
Update on Multiple Sclerosis Molecular Biomarkers to Monitor Treatment Effects



Abstract
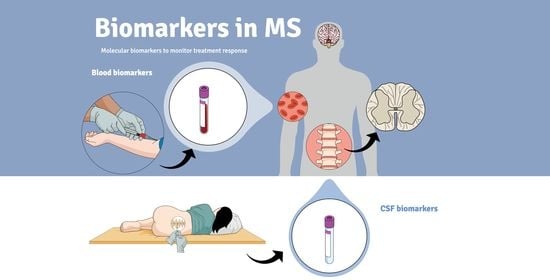
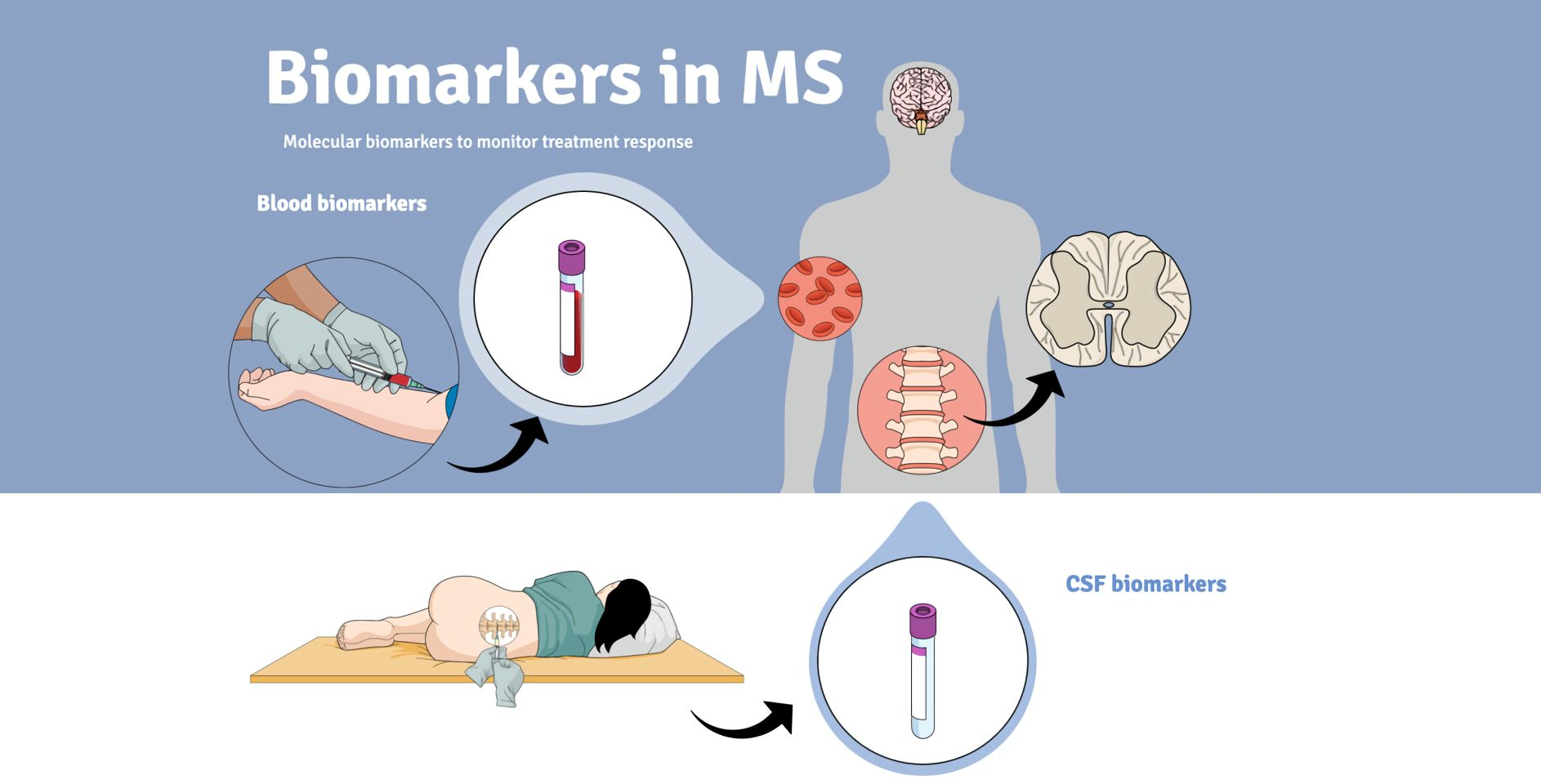
:
1. Introduction
2. Methods
3. Precision Medicine in Multiple Sclerosis
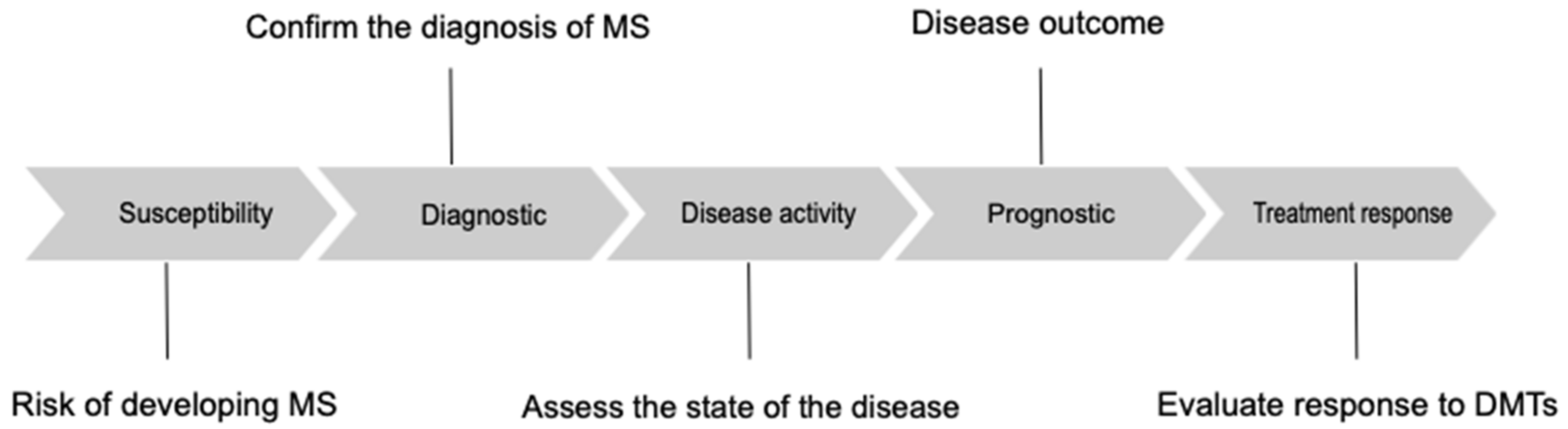
3.1. Definition and Categorization of Biomarkers
3.2. Biomarkers in Multiple Sclerosis
4. Treatment-Response Biomarkers in Multiple Sclerosis
4.1. Oligoclonal Bands
4.2. C-X-C Motif Chemokine 13
4.3. Osteopontin
4.4. Neutralizing Antibodies
4.4.1. Neutralizing Antibodies against Interferon-β
4.4.2. Neutralizing Antibodies against Natalizumab
4.5. Myxovirus Resistance Protein A
4.6. Neurofilaments
4.7. Chitinase 3-like Protein 1
5. Conclusions

{kind=link}
{kind=link}
| Biomarker | Function | References |
|---|---|---|
| OCBs | IgG or IgM antibodies synthesized intrathecally by plasma cells | [16,17,18,19,20,21,22,23,24] |
| CXCL13 | Chemokine expressed in lymphoid organs, essential for the recruitment of lymphocytes | [25,26,27,28,29,30,31,32,33,34,35,36,37] |
| Osteopontin | Pro-inflammatory cytokine secreted by activated immune cells | [38,39,40,41,42,43,44,45,46,47,48] |
| NAbs against IFN-ß | Serum antibodies against IFNβ | [49,50,51,52,53,54,55,56] |
| NAbs against natalizumab | Serum antibodies against natalizumab | [57,58,59,60,61,62,63,64,65,66,67,68,69,70,71,72] |
| MxA | Antiviral protein induced by IFNβ | [56,63,64,65] |
| Neurofilaments | Axonal cytoskeletal proteins | [66,67,68,69,70,71,72,73,74,75,76,77,78,79,80,81,82,83,84] |
| CHI3L1 | Chitinase-like glycoprotein, expressed by astrocytes and macrophages | [85,86,87,88,89,90,91,92,93,94,95,96,97,98] |
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Naegele, M.; Martin, R. The good and the bad of neuroinflammation in multiple sclerosis. Handb. Clin. Neurol. 2014, 122, 59–87. [Google Scholar] [CrossRef] [PubMed]
- Dendrou, C.A.; Fugger, L.; Friese, M.A. Immunopathology of multiple sclerosis. Nat. Rev. Immunol. 2015, 15, 545–558. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.; Baranzini, S.E.; Geurts, J.; Hemmer, B.; Ciccarelli, O. Multiple sclerosis. Lancet 2018, 391, 1622–1636. [Google Scholar] [CrossRef] [PubMed]
