
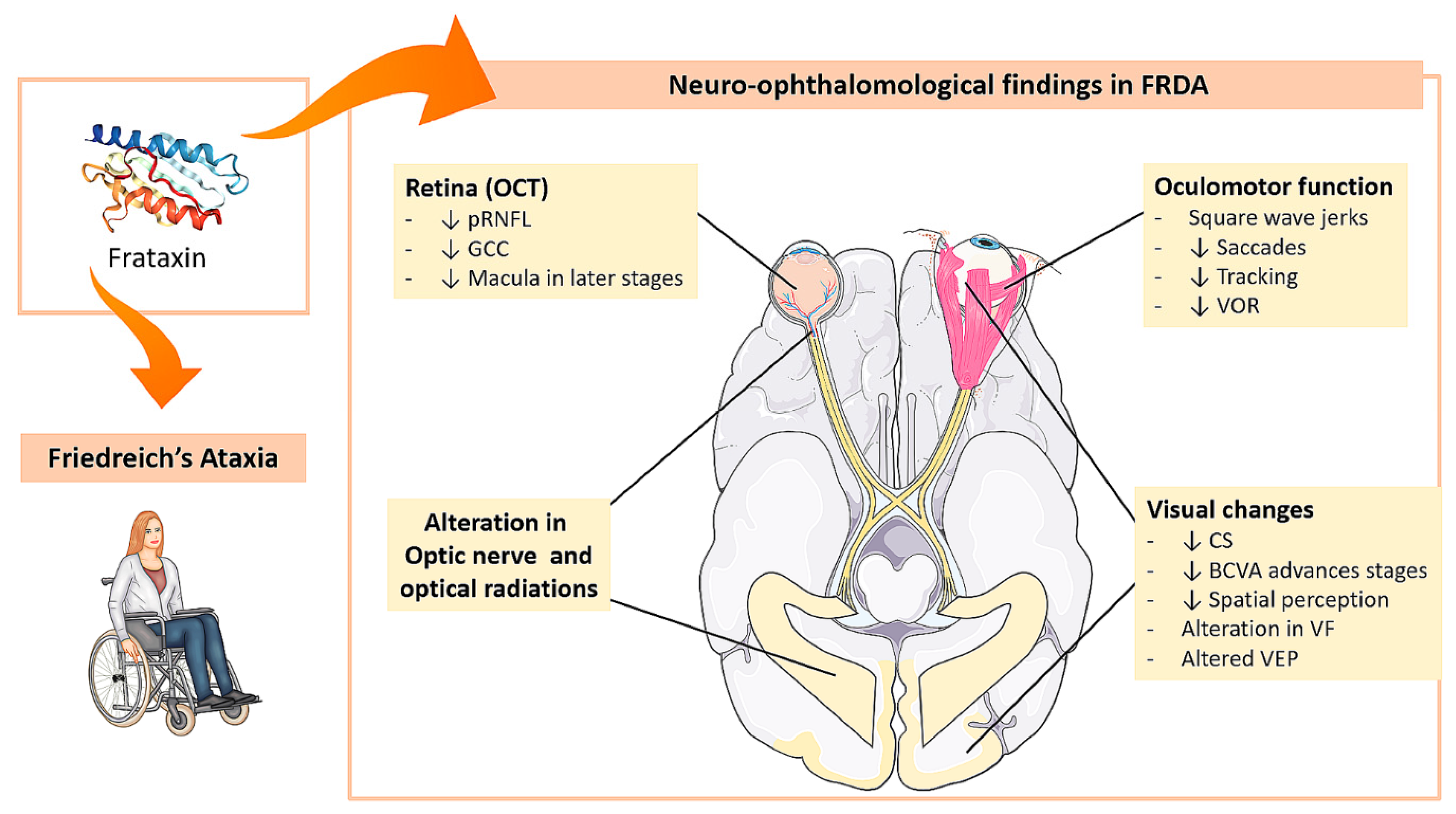
Neuro-Ophthalmological Findings in Friedreich’s Ataxia

 ,
,  ,
,  , ,
, ,  ,
, 
 ,
,  and
and 
Abstract
:1. Introduction: Overview of Friedreich Ataxia Disease
1.1. Epidemiology
1.2. Clinic
1.3. Genetics
2. Friedreich Ataxia and Eye
2.1. Oculomotor Function Alterations
2.1.1. Saccadic Movements
- Saccadic latency
- Saccade velocity
- Saccadic accuracy
2.1.2. Fixation
- Nystagmus
- Square wave saccades
2.1.3. Tracking Movements
2.1.4. Vestibulo-Ocular Reflexes (VOR)
2.1.5. Clinical Utility of Ocular Motility Testing in Patients with FRDA
2.2. Visual Pathway Disorders
2.2.1. Visual Evoked Potentials (VEP)
2.2.2. Electroretinogram (ERG)
2.2.3. Contrast Sensitivity (CS)
2.2.4. Spatial Perception
2.2.5. Best-Corrected Visual Acuity (BCVA)
2.2.6. Visual Field (VF)
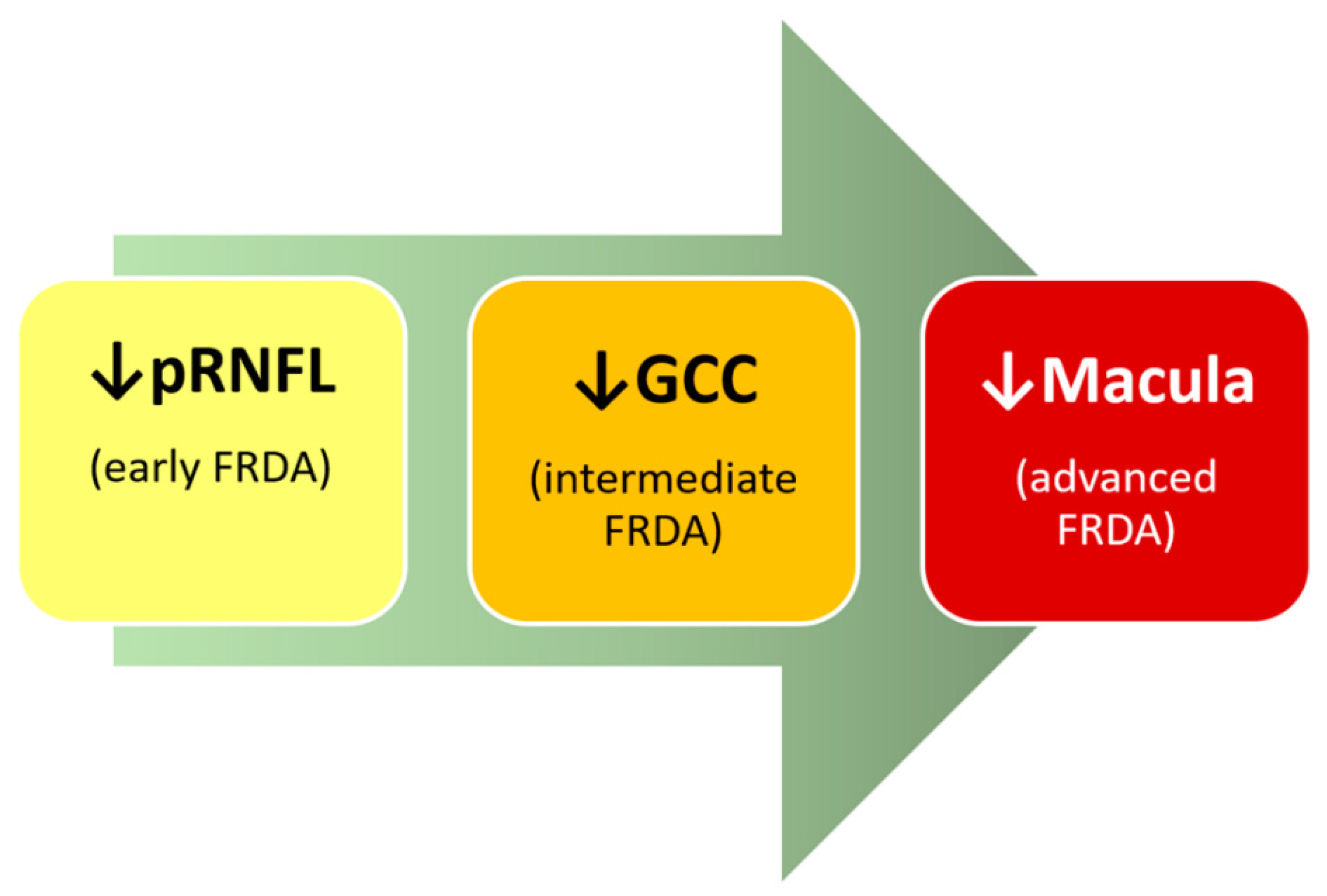
2.2.7. Optical Coherence Tomography (OCT)
- Peripapillary retinal nerve fiber layer (pRNFL)
- Ganglion Cell Complex (GCC)
- Macula
2.2.8. Histopathological Studies of the Retina of Patients and Experimental Models of FRDA
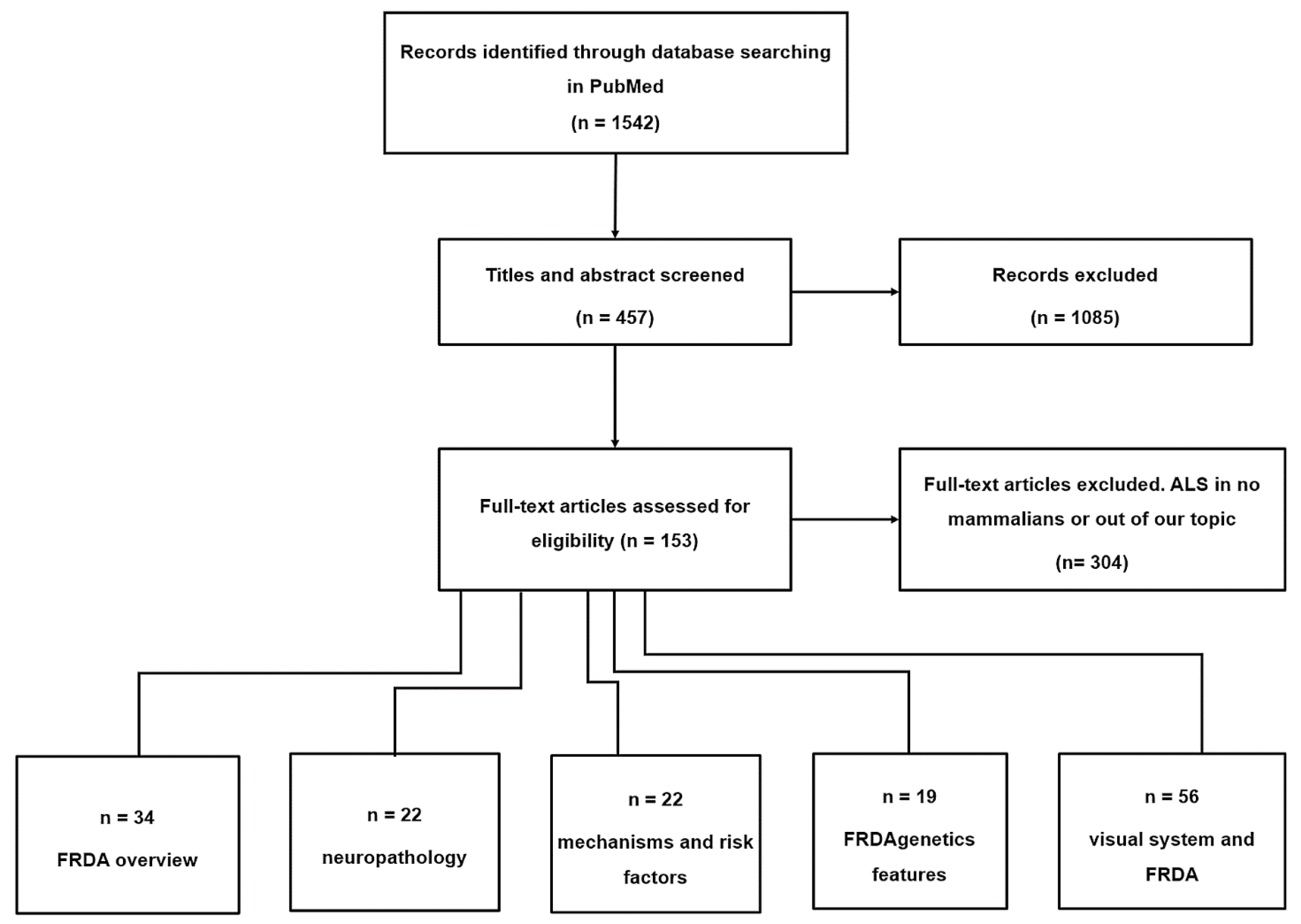
3. Material and Methods
4. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Lynch, D.R.; Farmer, J.M.; Balcer, L.J.; Wilson, R.B. Friedreich ataxia: Effects of genetic understanding on clinical evaluation and therapy. Arch. Neurol. 2002, 59, 743–747. [Google Scholar] [CrossRef] [Green Version]
- Campuzano, V.; Montermini, L.; Moltò, M.D.; Pianese, L.; Cossée, M.; Cavalcanti, F.; Monros, E.; Rodius, F.; Duclos, F.; Monticelli, A.; et al. Friedreich’s ataxia: Autosomal recessive disease caused by an intronic GAA triplet repeat expansion. Science 1996, 271, 1423–1427. [Google Scholar] [CrossRef] [PubMed]
- Tonon, C.; Lodi, R. Idebenone in Friedreich’s ataxia. Expert Opin. Pharmacother. 2008, 9, 2327–2337. [Google Scholar] [CrossRef]
- Dağ, E.; Örnek, N.; Örnek, K.; Erbahçeci-Timur, I.E. Optical Coherence Tomography and Visual Field Findings in Patients With Friedreich Ataxia. J. Neuro-Ophthalmology 2014, 34, 118–121. [Google Scholar] [CrossRef]
- Harding, A.E. Clinical features and classification of inherited ataxias. Adv. Neurol. 1993, 61, 1–14. [Google Scholar] [PubMed]
- Fraser, J.A.; Biousse, V.; Newman, N.J. The Neuro-ophthalmology of Mitochondrial Disease. Surv. Ophthalmol. 2010, 55, 299–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pandolfo, M. Friedreich ataxia: The clinical picture. J. Neurol. 2009, 256 (Suppl. 1), 3–8. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H.; Mazurkiewicz, J.E. Friedreich Ataxia: Neuropathology Revised. J Neuropathol Exp Neurol 2013, 72, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Cossée, M.; Dürr, A.; Schmitt, M.; Dahl, N.; Trouillas, P.; Allinson, P.; Kostrzewa, M.; Nivelon-Chevallier, A.; Gustavson, K.H.; Kohlschütter, A.; et al. Friedreich’s ataxia: Point mutations and clinical presentation of compound heterozygotes. Ann. Neurol. 1999, 45, 200–206. [Google Scholar] [CrossRef]
- Campuzano, V.; Montermini, L.; Lutz, Y.; Cova, L.; Hindelang, C.; Jiralerspong, S.; Trottier, Y.; Kish, S.J.; Faucheux, B.; Trouillas, P.; et al. Frataxin is reduced in Friedreich ataxia patients and is associated with mitochondrial membranes. Hum. Mol. Genet. 1997, 6, 1771–1780. [Google Scholar] [CrossRef] [Green Version]
