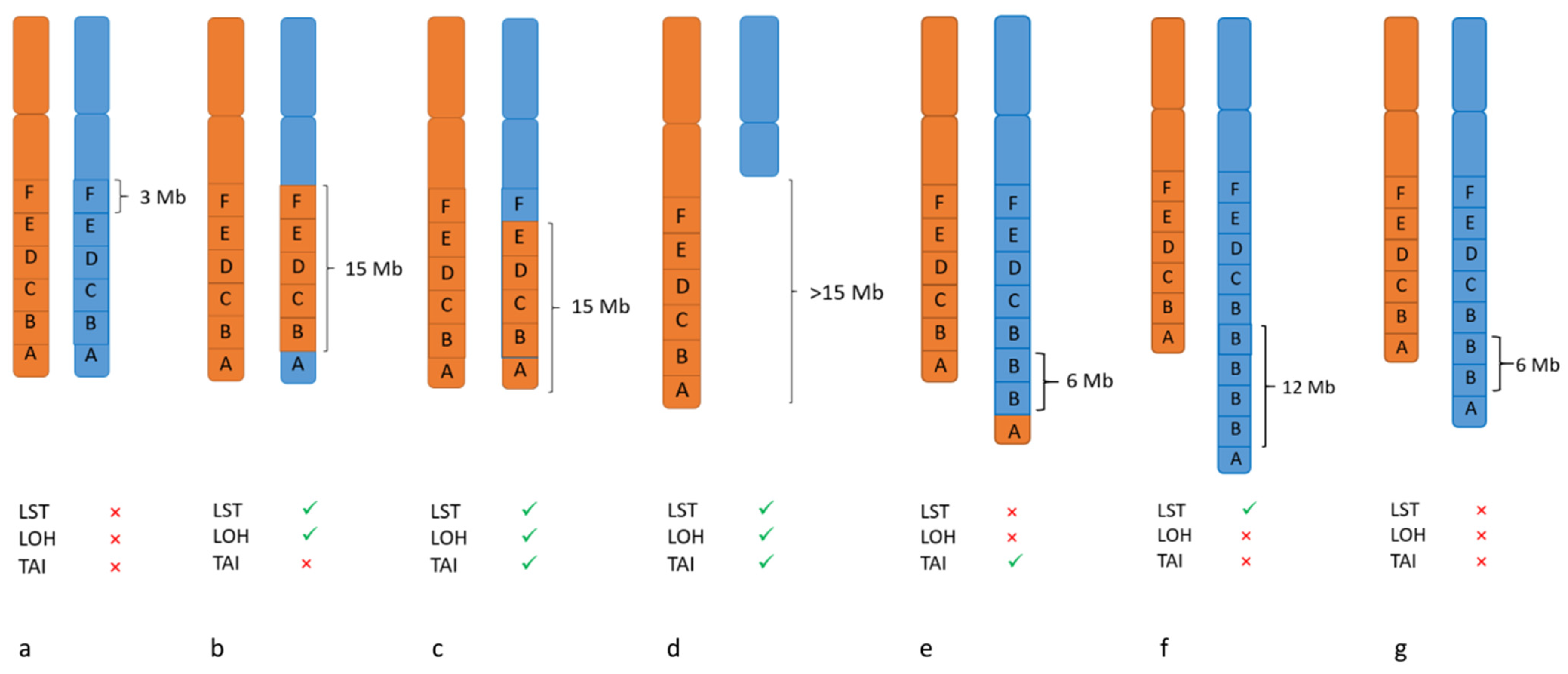
3.3. Biomarker for PARPi: Genomic Instability
LOH- TAI- LST
4. Methods for Detecting DNA Repair Defects as Biomarkers for PARP Inhibition
4.1. Single Nucleotide Polymorphism (SNP) Arrays
4.2. Whole Genome Sequencing
4.3. Targeted Panel Sequencing
5. Conclusions and Future Perspectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Hanahan, D.; Weinberg, R.A. Hallmarks of cancer: The next generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Beggs, R.; Yang, E.S. Targeting DNA repair in precision medicine. Adv. Protein Chem. Struct. Biol. 2019, 115, 135–155. [Google Scholar] [CrossRef] [PubMed]
- Jeggo, P.A.; Pearl, L.H.; Carr, A.M. DNA repair, genome stability and cancer: A historical perspective. Nat. Rev. Cancer 2016, 16, 35–42. [Google Scholar] [CrossRef] [Green Version]
- Soll, J.M.; Sobol, R.W.; Mosammaparast, N. Regulation of DNA Alkylation Damage Repair: Lessons and Therapeutic Opportunities. Trends Biochem. Sci. 2017, 42, 206–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chatterjee, N.; Walker, G.C. Mechanisms of DNA damage, repair, and mutagenesis. Environ. Mol. Mutagenesis 2017, 58, 235–263. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grundy, G.J.; Parsons, J.L. Base excision repair and its implications to cancer therapy. Essays Biochem. 2020, 64, 831–843. [Google Scholar] [CrossRef] [PubMed]
- Houtgraaf, J.H.; Versmissen, J.; van der Giessen, W.J. A concise review of DNA damage checkpoints and repair in mammalian cells. Cardiovasc. Revasc. Med. Incl. Mol. Interv. 2006, 7, 165–172. [Google Scholar] [CrossRef]
- Sancar, A.; Lindsey-Boltz, L.A.; Unsal-Kaçmaz, K.; Linn, S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu. Rev. Biochem. 2004, 73, 39–85. [Google Scholar] [CrossRef] [Green Version]
- San Filippo, J.; Sung, P.; Klein, H. Mechanism of eukaryotic homologous recombination. Annu. Rev. Biochem. 2008, 77, 229–257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Walsh, C.S. Two decades beyond BRCA1/2: Homologous recombination, hereditary cancer risk and a target for ovarian cancer therapy. Gynecol. Oncol. 2015, 137, 343–350. [Google Scholar] [CrossRef] [Green Version]
- De Almeida, L.C.; Calil, F.A.; Machado-Neto, J.A.; Costa-Lotufo, L.V. DNA damaging agents and DNA repair: From carcinogenesis to cancer therapy. Cancer Genet. 2021, 252, 6–24. [Google Scholar] [CrossRef] [PubMed]
- Brianese, R.C.; Nakamura, K.D.M.; Almeida, F.; Ramalho, R.F.; Barros, B.D.F.; Ferreira, E.N.E.; Formiga, M.; de Andrade, V.P.; de Lima, V.C.C.; Carraro, D.M. BRCA1 deficiency is a recurrent event in early-onset triple-negative breast cancer: A comprehensive analysis of germline mutations and somatic promoter methylation. Breast Cancer Res. Treat. 2018, 167, 803–814. [Google Scholar] [CrossRef] [PubMed]