- Thompson, A.J.; Banwell, B.L.; Barkhof, F.; Carroll, W.M.; Coetzee, T.; Comi, G.; Correale, J.; Fazekas, F.; Filippi, M.; Freedman, M.S.; et al. Diagnosis of multiple sclerosis: 2017 revisions of the McDonald criteria. Lancet Neurol. 2018, 17, 162–173. [Google Scholar] [CrossRef]
- Tur, C.; Moccia, M.; Barkhof, F.; Chataway, J.; Sastre-Garriga, J.; Thompson, A.J.; Ciccarelli, O. Assessing treatment outcomes in multiple sclerosis trials and in the clinical setting. Nat. Rev. Neurol. 2018, 14, 75–93. [Google Scholar] [CrossRef]
- Oh, J.; Vidal-Jordana, A.; Montalban, X. Multiple sclerosis: Clinical aspects. Curr. Opin. Neurol. 2018, 31, 752–759. [Google Scholar] [CrossRef]
- Pachner, A.R.; DiSano, K.; Royce, D.B.; Gilli, F. Clinical utility of a molecular signature in inflammatory demyelinating disease. Neurol.-Neuroimmunol. Neuroinflamm. 2019, 6, e520. [Google Scholar] [CrossRef] [Green Version]
- Paul, A.; Comabella, M.; Gandhi, R. Biomarkers in Multiple Sclerosis. Cold Spring Harb. Perspect. Med. 2019, 9, a029058. [Google Scholar] [CrossRef]
- Ziemssen, T.; Akgün, K.; Brück, W. Molecular biomarkers in multiple sclerosis. J Neuroinflamm. 2019, 16, 272. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization; International Programme on Chemical Safety. Biomarkers in Risk Assessment: Validity and Validation; World Health Organization: Geneva, Switzerland, 2001. [Google Scholar]
- BDW Group; Atkinson, A.J., Jr.; Colburn, W.A.; DeGruttola, W.G.; DeMets, D.L.; Downing, G.J.; Hoth, D.F.; Oates, J.A.; Peck, C.C.; Schooley, R.T.; et al. Biomarkers and surrogate endpoints: Preferred definitions and conceptual framework. Clin. Pharmacol. Ther. 2001, 69, 89–95. [Google Scholar] [CrossRef]
- Mishina, E.; FDA—NIH Biomarker Working Group. BEST (Biomarkers, EndpointS, and Other Tools) Resource [Internet]. Monitoring Biomarker. Silver Spring (MD): Food and Drug Administration (US); 2016; Co-Published by National Institutes of Health (US): Bethesda, MD, USA. 2020. Available online: https://www.ncbi.nlm.nih.gov/books/NBK326791/ (accessed on 27 March 2022).