- Fortuna, F.; Barboni, P.; Liguori, R.; Valentino, M.L.; Savini, G.; Gellera, C.; Mariotti, C.; Rizzo, G.; Tonon, C.; Manners, D.; et al. Visual system involvement in patients with Friedreich’s ataxia. Brain 2009, 132, 116–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carelli, V.; La Morgia, C.; Valentino, M.L.; Barboni, P.; Ross-Cisneros, F.N.; Sadun, A.A. Retinal ganglion cell neurodegeneration in mitochondrial inherited disorders. Biochim. Biophys. Acta Bioenerg. 2009, 1787, 518–528. [Google Scholar] [CrossRef] [Green Version]
- Alldredge, C.D.; Schlieve, C.R.; Miller, N.R.; Levin, L.A. Pathophysiology of the Optic Neuropathy Associated With Friedreich Ataxia. Arch. Ophthalmol. 2003, 121, 1582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ben Hamida, M.; Belal, S.; Sirugo, G.; Ben Hamida, C.; Panayides, K.; Ionannou, P.; Beckmann, J.; Mandel, J.L.; Hentati, F.; Koenig, M.; et al. Friedreich’s ataxia phenotype not linked to chromosome 9 and associated with selective autosomal recessive vitamin E deficiency in two inbred Tunisian families. Neurology 1993, 43, 2179–2183. [Google Scholar] [CrossRef] [PubMed]
- McCabe, D.J.H.; Ryan, F.; Moore, D.P.; McQuaid, S.; King, M.D.; Kelly, A.; Daly, K.; Barton, D.E.; Murphy, R.P. Typical Friedreich’s ataxia without GAA expansions and GAA expansions without typical Friedreich’s ataxia. J. Neurol. 2000, 247, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Harding, A.E. Friedreich’s ataxia: A clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain a J. Neurol. 1981, 104, 589–620. [Google Scholar] [CrossRef] [PubMed]
- Ormerod, I.E.C.; Harding, A.E.; Miller, D.H.; Johnson, G.; MacManus, D.; du Boulay, E.P.G.H.; Kendall, B.E.; Moseley, I.F.; McDonald, W.I.; Miller, G.; et al. Magnetic resonance imaging in degenerative ataxic disorders. Neurosurg. Psychiatry 1994, 57, 51–57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bhidayasiri, R.; Perlman, S.L.; Pulst, S.M.; Geschwind, D.H. Late-onset Friedreich Ataxia: Phenotypic analysis, magnetic resonance imaging findings, and review of the literature. Arch. Neurol. 2005, 62, 1865–1869. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akhlaghi, H.; Corben, L.; Georgiou-Karistianis, N.; Bradshaw, J.; Storey, E.; Delatycki, M.B.; Egan, G.F. Superior cerebellar peduncle atrophy in Friedreich’s ataxia correlates with disease symptoms. Cerebellum 2011, 10, 81–87. [Google Scholar] [CrossRef]
- Chevis, C.F.; Da Silva, C.B.; D’Abreu, A.; Lopes-Cendes, I.; Cendes, F.; Bergo, F.P.G.; França, M.C. Spinal cord atrophy correlates with disability in Friedreich’s ataxia. Cerebellum 2013, 12, 43–47. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, G.; Rey, A.; Sanchez-Dalmau, F.B.; Muñ Oz, E.; Ríos, J.; Adá, A. Optical coherence tomography findings in spinocerebellar ataxia-3. Eye 2013, 27, 1376–1381. [Google Scholar] [CrossRef]
- Seyer, L.A.; Galetta, K.; Wilson, J.; Sakai, R.; Perlman, S.; Mathews, K.; Wilmot, G.R.; Gomez, C.M.; Ravina, B.; Zesiewicz, T.; et al. Analysis of the visual system in Friedreich ataxia. J. Neurol. 2013, 260, 2362–2369. [Google Scholar] [CrossRef] [PubMed]