- Jiang, X.; Li, X.; Li, W.; Bai, H.; Zhang, Z. PARP inhibitors in ovarian cancer: Sensitivity prediction and resistance mechanisms. J. Cell. Mol. Med. 2019, 23, 2303–2313. [Google Scholar] [CrossRef] [Green Version]
- Heeke, A.L.; Pishvaian, M.J.; Lynce, F.; Xiu, J.; Brody, J.R.; Chen, W.J.; Baker, T.M.; Marshall, J.L.; Isaacs, C. Prevalence of Homologous Recombination-Related Gene Mutations Across Multiple Cancer Types. JCO Precis. Oncol. 2018, 2, 1–13. [Google Scholar] [CrossRef]
- Ray-Coquard, I.; Pautier, P.; Pignata, S.; Pérol, D.; González-Martín, A.; Berger, R.; Fujiwara, K.; Vergote, I.; Colombo, N.; Mäenpää, J.; et al. Olaparib plus Bevacizumab as First-Line Maintenance in Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2416–2428. [Google Scholar] [CrossRef]
- Hussain, M.; Mateo, J.; Fizazi, K.; Saad, F.; Shore, N.; Sandhu, S.; Chi, K.N.; Sartor, O.; Agarwal, N.; Olmos, D.; et al. Survival with Olaparib in Metastatic Castration-Resistant Prostate Cancer. N. Engl. J. Med. 2020, 383, 2345–2357. [Google Scholar] [CrossRef]
- Pommier, Y.; O’Connor, M.J.; de Bono, J. Laying a trap to kill cancer cells: PARP inhibitors and their mechanisms of action. Sci. Transl. Med. 2016, 8, 362ps317. [Google Scholar] [CrossRef]
- Hodgson, D.R.; Dougherty, B.A.; Lai, Z.; Fielding, A.; Grinsted, L.; Spencer, S.; O’Connor, M.J.; Ho, T.W.; Robertson, J.D.; Lanchbury, J.S.; et al. Candidate biomarkers of PARP inhibitor sensitivity in ovarian cancer beyond the BRCA genes. Br. J. Cancer 2018, 119, 1401–1409. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sunada, S.; Nakanishi, A.; Miki, Y. Crosstalk of DNA double-strand break repair pathways in poly(ADP-ribose) polymerase inhibitor treatment of breast cancer susceptibility gene 1/2-mutated cancer. Cancer Sci. 2018, 109, 893–899. [Google Scholar] [CrossRef]
- Ashworth, A. A synthetic lethal therapeutic approach: Poly(ADP) ribose polymerase inhibitors for the treatment of cancers deficient in DNA double-strand break repair. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2008, 26, 3785–3790. [Google Scholar] [CrossRef] [PubMed]
- Miller, R.E.; Leary, A.; Scott, C.L.; Serra, V.; Lord, C.J.; Bowtell, D.; Chang, D.K.; Garsed, D.W.; Jonkers, J.; Ledermann, J.A.; et al. ESMO recommendations on predictive biomarker testing for homologous recombination deficiency and PARP inhibitor benefit in ovarian cancer. Ann. Oncol. Off. J. Eur. Soc. Med. Oncol. 2020, 31, 1606–1622. [Google Scholar] [CrossRef]