- Califf, R.M. Biomarker definitions and their applications. Exp. Biol. Med. 2018, 243, 213–221. [Google Scholar] [CrossRef]
- Housley, W.J.; Pitt, D.; Hafler, D.A. Biomarkers in multiple sclerosis. Clin. Immunol. 2015, 161, 51–58. [Google Scholar] [CrossRef]
- Ziemssen, T.; Derfuss, T.; de Stefano, N.; Giovannoni, G.; Palavra, F.; Tomic, D.; Vollmer, T.; Schippling, S. Optimizing treatment success in multiple sclerosis. J. Neurol. 2016, 263, 1053–1065. [Google Scholar] [CrossRef] [Green Version]
- Arrambide, G.; Tintore, M.; Espejo, C.; Auger, C.; Castillo, M.; Río, J.; Castilló, J.; Vidal-Jordana, A.; Galán, I.; Nos, C.; et al. The value of oligoclonal bands in the multiple sclerosis diagnostic criteria. Brain 2018, 141, 1075–1084. [Google Scholar] [CrossRef] [Green Version]
- Dobson, R.; Ramagopalan, S.; Davis, A.; Giovannoni, G. Cerebrospinal fluid oligoclonal bands in multiple sclerosis and clinically isolated syndromes: A meta-analysis of prevalence, prognosis and effect of latitude. J. Neurol. Neurosurg. Psychiatry 2013, 84, 909–914. [Google Scholar] [CrossRef]
- Boyko, A. Radiologically isolated syndrome with oligoclonal bands in CSF (RIS + OCB) can be classified as high MS risk group. Mult. Scler. J. 2020, 26, 869–870. [Google Scholar] [CrossRef]
- Chu, A.B.; Sever, J.L.; Madden, D.L.; Iivanainen, M.; Leon, M.; Wallen, W.; Brooks, B.R.; Lee, Y.J.; Houff, S. Oligoclonal IgG bands in cerebrospinal fluid in various neurological diseases. Ann. Neurol. 1983, 13, 434–439. [Google Scholar] [CrossRef]
- Villar, L.M.; García-Sánchez, M.I.; Costa-Frossard, L.; Espiño, M.; Roldán, E.; Páramo, D.; Lucas, M.; Izquierdo, G.; Álvarez-Cermeño, J.C. Immunological Markers of Optimal Response to Natalizumab in Multiple Sclerosis. Arch. Neurol. 2012, 69, 191–197. [Google Scholar] [CrossRef] [Green Version]
- Mancuso, R.; Franciotta, D.; Rovaris, M.; Caputo, D.; Sala, A.; Hernis, A.; Agostini, S.; Calvo, M.; Clerici, M. Effects of natalizumab on oligoclonal bands in the cerebrospinal fluid of multiple sclerosis patients: A longitudinal study. Mult. Scler. J. 2014, 20, 1900–1903. [Google Scholar] [CrossRef]
- von Glehn, F.; Farias, A.S.; de Oliveira, A.C.; Damasceno, A.; Longhini, A.L.; Oliveira, E.C.; Damasceno, B.P.; Santos, L.M.; Brandão, C.O. Disappearance of cerebrospinal fluid oligoclonal bands after natalizumab treatment of multiple sclerosis patients. Mult. Scler. J. 2012, 18, 1038–1041. [Google Scholar] [CrossRef]
- Rejdak, K.; Stelmasiak, Z.; Grieb, P. Cladribine induces long lasting oligoclonal bands disappearance in relapsing multiple sclerosis patients: 10-year observational study. Mult. Scler. Relat. Disord. 2019, 27, 117–120. [Google Scholar] [CrossRef]
- Annunziata, P.; Giorgio, A.; De Santi, L.; Zipoli, V.; Portaccio, E.; Amato, M.P.; Clerici, R.; Scarpini, E.; Moscato, G.; Iudice, A.; et al. Absence of cerebrospinal fluid oligoclonal bands is associated with delayed disability progression in relapsing-remitting MS patients treated with interferon-beta. J. Neurol. Sci. 2006, 244, 97–102. [Google Scholar] [CrossRef]
- Legler, D.F.; Loetscher, M.; Roos, R.S.; Clark-Lewis, I.; Baggiolini, M.; Moser, B. B cell-attracting chemokine 1, a human CXC chemokine expressed in lymphoid tissues, selectively attracts B lymphocytes via BLR1/CXCR5. J. Exp. Med. 1998, 187, 655–660. [Google Scholar] [CrossRef] [PubMed]
- Pilz, G.; Sakic, I.; Wipfler, P.; Kraus, J.; Haschke-Becher, E.; Hitzl, W.; Trinka, E.; Harrer, A. Chemokine CXCL13 in serum, CSF and blood–CSF barrier function: Evidence of compartment restriction. Fluids Barriers CNS 2020, 17, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sellebjerg, F.; Börnsen, L.; Khademi, M.; Krakauer, M.; Olsson, T.; Frederiksen, J.L.; Sørensen, P.S. Increased cerebrospinal fluid concentrations of the chemokine CXCL13 in active MS. Neurology 2009, 73, 2003–2010. [Google Scholar] [CrossRef] [PubMed]