- Koeppen, A.H. Friedreich’s ataxia: Pathology, pathogenesis, and molecular genetics. J. Neurol. Sci. 2011, 303, 1–12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Corben, L.A.; Delatycki, M.B.; Bradshaw, J.L.; Horne, M.K.; Fahey, M.C.; Churchyard, A.J.; Georgiou-Karistianis, N. Impairment in motor reprogramming in Friedreich ataxia reflecting possible cerebellar dysfunction. J. Neurol. 2010, 257, 782–791. [Google Scholar] [CrossRef]
- Lynch, D.R.; Deutsch, E.C.; Wilson, R.B.; Tennekoon, G. Unanswered Questions in Friedreich Ataxia. J. Child Neurol. 2012, 27, 1223–1229. [Google Scholar] [CrossRef] [PubMed]
- Cnop, M.; Igoillo-Esteve, M.; Rai, M.; Begu, A.; Serroukh, Y.; Depondt, C.; Musuaya, A.E.; Marhfour, I.; Ladrière, L.; Moles Lopez, X.; et al. Central role and mechanisms of β-cell dysfunction and death in friedreich ataxia-associated diabetes. Ann. Neurol. 2012, 72, 971–982. [Google Scholar] [CrossRef] [Green Version]
- Filla, A.; De Michele, G.; Cavalcanti, F.; Pianese, L.; Monticelli, A.; Campanella, G.; Cocozza, S. The relationship between trinucleotide (GAA) repeat length and clinical features in Friedreich ataxia. Am. J. Hum. Genet. 1996, 59, 554–560. [Google Scholar] [PubMed]
- Warner, J.P.; Barron, L.H.; Goudie, D.; Kelly, K.; Dow, D.; Fitzpatrick, D.R.; Brock, D.J.H. A general method for the detection of large GAG repeat expansions by fluorescent PCR. J. Med. Genet. 1996, 33, 1022–1026. [Google Scholar] [CrossRef] [Green Version]
- Dürr, A.; Cossée, M.; Yves, A.; Victoria, C.; Claude, M.; Christiane, P.; Mandel, J.-L.; Alexis, B.; Michel, K. Clinical and genetic abnormalities in patients with Friedreich’s ataxia. N. Engl. J. Med. 1996, 335, 1169–1175. [Google Scholar] [CrossRef] [PubMed]
- Patel, P.; Isaya, G. Friedreich Ataxia: From GAA Triplet-Repeat Expansion to Frataxin Deficiency. Am. J. Hum. Genet 2001, 69, 15–24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- t Hart, L.M.; Ruige, J.B.; Dekker, J.M.; Stehouwer, C.D.; Maassen, J.A.; Heine, R.J. Altered β-cell characteristics in impaired glucose tolerant carriers of a GAA trinucleotide repeat polymorphism in the frataxin gene. Diabetes 1999, 48, 924–926. [Google Scholar] [CrossRef] [PubMed]
- Dalgaard, L.T.; Hansen, T.; Urhammer, S.A.; Clausen, J.O.; Eiberg, H.; Pedersen, O. Intermediate expansions of a GAA repeat in the frataxin gene are not associated with type 2 diabetes or altered glucose induced β-cell function in Danish Caucasians. Diabetes 1999, 48, 914–917. [Google Scholar] [CrossRef]
- Herman, D.; Jenssen, K.; Burnett, R.; Soragni, E.; Perlman, S.L.; Gottesfeld, J.M. Histone deacetylase inhibitors reverse gene silencing in Friedreich’s ataxia. Nat. Chem. Biol. 2006, 2, 551–558. [Google Scholar] [CrossRef] [PubMed]
- De Castro, M.; García-Planells, J.; Monrós, E.; Cañizares, J.; Vázquez-Manrique, R.; Vílchez, J.J.; Urtasun, M.; Lucas, M.; Navarro, G.; Izquierdo, G.; et al. Genotype and phenotype analysis of Friedreich’s ataxia compound heterozygous patients. Hum. Genet. 2000, 106, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Forrest, S.M.; Knight, M.; Delatycki, M.B.; Paris, D.; Williamson, R.; King, J.; Yeung, L.; Nassif, N.; Nicholson, G.A. The correlation of clinical phenotype in Friedreich ataxia with the site of point mutations in the FRDA gene. Neurogenetics 1998, 1, 253–257. [Google Scholar] [CrossRef] [PubMed]