- Lheureux, S.; Lai, Z.; Dougherty, B.A.; Runswick, S.; Hodgson, D.R.; Timms, K.M.; Lanchbury, J.S.; Kaye, S.; Gourley, C.; Bowtell, D.; et al. Long-Term Responders on Olaparib Maintenance in High-Grade Serous Ovarian Cancer: Clinical and Molecular Characterization. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2017, 23, 4086–4094. [Google Scholar] [CrossRef] [Green Version]
- Kondrashova, O.; Topp, M.; Nesic, K.; Lieschke, E.; Ho, G.-Y.; Harrell, M.I.; Zapparoli, G.V.; Hadley, A.; Holian, R.; Boehm, E.; et al. Methylation of all BRCA1 copies predicts response to the PARP inhibitor rucaparib in ovarian carcinoma. Nat. Commun. 2018, 9, 3970. [Google Scholar] [CrossRef] [Green Version]
- Tung, N.M.; Robson, M.E.; Ventz, S.; Santa-Maria, C.A.; Nanda, R.; Marcom, P.K.; Shah, P.D.; Ballinger, T.J.; Yang, E.S.; Vinayak, S.; et al. TBCRC 048: Phase II Study of Olaparib for Metastatic Breast Cancer and Mutations in Homologous Recombination-Related Genes. J. Clin. Oncol. Off. J. Am. Soc. Clin. Oncol. 2020, 38, 4274–4282. [Google Scholar] [CrossRef] [PubMed]
- Armstrong, S.A.; Schultz, C.W.; Azimi-Sadjadi, A.; Brody, J.R.; Pishvaian, M.J. ATM Dysfunction in Pancreatic Adenocarcinoma and Associated Therapeutic Implications. Mol. Cancer Ther. 2019, 18, 1899–1908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Javle, M.; Shacham-Shmueli, E.; Xiao, L.; Varadhachary, G.; Halpern, N.; Fogelman, D.; Boursi, B.; Uruba, S.; Margalit, O.; Wolff, R.A.; et al. Olaparib Monotherapy for Previously Treated Pancreatic Cancer with DNA Damage Repair Genetic Alterations Other Than Germline BRCA Variants: Findings from 2 Phase 2 Nonrandomized Clinical Trials. JAMA Oncol. 2021, 7, 693–699. [Google Scholar] [CrossRef]
- Caracciolo, D.; Riillo, C.; Di Martino, M.T.; Tagliaferri, P.; Tassone, P. Alternative Non-Homologous End-Joining: Error-Prone DNA Repair as Cancer’s Achilles’ Heel. Cancers 2021, 13, 1392. [Google Scholar] [CrossRef] [PubMed]
- Plon, S.E.; Eccles, D.M.; Easton, D.; Foulkes, W.D.; Genuardi, M.; Greenblatt, M.S.; Hogervorst, F.B.; Hoogerbrugge, N.; Spurdle, A.B.; Tavtigian, S.V. Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008, 29, 1282–1291. [Google Scholar] [CrossRef] [Green Version]
- Cline, M.S.; Liao, R.G.; Parsons, M.T.; Paten, B.; Alquaddoomi, F.; Antoniou, A.; Baxter, S.; Brody, L.; Cook-Deegan, R.; Coffin, A.; et al. BRCA Challenge: BRCA Exchange as a global resource for variants in BRCA1 and BRCA2. PLoS Genet. 2018, 14, e1007752. [Google Scholar] [CrossRef] [Green Version]
- Hauke, J.; Horvath, J.; Groß, E.; Gehrig, A.; Honisch, E.; Hackmann, K.; Schmidt, G.; Arnold, N.; Faust, U.; Sutter, C.; et al. Gene panel testing of 5589 BRCA1/2-negative index patients with breast cancer in a routine diagnostic setting: Results of the German Consortium for Hereditary Breast and Ovarian Cancer. Cancer Med. 2018, 7, 1349–1358. [Google Scholar] [CrossRef]