- Alvarez, E.; Piccio, L.; Mikesell, R.J.; Klawiter, E.C.; Parks, B.J.; Naismith, R.T.; Cross, A.H. CXCL13 is a biomarker of inflammation in multiple sclerosis, neuromyelitis optica, and other neurological conditions. Mult. Scler. 2013, 19, 1204–1208. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krumbholz, M.; Theil, D.; Cepok, S.; Hemmer, B.; Kivisäkk, P.; Ransohoff, R.M.; Hofbauer, M.; Farina, C.; Derfuss, T.; Hartle, C.; et al. Chemokines in multiple sclerosis: CXCL12 and CXCL13 up-regulation is differentially linked to CNS immune cell recruitment. Brain 2006, 129, 200–211. [Google Scholar] [CrossRef] [Green Version]
- Brettschneider, J.; Czerwoniak, A.; Senel, M.; Fang, L.; Kassubek, J.; Pinkhardt, E.; Lauda, F.; Kapfer, T.; Jesse, S.; Lehmensiek, V.; et al. The chemokine CXCL13 is a prognostic marker in clinically isolated syndrome (CIS). PLoS ONE 2010, 5, e11986. [Google Scholar] [CrossRef]
- Khademi, M.; Kockum, I.; Andersson, M.L.; Iacobaeus, E.; Brundin, L.; Sellebjerg, F.; Hillert, J.; Piehl, F.; Olsson, T. Cerebrospinal fluid CXCL13 in multiple sclerosis: A suggestive prognostic marker for the disease course. Mult. Scler. J. 2011, 17, 335–343. [Google Scholar] [CrossRef]
- Festa, E.D.; Hankiewicz, K.; Kim, S.; Skurnick, J.; Wolansky, L.J.; Cook, S.D.; Cadavid, D. Serum levels of CXCL13 are elevated in active multiple sclerosis. Mult. Scler. J. 2009, 15, 1271–1279. [Google Scholar] [CrossRef]
- Rupprecht, T.A.; Pfister, H.W.; Angele, B.; Kastenbauer, S.; Wilske, B.; Koedel, U. The chemokine CXCL13 (BLC): A putative diagnostic marker for neuroborreliosis. Neurology 2005, 65, 448. [Google Scholar] [CrossRef]
- Kowarik, M.C.; Cepok, S.; Sellner, J.; Grummel, V.; Weber, M.S.; Korn, T.; Berthele, A.; Hemmer, B. CXCL13 is the major determinant for B cell recruitment to the CSF during neuroinflammation. J. Neuroinflamm. 2012, 9, 93. [Google Scholar] [CrossRef] [Green Version]
- Novakova, L.; Axelsson, M.; Khademi, M.; Zetterberg, H.; Blennow, K.; Malmeström, C.; Piehl, F.; Olsson, T.; Lycke, J. Cerebrospinal fluid biomarkers as a measure of disease activity and treatment efficacy in relapsing-remitting multiple sclerosis. J. Neurochem. 2017, 141, 296–304. [Google Scholar] [CrossRef] [Green Version]
- Alvarez, E.; Piccio, L.; Mikesell, R.J.; Trinkaus, K.; Parks, B.J.; Naismith, R.T.; Cross, A.H. Predicting optimal response to B-cell depletion with rituximab in multiple sclerosis using CXCL13 index, magnetic resonance imaging and clinical measures. Mult. Scler. J. Exp. Transl. Clin. 2015, 1, 2055217315623800. [Google Scholar] [CrossRef] [Green Version]
- Piccio, L.; Naismith, R.T.; Trinkaus, K.; Klein, R.S.; Parks, B.J.; Lyons, J.A.; Cross, A.H. Changes in B- and T-lymphocyte and chemokine levels with rituximab treatment in multiple sclerosis. Arch. Neurol. 2010, 67, 707–714. [Google Scholar] [CrossRef] [Green Version]
- Denhardt, D.T.; Noda, M.; O’Regan, A.W.; Pavlin, D.; Berman, J.S. Osteopontin as a means to cope with environmental insults: Regulation of inflammation, tissue remodeling, and cell survival. J. Clin. Invest. 2001, 107, 1055–1061. [Google Scholar] [CrossRef] [Green Version]
- Lund, S.A.; Giachelli, C.M.; Scatena, M. The role of osteopontin in inflammatory processes. J. Cell Commun. Signal. 2009, 3, 311–322. [Google Scholar] [CrossRef] [Green Version]
- Shimizu, Y.; Ota, K.; Ikeguchi, R.; Kubo, S.; Kabasawa, C.; Uchiyama, S. Plasma osteopontin levels are associated with disease activity in the patients with multiple sclerosis and neuromyelitis optica. J. Neuroimmunol. 2013, 263, 148–151. [Google Scholar] [CrossRef]
- Agah, E.; Zardoui, A.; Saghazadeh, A.; Ahmadi, M.; Tafakhori, A.; Rezaei, N. Osteopontin (OPN) as a CSF and blood biomarker for multiple sclerosis: A systematic review and meta-analysis. PLoS ONE 2018, 13, e0190252. [Google Scholar] [CrossRef]
- Braitch, M.; Nunan, R.; Niepel, G.; Edwards, L.J.; Constantinescu, C.S. Increased Osteopontin Levels in the Cerebrospinal Fluid of Patients With Multiple Sclerosis. Arch. Neurol. 2008, 65, 633–635. [Google Scholar] [CrossRef] [Green Version]
- Szalardy, L.; Zadori, D.; Simu, M.; Bencsik, K.; Vecsei, L.; Klivenyi, P. Evaluating biomarkers of neuronal degeneration and neuroinflammation in CSF of patients with multiple sclerosis-osteopontin as a potential marker of clinical severity. J. Neurol. Sci. 2013, 331, 38–42. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Börnsen, L.; Khademi, M.; Olsson, T.; Sørensen, P.S.; Sellebjerg, F. Osteopontin concentrations are increased in cerebrospinal fluid during attacks of multiple sclerosis. Mult. Scler. J. 2011, 17, 32–42. [Google Scholar] [CrossRef] [PubMed]