- Mccormack, M.L.; Guttmann, R.P.; Schumann, M.; Farmer, J.M.; Stolle, C.A.; Campuzano, V.; Koenig, M.; Lynch, D.R. Frataxin point mutations in two patients with Friedreich’s ataxia and unusual clinical features. J. Neurol. Neurosurg. Psychiatry 2000, 68, 661–664. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delatycki, M.B.; Knight, M.; Koenig, M.; Cossée, M.; Williamson, R.; Forrest, S.M. G130V, a common FRDA point mutation, appears to have arisen from a common founder. Hum. Genet. 1999, 105, 343–346. [Google Scholar] [CrossRef] [PubMed]
- La Pean, A.; Jeffries, N.; Grow, C.; Ravina, B.; di Prospero, N.A. Predictors of progression in patients with Friedreich ataxia. Mov. Disord. 2008, 23, 2026–2032. [Google Scholar] [CrossRef] [Green Version]
- Carroll, W.M.; Kriss, A.; Baraitser, M.; Barrett, G.; Halliday, A.M. The incidence and nature of visual pathway involvement in Friedreich’s ataxia. A clinical and visual evoked potential study of 22 patients. Brain 1980, 103, 413–434. [Google Scholar] [CrossRef] [PubMed]
- Livingstone, I.R.; Mastaglia, F.L.; Edis, R.; Howe, J.W. Visual Involvement in Friedreich’s Ataxia and Hereditary Spastic Ataxia: A Clinical and Visual Evoked Response Study. Arch. Neurol. 1981, 38, 75–79. [Google Scholar] [CrossRef] [PubMed]
- Newman, N.J.; Biousse, V. Hereditary optic neuropathies. Eye 2004, 18, 1144–1160. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carelli, V.; Ross-Cisneros, F.N.; Sadun, A.A. Optic nerve degeneration and mitochondrial dysfunction: Genetic and acquired optic neuropathies. Neurochem. Int. 2002, 40, 573–584. [Google Scholar] [CrossRef]
- Pinto, F.; Amantini, A.; de Scisciolo, G.; Scaioli, V.; Guidi, L.; Frosini, R. Visual Involvement in Friedreich’s Ataxia: PERG and VEP Study. Eur. Neurol. 1988, 28, 246–251. [Google Scholar] [CrossRef] [PubMed]
- Fahey, M.C.; Cremer, P.D.; Aw, S.T.; Millist, L.; Todd, M.J.; White, O.B.; Halmagyi, M.; Corben, L.A.; Collins, V.; Churchyard, A.J.; et al. Vestibular, saccadic and fixation abnormalities in genetically confirmed Friedreich ataxia. Brain 2008, 131, 1035–1045. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dale, R.T.; Kirby, A.W.; Jampel, R.S. Square wave jerks in Friedreich’s ataxia. Am. J. Ophthalmol. 1978, 85, 400–406. [Google Scholar] [CrossRef]
- Furman, J.M.; Perlman, S.; Baloh, R.W. Eye movements in Friedreich’s ataxia. Arch. Neurol. 1983, 40, 343–346. [Google Scholar] [CrossRef] [PubMed]
- Hocking, D.R.; Fielding, J.; Corben, L.A.; Cremer, P.D.; Millist, L.; White, O.B.; Delatycki, M.B. Ocular Motor Fixation Deficits in Friedreich Ataxia. Cerebellum 2010, 9, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Kirkham, T.H.; Guitton, D.; Katsarkas, A.; Kline, L.B.; Andermann, E. Oculomotor abnormalities in Friedreich’s ataxia. Can. J. Neurol. Sci. 1979, 6, 167–172. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Spieker, S.; Schulz, J.B.; Petersen, D.; Fetter, M.; Klockgether, T.; Dichgans, J. Fixation instability and oculomotor abnormalities in Friedreich’s ataxia. J. Neurol. 1995, 242, 517–521. [Google Scholar] [CrossRef] [PubMed]