- Hoppe, M.M.; Sundar, R.; Tan, D.S.P.; Jeyasekharan, A.D. Biomarkers for Homologous Recombination Deficiency in Cancer. J. Natl. Cancer Inst. 2018, 110, 704–713. [Google Scholar] [CrossRef] [Green Version]
- Watkins, J.A.; Irshad, S.; Grigoriadis, A.; Tutt, A.N. Genomic scars as biomarkers of homologous recombination deficiency and drug response in breast and ovarian cancers. Breast Cancer Res. BCR 2014, 16, 211. [Google Scholar] [CrossRef] [Green Version]
- Abkevich, V.; Timms, K.M.; Hennessy, B.T.; Potter, J.; Carey, M.S.; Meyer, L.A.; Smith-McCune, K.; Broaddus, R.; Lu, K.H.; Chen, J.; et al. Patterns of genomic loss of heterozygosity predict homologous recombination repair defects in epithelial ovarian cancer. Br. J. Cancer 2012, 107, 1776–1782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Birkbak, N.J.; Wang, Z.C.; Kim, J.Y.; Eklund, A.C.; Li, Q.; Tian, R.; Bowman-Colin, C.; Li, Y.; Greene-Colozzi, A.; Iglehart, J.D.; et al. Telomeric allelic imbalance indicates defective DNA repair and sensitivity to DNA-damaging agents. Cancer Discov. 2012, 2, 366–375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Popova, T.; Manié, E.; Rieunier, G.; Caux-Moncoutier, V.; Tirapo, C.; Dubois, T.; Delattre, O.; Sigal-Zafrani, B.; Bollet, M.; Longy, M.; et al. Ploidy and large-scale genomic instability consistently identify basal-like breast carcinomas with BRCA1/2 inactivation. Cancer Res. 2012, 72, 5454–5462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Timms, K.M.; Abkevich, V.; Hughes, E.; Neff, C.; Reid, J.; Morris, B.; Kalva, S.; Potter, J.; Tran, T.V.; Chen, J.; et al. Association of BRCA1/2defects with genomic scores predictive of DNA damage repair deficiency among breast cancer subtypes. Breast Cancer Res. 2014, 16, 475. [Google Scholar] [CrossRef] [Green Version]
- Telli, M.L.; Timms, K.M.; Reid, J.; Hennessy, B.; Mills, G.B.; Jensen, K.C.; Szallasi, Z.; Barry, W.T.; Winer, E.P.; Tung, N.M.; et al. Homologous Recombination Deficiency (HRD) Score Predicts Response to Platinum-Containing Neoadjuvant Chemotherapy in Patients with Triple-Negative Breast Cancer. Clin. Cancer Res. Off. J. Am. Assoc. Cancer Res. 2016, 22, 3764–3773. [Google Scholar] [CrossRef] [Green Version]
- Fuh, K.; Mullen, M.; Blachut, B.; Stover, E.; Konstantinopoulos, P.; Liu, J.; Matulonis, U.; Khabele, D.; Mosammaparast, N.; Vindigni, A. Homologous recombination deficiency real-time clinical assays, ready or not? Gynecol. Oncol. 2020, 159, 877–886. [Google Scholar] [CrossRef] [PubMed]