- Marastoni, D.; Magliozzi, R.; Bolzan, A.; Pisani, A.I.; Rossi, S.; Crescenzo, F.; Montemezzi, S.; Pizzini, F.B.; Calabrese, M. CSF Levels of CXCL12 and Osteopontin as Early Markers of Primary Progressive Multiple Sclerosis. Neurol. Neuroimmunol. Neuroinflamm. 2021, 8. [Google Scholar] [CrossRef] [PubMed]
- Comabella, M.; Pericot, I.; Goertsches, R.; Nos, C.; Castillo, M.; Blas Navarro, J.; Río, J.; Montalban, X. Plasma osteopontin levels in multiple sclerosis. J. Neuroimmunol. 2005, 158, 231–239. [Google Scholar] [CrossRef] [PubMed]
- Kivisäkk, P.; Healy, B.C.; Francois, K.; Gandhi, R.; Gholipour, T.; Egorova, S.; Sevdalinova, V.; Quintana, F.; Chitnis, T.; Weiner, H.L.; et al. Evaluation of circulating osteopontin levels in an unselected cohort of patients with multiple sclerosis: Relevance for biomarker development. Mult. Scler. J. 2014, 20, 438–444. [Google Scholar] [CrossRef] [PubMed]
- Romme Christensen, J.; Ratzer, R.; Börnsen, L.; Lyksborg, M.; Garde, E.; Dyrby, T.B.; Siebner, H.R.; Sorensen, P.S.; Sellebjerg, F. Natalizumab in progressive MS: Results of an open-label, phase 2A, proof-of-concept trial. Neurology 2014, 82, 1499–1507. [Google Scholar] [CrossRef] [PubMed]
- Rudick, R.A.; Lee, J.C.; Simon, J.; Ransohoff, R.M.; Fisher, E. Defining interferon beta response status in multiple sclerosis patients. Ann. Neurol. 2004, 56, 548–555. [Google Scholar] [CrossRef]
- Duquette, P.; Girard, M.; Dubois, R.; Kobler, R.L.; Lublin, F.; Kelley, L.; Francis, C.S.; Freedman, M.; Greenstein, J.I.; Mishra, B.; et al. Neutralizing antibodies during treatment of multiple sclerosis with interferon beta-1b. Exp. Dur. First Three Years 1996, 47, 889–894. [Google Scholar] [CrossRef]
- Bertolotto, A.; Deisenhammer, F.; Gallo, P.; Sölberg Sørensen, P. Immunogenicity of interferon beta: Differences among products. J. Neurol. 2004, 251 (Suppl. 2), ii15–ii24. [Google Scholar] [CrossRef]
- Link, J.; Ramanujam, R.; Auer, M.; Ryner, M.; Hässler, S.; Bachelet, D.; Mbogning, C.; Warnke, C.; Buck, D.; Hyldgaard Jensen, P.E.; et al. Clinical practice of analysis of anti-drug antibodies against interferon beta and natalizumab in multiple sclerosis patients in Europe: A descriptive study of test results. PLoS ONE 2017, 12, e0170395. [Google Scholar] [CrossRef]
- Sorensen, P.S.; Ross, C.; Clemmesen, K.M.; Bendtzen, K.; Frederiksen, J.L.; Jensen, K.; Kristensen, O.; Petersen, T.; Rasmussen, S.; Ravnborg, M.; et al. Clinical importance of neutralising antibodies against interferon beta in patients with relapsing-remitting multiple sclerosis. Lancet 2003, 362, 1184–1191. [Google Scholar] [CrossRef]
- Kappos, L.; Clanet, M.; Sandberg-Wollheim, M.; Radue, E.W.; Hartung, H.P.; Hohlfeld, R.; Xu, J.; Bennett, D.; Sandrock, A.; Goelz, S. Neutralizing antibodies and efficacy of interferon beta-1a: A 4-year controlled study. Neurology 2005, 65, 40–47. [Google Scholar] [CrossRef]
- Tomassini, V.; Paolillo, A.; Russo, P.; Giugni, E.; Prosperini, L.; Gasperini, C.; Antonelli, G.; Bastianello, S.; Pozzilli, C. Predictors of long-term clinical response to interferon beta therapy in relapsing multiple sclerosis. J. Neurol. 2006, 253, 287–293. [Google Scholar] [CrossRef]
- Polman, C.H.; Bertolotto, A.; Deisenhammer, F.; Giovannoni, G.; Hartung, H.P.; Hemmer, B.; Killestein, J.; McFarland, H.F.; Oger, J.; Pachner, A.R.; et al. Recommendations for clinical use of data on neutralising antibodies to interferon-beta therapy in multiple sclerosis. Lancet Neurol. 2010, 9, 740–750. [Google Scholar] [CrossRef]
- Calabresi, P.A.; Giovannoni, G.; Confavreux, C.; Galetta, S.L.; Havrdova, E.; Hutchinson, M.; Kappos, L.; Miller, D.H.; Connor, P.W.; Phillips, J.T.; et al. The incidence and significance of anti-natalizumab antibodies. Neurology 2007, 69, 1391. [Google Scholar] [CrossRef]