- Rabiah, P.K.; Bateman, J.B.; Demer, J.L.; Perlman, S. Ophthalmologic findings in patients with ataxia. Am. J. Ophthalmol. 1997, 123, 108–117. [Google Scholar] [CrossRef]
- Bird, T.D.; Crill, W.E. Pattern-reversal visual evoked potentials in the hereditary ataxias and spinal degenerations. Ann. Neurol. 1981, 9, 243–250. [Google Scholar] [CrossRef] [PubMed]
- Wenzel, W.; Camacho, L.; Claus, D.; Aschoff, J. Visually evoked potentials in Friedreich’s ataxia. Adv. Neurol. 1982, 32, 131–139. [Google Scholar] [PubMed]
- Lynch, D.R.; Farmer, J.M.; Rochestie, D.; Balcer, L.J. Contrast letter acuity as a measure of visual dysfunction in patients with Friedreich ataxia. J. Neuro-Ophthalmol. 2002, 22, 270–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balcer, L.J.; Baier, M.L.; Pelak, V.S.; Fox, R.J.; Shuwairi, S.; Galetta, S.L.; Cutter, G.R.; Maguire, M.G. New low-contrast vision charts: Reliability and test characteristics in patients with multiple sclerosis. Mult. Scler. J. 2000, 6, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Noval, S.; Contreras, I.; Sanz-Gallego, I.; Manrique, R.K.; Arpa, J. Ophthalmic features of Friedreich ataxia. Eye 2012, 26, 315–320. [Google Scholar] [CrossRef] [PubMed]
- Hamedani, A.G.; Hauser, L.A.; Perlman, S.; Mathews, K.; Wilmot, G.R.; Zesiewicz, T.; Subramony, S.H.; Ashizawa, T.; Delatycki, M.B.; Brocht, A.; et al. Longitudinal analysis of contrast acuity in Friedreich ataxia. Neurol. Genet. 2018, 4, e250. [Google Scholar] [CrossRef] [Green Version]
- Fehrenbach, R.A.; Wallesch, C.W.; Claus, D. Neuropsychologic Findings in Friedreich’s Ataxia. Arch. Neurol. 1984, 41, 306–308. [Google Scholar] [CrossRef] [PubMed]
- Waldvogel, D.; Van Gelderen, P.; Hallett, M. Increased iron in the dentate nucleus of patients with Friedreich’s ataxia. Ann. Neurol. 1999, 46, 123–125. [Google Scholar] [CrossRef]
- Optican, L.M.; Quaia, C. Distributed model of collicular and cerebellar function during saccades. Ann. N. Y. Acad. Sci. 2002, 956, 164–177. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blekher, T.; Johnson, S.A.; Marshall, J.; White, K.; Hui, S.; Weaver, M.; Gray, J.; Yee, R.; Stout, J.C.; Beristain, X.; et al. Saccades in presymptomatic and early stages of Huntington disease. Neurology 2006, 67, 394–399. [Google Scholar] [CrossRef]
- Irving, E.L.; Steinbach, M.J.; Lillakas, L.; Babu, R.J.; Hutchings, N. Horizontal saccade dynamics across the human life span. Invest. Ophthalmol. Vis. Sci. 2006, 47, 2478–2484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leigh, R.J.; Zee, D.S. The Neurology of Eye Movements, 5th ed.; Oxford University Press: Oxford, UK, 2015. [Google Scholar]
- Zee, D.S.; Robinson, D.A. A hypothetical explanation of saccadic oscillations. Ann. Neurol. 1979, 5, 405–414. [Google Scholar] [CrossRef] [PubMed]
- Schon, F.; Hodgson, T.L.; Mort, D.; Kennard, C. Ocular flutter associated with a localized lesion in the paramedian pontine reticular formation. Ann. Neurol. 2001, 50, 413–416. [Google Scholar] [CrossRef] [PubMed]
- Fukazawa, T.; Tashiro, K.; Hamada, T.; Kase, M. Multisystem degeneration: Drugs and square wave jerks. Neurology 1986, 36, 1230–1233. [Google Scholar] [CrossRef]