- Stover, E.H.; Fuh, K.; Konstantinopoulos, P.A.; Matulonis, U.A.; Liu, J.F. Clinical assays for assessment of homologous recombination DNA repair deficiency. Gynecol. Oncol. 2020, 159, 887–898. [Google Scholar] [CrossRef] [PubMed]
- LaFramboise, T. Single nucleotide polymorphism arrays: A decade of biological, computational and technological advances. Nucleic Acids Res. 2009, 37, 4181–4193. [Google Scholar] [CrossRef] [Green Version]
- Gresham, D.; Dunham, M.J.; Botstein, D. Comparing whole genomes using DNA microarrays. Nat. Rev. Genet. 2008, 9, 291–302. [Google Scholar] [CrossRef] [PubMed]
- Foster, J.M.; Oumie, A.; Togneri, F.S.; Vasques, F.R.; Hau, D.; Taylor, M.; Tinkler-Hundal, E.; Southward, K.; Medlow, P.; McGreeghan-Crosby, K.; et al. Cross-laboratory validation of the OncoScan® FFPE Assay, a multiplex tool for whole genome tumour profiling. BMC Med. Genom. 2015, 8, 5. [Google Scholar] [CrossRef] [Green Version]
- Illumina. Available online: https://www.illumina.com/products/by-type/clinical-research-products/infinium-cytosnp-850k.html (accessed on 19 April 2021).
- Marquard, A.M.; Eklund, A.C.; Joshi, T.; Krzystanek, M.; Favero, F.; Wang, Z.C.; Richardson, A.L.; Silver, D.P.; Szallasi, Z.; Birkbak, N.J. Pan-cancer analysis of genomic scar signatures associated with homologous recombination deficiency suggests novel indications for existing cancer drugs. Biomark. Res. 2015, 3, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Y.; Cottman, M.; Schiffman, J.D. Molecular inversion probes: A novel microarray technology and its application in cancer research. Cancer Genet. 2012, 205, 341–355. [Google Scholar] [CrossRef] [PubMed]
- Cristescu, R.; Liu, X.Q.; Arreaza, G.; Chen, C.; Albright, A.; Qiu, P.; Marton, M. 428 Genomic instability metric concordance between oncoscan™, cytosnp and an fda-approved HRD test. Int. J. Gynecol. Cancer 2020, 30, A130–A132. [Google Scholar] [CrossRef]
- Chao, A.; Lai, C.H.; Wang, T.H.; Jung, S.M.; Lee, Y.S.; Chang, W.Y.; Yang, L.Y.; Ku, F.C.; Huang, H.J.; Chao, A.S.; et al. Genomic scar signatures associated with homologous recombination deficiency predict adverse clinical outcomes in patients with ovarian clear cell carcinoma. J. Mol. Med. 2018, 96, 527–536. [Google Scholar] [CrossRef] [PubMed]
- Sztupinszki, Z.; Diossy, M.; Krzystanek, M.; Reiniger, L.; Csabai, I.; Favero, F.; Birkbak, N.J.; Eklund, A.C.; Syed, A.; Szallasi, Z. Migrating the SNP array-based homologous recombination deficiency measures to next generation sequencing data of breast cancer. NPJ Breast Cancer 2018, 4, 16. [Google Scholar] [CrossRef] [PubMed]