- Lundkvist, M.; Engdahl, E.; Holmén, C.; Movérare, R.; Olsson, T.; Hillert, J.; Fogdell-Hahn, A. Characterization of anti-natalizumab antibodies in multiple sclerosis patients. Mult. Scler. J. 2013, 19, 757–764. [Google Scholar] [CrossRef] [Green Version]
- Deisenhammer, F.; Jank, M.; Lauren, A.; Sjödin, A.; Ryner, M.; Fogdell-Hahn, A.; Sievers, C.; Lindberg, R.; Jensen, P.E.; Sellebjerg, F.; et al. Prediction of natalizumab anti-drug antibodies persistency. Mult. Scler. J. 2019, 25, 392–398. [Google Scholar] [CrossRef]
- Vennegoor, A.; Rispens, T.; Strijbis, E.M.; Seewann, A.; Uitdehaag, B.M.; Balk, L.J.; Barkhof, F.; Polman, C.H.; Wolbink, G.; Killestein, J. Clinical relevance of serum natalizumab concentration and anti-natalizumab antibodies in multiple sclerosis. Mult. Scler. J. 2013, 19, 593–600. [Google Scholar] [CrossRef]
- Berger, J.R.; Fox, R.J. Reassessing the risk of natalizumab-associated PML. J. Neurovirol. 2016, 22, 533–535. [Google Scholar] [CrossRef]
- Sorensen, P.S.; Koch-Henriksen, N.; Petersen, T.; Ravnborg, M.; Oturai, A.; Sellebjerg, F. Recurrence or rebound of clinical relapses after discontinuation of natalizumab therapy in highly active MS patients. J. Neurol. 2014, 261, 1170–1177. [Google Scholar] [CrossRef]
- Dick, A.; Graf, L.; Olal, D.; von der Malsburg, A.; Gao, S.; Kochs, G.; Daumke, O. Role of nucleotide binding and GTPase domain dimerization in dynamin-like myxovirus resistance protein A for GTPase activation and antiviral activity. J. Biol. Chem. 2015, 290, 12779–12792. [Google Scholar] [CrossRef] [Green Version]
- Bertolotto, A. Implications of neutralising antibodies on therapeutic efficacy. J. Neurol. Sci. 2009, 277, S29–S32. [Google Scholar] [CrossRef]
- Furuyama, H.; Chiba, S.; Okabayashi, T.; Yokota, S.; Nonaka, M.; Imai, T.; Fujii, N.; Matsumoto, H. Single nucleotide polymorphisms and functional analysis of MxA promoter region in multiple sclerosis. J. Neurol. Sci. 2006, 249, 153–157. [Google Scholar] [CrossRef]
- Yuan, A.; Rao, M.V.; Veeranna; Nixon, R.A. Neurofilaments and Neurofilament Proteins in Health and Disease. Cold Spring Harb. Perspect. Biol. 2017, 9, a018309. [Google Scholar] [CrossRef] [Green Version]
- Petzold, A. Neurofilament phosphoforms: Surrogate markers for axonal injury, degeneration and loss. J. Neurol. Sci. 2005, 233, 183–198. [Google Scholar] [CrossRef] [Green Version]
- Khalil, M.; Teunissen, C.E.; Otto, M.; Piehl, F.; Sormani, M.P.; Gattringer, T.; Barro, C.; Kappos, L.; Comabella, M.; Fazekas, F.; et al. Neurofilaments as biomarkers in neurological disorders. Nat. Rev. Neurol. 2018, 14, 577–589. [Google Scholar] [CrossRef]
- Norgren, N.; Sundström, P.; Svenningsson, A.; Rosengren, L.; Stigbrand, T.; Gunnarsson, M. Neurofilament and glial fibrillary acidic protein in multiple sclerosis. Neurology 2004, 63, 1586–1590. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Iacobaeus, E.; Khademi, M.; Brundin, L.; Norgren, N.; Koel-Simmelink, M.J.; Schepens, M.; Bouwman, F.; Twaalfhoven, H.A.; Blom, H.J.; et al. Combination of CSF N-acetylaspartate and neurofilaments in multiple sclerosis. Neurology 2009, 72, 1322–1329. [Google Scholar] [CrossRef]
- Disanto, G.; Barro, C.; Benkert, P.; Naegelin, Y.; Schädelin, S.; Giardiello, A.; Zecca, C.; Blennow, K.; Zetterberg, H.; Leppert, D.; et al. Serum Neurofilament light: A biomarker of neuronal damage in multiple sclerosis. Ann. Neurol. 2017, 81, 857–870. [Google Scholar] [CrossRef]
- Thebault, S.; Abdoli, M.; Fereshtehnejad, S.-M.; Tessier, D.; Tabard-Cossa, V.; Freedman, M.S. Serum neurofilament light chain predicts long term clinical outcomes in multiple sclerosis. Sci. Rep. 2020, 10, 10381. [Google Scholar] [CrossRef]
- Siller, N.; Kuhle, J.; Muthuraman, M.; Barro, C.; Uphaus, T.; Groppa, S.; Kappos, L.; Zipp, F.; Bittner, S. Serum neurofilament light chain is a biomarker of acute and chronic neuronal damage in early multiple sclerosis. Mult. Scler. J. 2019, 25, 678–686. [Google Scholar] [CrossRef] [PubMed]
- Kuhle, J.; Leppert, D.; Petzold, A.; Regeniter, A.; Schindler, C.; Mehling, M.; Anthony, D.C.; Kappos, L.; Lindberg, R.L. Neurofilament heavy chain in CSF correlates with relapses and disability in multiple sclerosis. Neurology 2011, 76, 1206–1213. [Google Scholar] [CrossRef]