- Hotson, J.R. Cerebellar control of fixation eye movements. Neurology 1982, 32, 31–36. [Google Scholar] [CrossRef] [PubMed]
- Moschner, C.; Perlman, S.; Baloh, R.W. Comparison of oculomotor findings in the progressive ataxia syndromes. Brain 1994, 117, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Ramat, S.; Leigh, R.J.; Zee, D.S.; Optican, L.M. Ocular oscillations generated by coupling of brainstem excitatory and inhibitory saccadic burst neurons. Exp. Brain Res. 2005, 160, 89–106. [Google Scholar] [CrossRef] [PubMed]
- Abadi, R.V.; Gowen, E. Characteristics of saccadic intrusions. Vis. Res. 2004, 44, 2675–2690. [Google Scholar] [CrossRef] [PubMed]
- Monday, L.; Lespérance, J.; Lemieux, B.; Saint-Vincent, H. Follow-up study of electronystagmographic findings in friedreich’s ataxia patients and evaluation of their relatives. Can. J. Neurol. Sci. 1984, 11, 570–573. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ell, J.; Prasher, D.; Rudge, P. Neuro-otological abnormalities in Friedreich’s ataxia. J. Neurol. Neurosurg. Psychiatry 1984, 47, 26–32. [Google Scholar] [CrossRef] [Green Version]
- Wessel, K.; Moschner, C.; Wandinger, K.P.; Kömpf, D.; Heide, W. Oculomotor testing in the differential diagnosis of degenerative ataxic disorders. Arch. Neurol. 1998, 55, 949–956. [Google Scholar] [CrossRef] [Green Version]
- Bronstein, A.M. Vision and vertigo: Some visual aspects of vestibular disorders. J. Neurol. 2004, 251, 381–387. [Google Scholar] [CrossRef] [PubMed]
- Biglan, K.M.; Halmagyi, M. The eyes as a window into disease prevention. Neurology 2006, 67, 376–377. [Google Scholar] [CrossRef] [PubMed]
- Pulst, S.M. Ataxia rating scales in the balance. Nat. Clin. Pract. Neurol. 2007, 3, 119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carelli, V.; Ross-Cisneros, F.N.; Sadun, A.A. Mitochondrial dysfunction as a cause of optic neuropathies. Prog. Retin. Eye Res. 2004, 23, 53–89. [Google Scholar] [CrossRef] [PubMed]
- Yu-Wai-Man, P.; Griffiths, P.G.; Hudson, G.; Chinnery, P.F. Inherited mitochondrial optic neuropathies. J. Med. Genet. 2008, 46, 145–158. [Google Scholar] [CrossRef] [Green Version]
- Hwang, T.J.; Karanjia, R.; Moraes-Filho, M.N.; Gale, J.; Tran, J.S.; Chu, E.R.; Salomao, S.R.; Berezovsky, A.; Belfort, R.; Moraes, M.N.; et al. Natural History of Conversion of Leber’s Hereditary Optic Neuropathy. Ophthalmology 2017, 124, 843–850. [Google Scholar] [CrossRef] [PubMed]
- Rojas, P.; Ramírez, A.I.; de Hoz, R.; Cadena, M.; Ferreras, A.; Monsalve, B.; Salobrar-García, E.; Muñoz-Blanco, J.L.; Urcelay-Segura, J.L.; Salazar, J.J.; et al. Ocular Involvement in Friedreich Ataxia Patients and Its Relationship with Neurological Disability, a Follow-Up Study. Diagnostics 2020, 10, 75. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thomas-Black, G.J.; Parkinson, M.H.; Bremner, F.; Giunti, P. Peripapillary retinal nerve fibre layer thickness in Friedreich’s ataxia: A biomarker for trials? Brain 2019, 142, e23. [Google Scholar] [CrossRef]
- Curcio, C.A.; Allen, K.A. Topography of ganglion cells in human retina. J. Comp. Neurol. 1990, 300, 5–25. [Google Scholar] [CrossRef]
- Fitzgibbon, T.; Taylor, S.F. Retinotopy of the human retinal nerve fibre layer and optic nerve head. J. Comp. Neurol. 1996, 375, 238–251. [Google Scholar] [CrossRef]