- Bentley, D.R.; Balasubramanian, S.; Swerdlow, H.P.; Smith, G.P.; Milton, J.; Brown, C.G.; Hall, K.P.; Evers, D.J.; Barnes, C.L.; Bignell, H.R.; et al. Accurate whole human genome sequencing using reversible terminator chemistry. Nature 2008, 456, 53–59. [Google Scholar] [CrossRef]
- Davies, H.; Glodzik, D.; Morganella, S.; Yates, L.R.; Staaf, J.; Zou, X.; Ramakrishna, M.; Martin, S.; Boyault, S.; Sieuwerts, A.M.; et al. HRDetect is a predictor of BRCA1 and BRCA2 deficiency based on mutational signatures. Nat. Med. 2017, 23, 517–525. [Google Scholar] [CrossRef]
- Nguyen, L.; Martens, J.W.M.; Van Hoeck, A.; Cuppen, E. Pan-cancer landscape of homologous recombination deficiency. Nat. Commun. 2020, 11, 5584. [Google Scholar] [CrossRef] [PubMed]
- Eeckhoutte, A.; Houy, A.; Manié, E.; Reverdy, M.; Bièche, I.; Marangoni, E.; Goundiam, O.; Vincent-Salomon, A.; Stoppa-Lyonnet, D.; Bidard, F.-C.; et al. ShallowHRD: Detection of homologous recombination deficiency from shallow whole genome sequencing. Bioinformatics 2020, 36, 3888–3889. [Google Scholar] [CrossRef] [PubMed]
- De Luca, X.M.; Newell, F.; Kazakoff, S.H.; Hartel, G.; McCart Reed, A.E.; Holmes, O.; Xu, Q.; Wood, S.; Leonard, C.; Pearson, J.V.; et al. Using whole-genome sequencing data to derive the homologous recombination deficiency scores. NPJ Breast Cancer 2020, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Mamanova, L.; Coffey, A.J.; Scott, C.E.; Kozarewa, I.; Turner, E.H.; Kumar, A.; Howard, E.; Shendure, J.; Turner, D.J. Target-enrichment strategies for next-generation sequencing. Nat. Methods 2010, 7, 111–118. [Google Scholar] [CrossRef]
- ThermoFisher. Available online: https://www.thermofisher.com/de/de/home/clinical/preclinical-companion-diagnostic-development/oncomine-oncology/oncomine-cancer-research-panel-workflow/oncomine-comprehensive-assay-plus.html (accessed on 19 April 2021).
- Myriad. Available online: https://myriad.com/products-services/precision-medicine/mychoice-cdx/ (accessed on 19 April 2021).
- González-Martín, A.; Pothuri, B.; Vergote, I.; DePont Christensen, R.; Graybill, W.; Mirza, M.R.; McCormick, C.; Lorusso, D.; Hoskins, P.; Freyer, G.; et al. Niraparib in Patients with Newly Diagnosed Advanced Ovarian Cancer. N. Engl. J. Med. 2019, 381, 2391–2402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirza, M.R.; Monk, B.J.; Herrstedt, J.; Oza, A.M.; Mahner, S.; Redondo, A.; Fabbro, M.; Ledermann, J.A.; Lorusso, D.; Vergote, I.; et al. Niraparib Maintenance Therapy in Platinum-Sensitive, Recurrent Ovarian Cancer. N. Engl. J. Med. 2016, 375, 2154–2164. [Google Scholar] [CrossRef]
- AmoyDiagnostics. Available online: http://www.amoydiagnostics.com/productDetail_38.html (accessed on 19 April 2021).