- Petzold, A.; Steenwijk, M.D.; Eikelenboom, J.M.; Wattjes, M.P.; Uitdehaag, B.M. Elevated CSF neurofilament proteins predict brain atrophy: A 15-year follow-up study. Mult. Scler. J. 2016, 22, 1154–1162. [Google Scholar] [CrossRef] [Green Version]
- Teunissen, C.E.; Khalil, M. Neurofilaments as biomarkers in multiple sclerosis. Mult. Scler. J. 2012, 18, 552–556. [Google Scholar] [CrossRef] [PubMed]
- Varhaug, K.N.; Barro, C.; Bjørnevik, K.; Myhr, K.-M.; Torkildsen, Ø.; Wergeland, S.; Bindoff, L.A.; Kuhle, J.; Vedeler, C. Neurofilament light chain predicts disease activity in relapsing-remitting MS. Neurol.-Neuroimmunol. Neuroinflamm. 2018, 5, e422. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Piehl, F.; Kockum, I.; Khademi, M.; Blennow, K.; Lycke, J.; Zetterberg, H.; Olsson, T. Plasma neurofilament light chain levels in patients with MS switching from injectable therapies to fingolimod. Mult. Scler. J. 2018, 24, 1046–1054. [Google Scholar] [CrossRef] [PubMed]
- Gunnarsson, M.; Malmeström, C.; Axelsson, M.; Sundström, P.; Dahle, C.; Vrethem, M.; Olsson, T.; Piehl, F.; Norgren, N.; Rosengren, L.; et al. Axonal damage in relapsing multiple sclerosis is markedly reduced by natalizumab. Ann. Neurol. 2011, 69, 83–89. [Google Scholar] [CrossRef]
- Axelsson, M.; Malmeström, C.; Gunnarsson, M.; Zetterberg, H.; Sundström, P.; Lycke, J.; Svenningsson, A. Immunosuppressive therapy reduces axonal damage in progressive multiple sclerosis. Mult. Scler. J. 2014, 20, 43–50. [Google Scholar] [CrossRef]
- de Flon, P.; Gunnarsson, M.; Laurell, K.; Söderström, L.; Birgander, R.; Lindqvist, T.; Krauss, W.; Dring, A.; Bergman, J.; Sundström, P.; et al. Reduced inflammation in relapsing-remitting multiple sclerosis after therapy switch to rituximab. Neurology 2016, 87, 141. [Google Scholar] [CrossRef]
- Kuhle, J.; Disanto, G.; Lorscheider, J.; Stites, T.; Chen, Y.; Dahlke, F.; Francis, G.; Shrinivasan, A.; Radue, E.W.; Giovannoni, G.; et al. Fingolimod and CSF neurofilament light chain levels in relapsing-remitting multiple sclerosis. Neurology 2015, 84, 1639–1643. [Google Scholar] [CrossRef] [Green Version]
- Hauser, S.L.; Bar-Or, A.; Cohen, J.A.; Comi, G.; Correale, J.; Coyle, P.K.; Cross, A.H.; de Seze, J.; Leppert, D.; Montalban, X.; et al. Ofatumumab versus Teriflunomide in Multiple Sclerosis. N. Engl. J. Med. 2020, 383, 546–557. [Google Scholar] [CrossRef]
- Kuhle, J.; Barro, C.; Andreasson, U.; Derfuss, T.; Lindberg, R.; Sandelius, Å.; Liman, V.; Norgren, N.; Blennow, K.; Zetterberg, H. Comparison of three analytical platforms for quantification of the neurofilament light chain in blood samples: ELISA, electrochemiluminescence immunoassay and Simoa. Clin. Chem. Lab. Med. (CCLM) 2016, 54, 1655–1661. [Google Scholar] [CrossRef]
- Lee, C.G.; Da Silva, C.A.; Dela Cruz, C.S.; Ahangari, F.; Ma, B.; Kang, M.-J.; He, C.-H.; Takyar, S.; Elias, J.A. Role of chitin and chitinase/chitinase-like proteins in inflammation, tissue remodeling, and injury. Annu. Rev. Physiol. 2011, 73, 479–501. [Google Scholar] [CrossRef] [Green Version]
- Cubas-Núñez, L.; Gil-Perotín, S.; Castillo-Villalba, J.; López, V.; Solís Tarazona, L.; Gasqué-Rubio, R.; Carratalá-Boscá, S.; Alcalá-Vicente, C.; Pérez-Miralles, F.; Lassmann, H.; et al. Potential Role of CHI3L1+ Astrocytes in Progression in MS. Neurol.-Neuroimmunol. Neuroinflamm. 2021, 8, e972. [Google Scholar] [CrossRef]
- Comabella, M.; Fernández, M.; Martin, R.; Rivera-Vallvé, S.; Borrás, E.; Chiva, C.; Julià, E.; Rovira, A.; Cantó, E.; Alvarez-Cermeño, J.C.; et al. Cerebrospinal fluid chitinase 3-like 1 levels are associated with conversion to multiple sclerosis. Brain 2010, 133, 1082–1093. [Google Scholar] [CrossRef] [Green Version]
- Hinsinger, G.; Galéotti, N.; Nabholz, N.; Urbach, S.; Rigau, V.; Demattei, C.; Lehmann, S.; Camu, W.; Labauge, P.; Castelnovo, G.; et al. Chitinase 3-like proteins as diagnostic and prognostic biomarkers of multiple sclerosis. Mult. Scler. J. 2015, 21, 1251–1261. [Google Scholar] [CrossRef]
- Malmeström, C.; Axelsson, M.; Lycke, J.; Zetterberg, H.; Blennow, K.; Olsson, B. CSF levels of YKL-40 are increased in MS and replaces with immunosuppressive treatment. J. Neuroimmunol. 2014, 269, 87–89. [Google Scholar] [CrossRef]