- Ogden, T.E. Nerve fiber layer of the macaque retina: Retinotopic organization. Investig. Ophthalmol. Vis. Sci. 1983, 24, 85–98. [Google Scholar]
- La Morgia, C.; Di Vito, L.; Carelli, V.; Carbonelli, M. Patterns of Retinal Ganglion Cell Damage in Neurodegenerative Disorders: Parvocellular vs Magnocellular Degeneration in Optical Coherence Tomography Studies. Front. Neurol. 2017, 8, 710. [Google Scholar] [CrossRef] [PubMed]
- Crombie, D.E.; Van Bergen, N.; Davidson, K.C.; Anjomani Virmouni, S.; Mckelvie, P.A.; Chrysostomou, V.; Conquest, A.; Corben, L.A.; Pook, M.A.; Kulkarni, T.; et al. Characterization of the retinal pigment epithelium in Friedreich ataxia. Biochem. Biophys. Rep. 2015, 4, 141–147. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chandran, V.; Gao, K.; Swarup, V.; Versano, R.; Dong, H.; Jordan, M.C.; Geschwind, D.H. Inducible and reversible phenotypes in a novel mouse model of Friedreich’s ataxia. eLife 2017, 6, e30054. [Google Scholar] [CrossRef] [PubMed]
- Saxena, S.; Srivastav, K.; Cheung, C.M.; Ng Yui Wing, J.; Lai, T. Photoreceptor inner segment ellipsoid band integrity on spectral domain optical coherence tomography. Clin. Ophthalmol. 2014, 8, 2507. [Google Scholar] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Clinical Symptoms | Mean Length of GAA Repeat Expansions | References |
|---|---|---|
| Horizontal nystagmus | Between 630 ± 230 and 890 ± 230 | [29] |
| Saccadic-pursuit eye movements | ||
| Reduced visual acuity | ||
| Axonal neuropathy | ||
| Abnormal visual evoked potentials |
| Changes in FRDA Patients | Main Features | References |
|---|---|---|
| Oculomotor function | Dysmetria in saccadic movements. Interruption of tracking movements. Vestibular ocular reflexes altered. Fixation instability with frequent square wave jerks | [11,13,43,45,46,47,48,49,50] |
| Neurophysiology | VEP: altered | [11,39,43,51,52] |
| ERG: Usually normal or minimally abnormal | [39,43] | |
| Color vision | Normal | [39] |
| Contrast sensitivity | Supplementary role of low-contrast Sloan Chart testing in the assessment of disease status and visual function in FRDA | [53,54] |
| Correlation with mean pRNFL thickness and binocular VA with 1.25% and 2.5% contrast | [55] | |
| Low-contrast visual acuity drop-off linearly with time in FRDA, especially at longer GAA repeat lengths | [56] | |
| Spatial perception | Impairment in spatial construction tests | [57] |
| Visual acuity | Loss of visual acuity is rare | [11,53] |
| 30% of cases with optic nerve atrophy | [16] | |
| Chronic and progressive impairment, late-stage effects occur | [4,11,14,23,58] | |
| Correlation with mean pRNFL thickness and high-contrast VA | [55] | |
| Reduced BCVA can range from mild to severe and is caused by optic atrophy, retinal degeneration, or both | [50] | |
| There is a subgroup mimicking Leber’s hereditary optic neuropathy with severe BCVA affectation | [41,42] | |
| Visual field | Three patterns of involvement ranging from reduced sensitivity in a paracentral area, followed by concentric superior and/or inferior defects, to a general and concentric reduction in sensitivity in later stages | [4,11,58] |
| OCT | In detail in Table 3 | |