{kind=link}
{kind=link}
{kind=link}
| Panel | Provider | Applicable in Lab | Focus | Number of Genes | LOH | TAI | LST |
|---|---|---|---|---|---|---|---|
| BROCA Cancer Risk Panel | UW Medical Center Laboratory Medicine—Genetics Lab | no | Hereditary cancer predisposition syndromes/Ovarian cancer/Breast cancer | 74 | no | no | no |
| BRCANextTM | AmbryGenetics | no | Hereditary Gynecological carcinomas and/or Breast cancer | 18 | no | no | no |
| BRCANext-Expanded™ | AmbryGenetics | no | Hereditary Gynecological carcinomas and/or Breast cancer | 23 | no | no | no |
| CAN02 | CeGaT | no | Ovarian cancer/Breast cancer | 13 | no | no | no |
| CAN21 | CeGaT | no | Ovarian cancer/Breast cancer | 41 | no | no | no |
| Myriad myRisk® Hereditary Cancer | Myriad | no | Hereditary cancer predisposition syndromes/Ovarian cancer/Breast cancer | 36 | no | no | no |
| Myriad’s myChoice® CDx | Myriad | no | homologous recombination deficiency score/BRCA1,2 | 2 | yes | yes | yes |
| Oncomine BRCA Expanded Panel | Thermo Fisher Scientific | Yes | Ovarian cancer/Breast cancer and Prostate carcinomas | 15 | no | no | no |
| Oncomine HRR Pathway Predesigned Panel | Thermo Fisher Scientific | Yes | Homologous recombination repair genes and other | 28 | no | no | no |
| Oncomine Comprehensive Plus | Thermo Fisher Scientific | Yes | Pan-Cancer | 500+ | yes | no | no |
| Trusight Tumor 170 | Illumina | Yes | Pan-Cancer | 170 | no | no | no |
| TrueSight Oncology 500 | Illumina | Yes | Pan-Cancer | 500+ | no | no | no |
| CentoBreast® | Centogene | no | Ovarian cancer/Breast cancer | 30 | no | no | no |
| CentoCancer® | Centogene | no | Pan-Cancer | 70 | no | no | no |
| HANDLE HRR NGS Panel | AmoyDx | yes | Homologous recombination repair genes and other | 32 | no | no | no |
| HRD Focus Panel | AmoyDx | yes | homologous recombination deficiency score/BRCA1,2 | 2 | yes | yes | yes |
| Invitae Common Hereditary Cancers Panel | Invitae | Yes | Hereditary cancer predisposition syndromes/Ovarian cancer/Breast cancer | 47 | |||
| FoundationOne®CD | Foundation Medicine | no | Pan-Cancer | 324 | |||
| FoundationFocus CDxBRCA LOH | Foundation Medicine | no | 2 | yes | no | no | |
| NeoTYPE® HRD+ Profile | NeoGenomics | no | homologous recombination deficiency/Homologous recombination repair genes | 30 | no | no | no |
| QIAseq Homologous Recombination Repair (HRR) Panel | Qiagen | yes | Homologous recombination repair genes | 15 | no | no | no |
| SureMASTR HRR | Agilent | yes | Homologous recombination repair genes | 17 | no | no | no |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wagener-Ryczek, S.; Merkelbach-Bruse, S.; Siemanowski, J. Biomarkers for Homologous Recombination Deficiency in Cancer. J. Pers. Med. 2021, 11, 612. https://doi.org/10.3390/jpm11070612
Wagener-Ryczek S, Merkelbach-Bruse S, Siemanowski J. Biomarkers for Homologous Recombination Deficiency in Cancer. Journal of Personalized Medicine. 2021; 11(7):612. https://doi.org/10.3390/jpm11070612
Chicago/Turabian StyleWagener-Ryczek, Svenja, Sabine Merkelbach-Bruse, and Janna Siemanowski. 2021. "Biomarkers for Homologous Recombination Deficiency in Cancer" Journal of Personalized Medicine 11, no. 7: 612. https://doi.org/10.3390/jpm11070612
APA StyleWagener-Ryczek, S., Merkelbach-Bruse, S., & Siemanowski, J. (2021). Biomarkers for Homologous Recombination Deficiency in Cancer. Journal of Personalized Medicine, 11(7), 612. https://doi.org/10.3390/jpm11070612