- Burman, J.; Raininko, R.; Blennow, K.; Zetterberg, H.; Axelsson, M.; Malmeström, C. YKL-40 is a CSF biomarker of intrathecal inflammation in secondary progressive multiple sclerosis. J. Neuroimmunol. 2016, 292, 52–57. [Google Scholar] [CrossRef] [Green Version]
- Correale, J.; Fiol, M. Chitinase effects on immune cell response in neuromyelitis optica and multiple sclerosis. Mult. Scler. J. 2011, 17, 521–531. [Google Scholar] [CrossRef]
- Gil-Perotin, S.; Castillo-Villalba, J.; Cubas-Nuñez, L.; Gasque, R.; Hervas, D.; Gomez-Mateu, J.; Alcala, C.; Perez-Miralles, F.; Gascon, F.; Dominguez, J.A.; et al. Combined Cerebrospinal Fluid Neurofilament Light Chain Protein and Chitinase-3 Like-1 Levels in Defining Disease Course and Prognosis in Multiple Sclerosis. Front. Neurol. 2019, 10, 1008. [Google Scholar] [CrossRef]
- Modvig, S.; Degn, M.; Horwitz, H.; Cramer, S.P.; Larsson, H.B.; Wanscher, B.; Sellebjerg, F.; Frederiksen, J.L. Relationship between cerebrospinal fluid biomarkers for inflammation, demyelination and neurodegeneration in acute optic neuritis. PLoS ONE 2013, 8, e77163. [Google Scholar] [CrossRef] [Green Version]
- Cantó, E.; Reverter, F.; Morcillo-Suárez, C.; Matesanz, F.; Fernández, O.; Izquierdo, G.; Vandenbroeck, K.; Rodríguez-Antigüedad, A.; Urcelay, E.; Arroyo, R.; et al. Chitinase 3-like 1 plasma levels are increased in patients with progressive forms of multiple sclerosis. Mult. Scler. J. 2012, 18, 983–990. [Google Scholar] [CrossRef] [Green Version]
- Thouvenot, E.; Hinsinger, G.; Demattei, C.; Uygunoglu, U.; Castelnovo, G.; Pittion-Vouyovitch, S.; Okuda, D.; Kantarci, O.; Pelletier, D.; Lehmann, S.; et al. Cerebrospinal fluid chitinase-3-like protein 1 level is not an independent predictive factor for the risk of clinical conversion in radiologically isolated syndrome. Mult. Scler. J. 2019, 25, 669–677. [Google Scholar] [CrossRef]
- Matute-Blanch, C.; Río, J.; Villar, L.M.; Midaglia, L.; Malhotra, S.; Álvarez-Cermeño, J.C.; Vidal-Jordana, A.; Montalban, X.; Comabella, M. Chitinase 3-like 1 is associated with the response to interferon-beta treatment in multiple sclerosis. J. Neuroimmunol. 2017, 303, 62–65. [Google Scholar] [CrossRef]
- Stoop, M.P.; Singh, V.; Stingl, C.; Martin, R.; Khademi, M.; Olsson, T.; Hintzen, R.Q.; Luider, T.M. Effects of natalizumab treatment on the cerebrospinal fluid proteome of multiple sclerosis patients. J. Proteome Res. 2013, 12, 1101–1107. [Google Scholar] [CrossRef]
- Novakova, L.; Axelsson, M.; Khademi, M.; Zetterberg, H.; Blennow, K.; Malmeström, C.; Piehl, F.; Olsson, T.; Lycke, J. Cerebrospinal fluid biomarkers of inflammation and degeneration as measures of fingolimod efficacy in multiple sclerosis. Mult. Scler. J. 2017, 23, 62–71. [Google Scholar] [CrossRef]
- Teunissen, C.E.; Malekzadeh, A.; Leurs, C.; Bridel, C.; Killestein, J. Body fluid biomarkers for multiple sclerosis—The long road to clinical application. Nat. Rev. Neurol. 2015, 11, 585–596. [Google Scholar] [CrossRef]

| Advantages | Disadvantages | Molecular Biomarkers | |
|---|---|---|---|
| Blood |
|
| NFL CHI3L1 Osteopontin MxA NAbs against natalizumab and INF-ß |
| CSF |
|
| NFL CXCL13 CHI3L1 OCBs Osteopontin |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Nociti, V.; Romozzi, M.; Mirabella, M. Update on Multiple Sclerosis Molecular Biomarkers to Monitor Treatment Effects. J. Pers. Med. 2022, 12, 549. https://doi.org/10.3390/jpm12040549
Nociti V, Romozzi M, Mirabella M. Update on Multiple Sclerosis Molecular Biomarkers to Monitor Treatment Effects. Journal of Personalized Medicine. 2022; 12(4):549. https://doi.org/10.3390/jpm12040549
Chicago/Turabian StyleNociti, Viviana, Marina Romozzi, and Massimiliano Mirabella. 2022. "Update on Multiple Sclerosis Molecular Biomarkers to Monitor Treatment Effects" Journal of Personalized Medicine 12, no. 4: 549. https://doi.org/10.3390/jpm12040549
APA StyleNociti, V., Romozzi, M., & Mirabella, M. (2022). Update on Multiple Sclerosis Molecular Biomarkers to Monitor Treatment Effects. Journal of Personalized Medicine, 12(4), 549. https://doi.org/10.3390/jpm12040549