| Visual pathway involvement | Retina: retinosis pigmentaria-like syndrome | [11,14,22,23] |
| Anterior (optic nerve) | [11] | |
| Posterior (optical radiations) | ||
| Author | Study Type | OCT | Mean Age ± SD | pRNFL | Macular Thickness | GCC | Correlation OCT with Neurological Disability (Clinical Scale) |
|---|---|---|---|---|---|---|---|
| Fortuna et al. 2009 [11] | Cross-sectional study | TD- Stratus | 32.00 ± 8.00 | ↓ mean RNFL | Not documented | Not documented | pRNFL with ICARS r = –0.576 |
| ↓ 4 quadrants | |||||||
| Noval et al. 2012 [55] | Cross-sectional study | TD- Stratus | 25.22 ± 6.69 | ↓ 75% pRNFL | Normal foveal thickness and macular volume | Not documented | pRNFL with ICARS (RE r = 0.638 and LE r = 0.695) |
| Normal temporal quadrant | pRNFL with FARS (RE r = 0.531 and LE non-significant) | ||||||
| Seyer et al. 2013 [22] | Cross-sectional study | TD- Stratus for RNFL | 28.20 ± 15.90 | ↓ pRNFL | Not documented | Not documented | pRNFL with FARS (r = −0.72) |
| ↓ 4 quadrants | |||||||
| SD-Cirrus for macula | Not documented | ↓ Macular thickness (20.7%) | Not documented | Not documented | |||
| Dağ et al. 2014 [4] | Cross-sectional study | SD RS-3000 | 32.10 ± 10.46 | ↓ pRNFL | ↓ CMT | ↓GCC (S and I) | pRNFL with ICARS (r not documented) |
| ↓ 4 quadrants | |||||||
| Thomas-Black et al. 2019 [80] | Cross-sectional study | TD-Stratus | 32.0 ± 11.80 | Values not compared with control | Not documented | Not documented | pRNFL with SARA r = −0.457 |
| Rojas et al. 2020 [79] | 6-month follow-up study | SD-Cirrus | 35.00 ± 10.36 | ↓ RNFL | Normal CMT | ↓GCC | Several OCT parameters with SARA (most relevants): |
| ↓ 4 quadrants (I > S > N > T) | ↓ Macular thickness (IMR: S, N, I; OMR: N, Cube Vol.) | pRNFL (r = −0.693), T-Q (r = −0.803); H10 (r = −0.783) | |||||
| ↓ 4 H5-H11 | AMI-S (r = −0.507) | ||||||
| GCC [I-T (r = −0.679)] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rojas, P.; de Hoz, R.; Cadena, M.; Salobrar-García, E.; Fernández-Albarral, J.A.; López-Cuenca, I.; Elvira-Hurtado, L.; Urcelay-Segura, J.L.; Salazar, J.J.; Ramírez, J.M.; et al. Neuro-Ophthalmological Findings in Friedreich’s Ataxia. J. Pers. Med. 2021, 11, 708. https://doi.org/10.3390/jpm11080708
Rojas P, de Hoz R, Cadena M, Salobrar-García E, Fernández-Albarral JA, López-Cuenca I, Elvira-Hurtado L, Urcelay-Segura JL, Salazar JJ, Ramírez JM, et al. Neuro-Ophthalmological Findings in Friedreich’s Ataxia. Journal of Personalized Medicine. 2021; 11(8):708. https://doi.org/10.3390/jpm11080708
Chicago/Turabian StyleRojas, Pilar, Rosa de Hoz, Manuel Cadena, Elena Salobrar-García, José A. Fernández-Albarral, Inés López-Cuenca, Lorena Elvira-Hurtado, José L. Urcelay-Segura, Juan J. Salazar, José M. Ramírez, and et al. 2021. "Neuro-Ophthalmological Findings in Friedreich’s Ataxia" Journal of Personalized Medicine 11, no. 8: 708. https://doi.org/10.3390/jpm11080708
APA StyleRojas, P., de Hoz, R., Cadena, M., Salobrar-García, E., Fernández-Albarral, J. A., López-Cuenca, I., Elvira-Hurtado, L., Urcelay-Segura, J. L., Salazar, J. J., Ramírez, J. M., & Ramírez, A. I. (2021). Neuro-Ophthalmological Findings in Friedreich’s Ataxia. Journal of Personalized Medicine, 11(8), 708. https://doi.org/10.3390/jpm11080708