

Elucidating the Potential Inhibitor against Type 2 Diabetes Mellitus Associated Gene of GLUT4

 , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Protein Selection and GLUT4 Sequence Retrieval
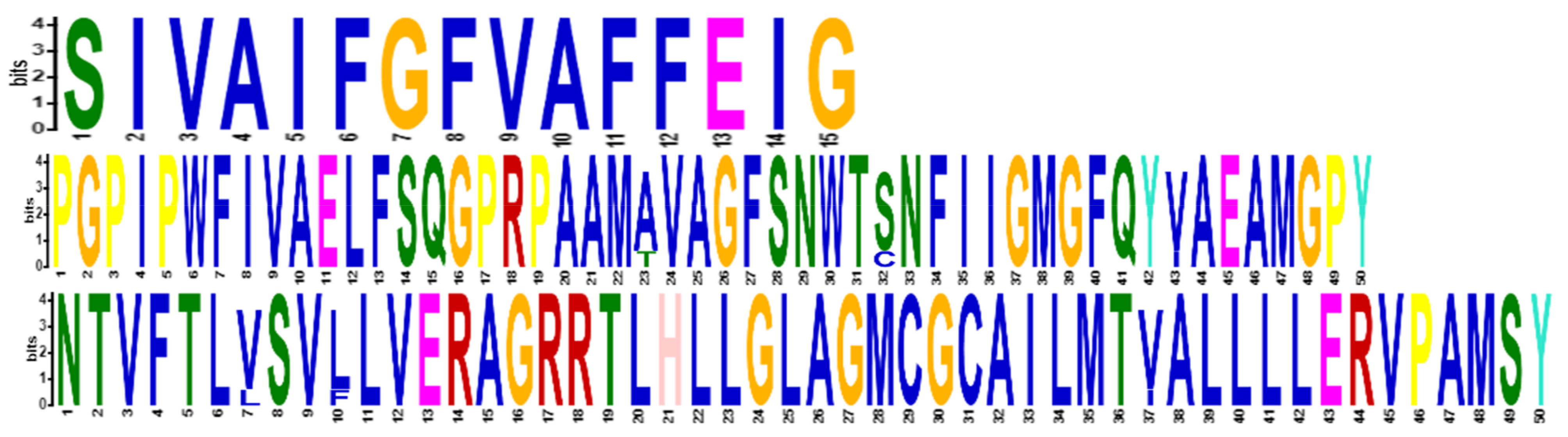
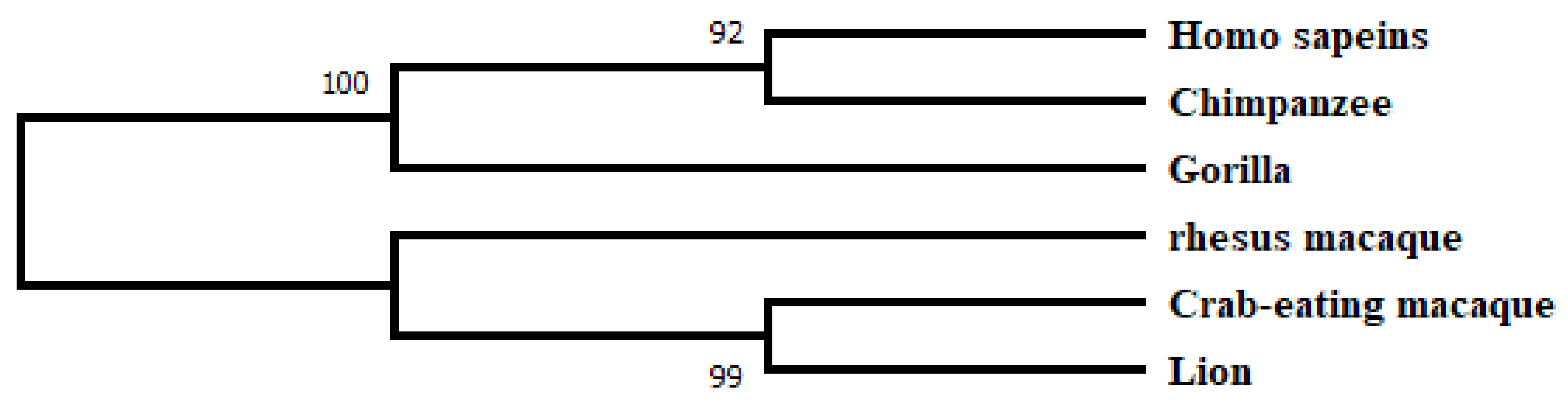
2.2. Phylogenetic Analysis and Motif Detection
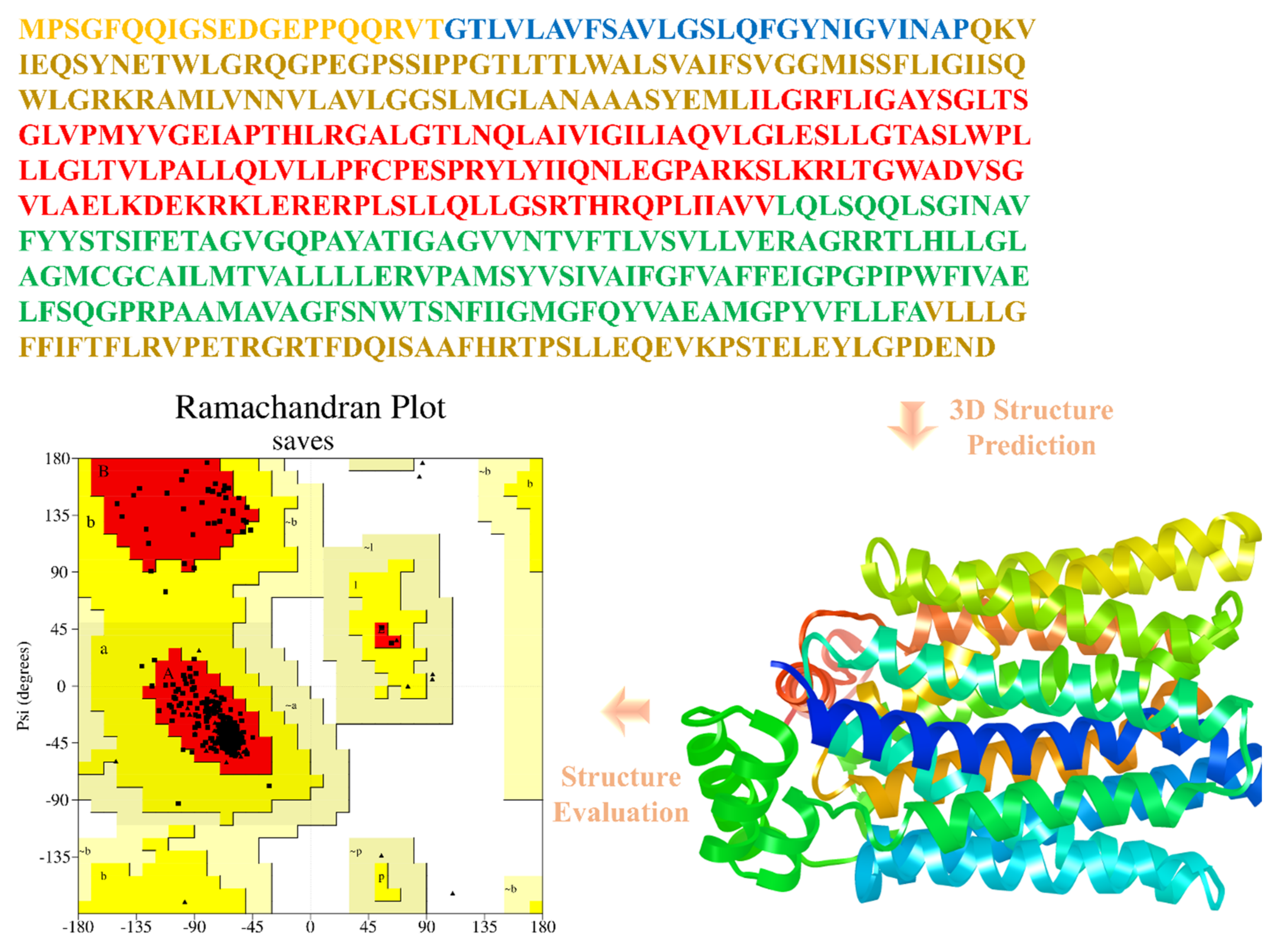
2.3. Protein 3D Structure Prediction and Evaluation
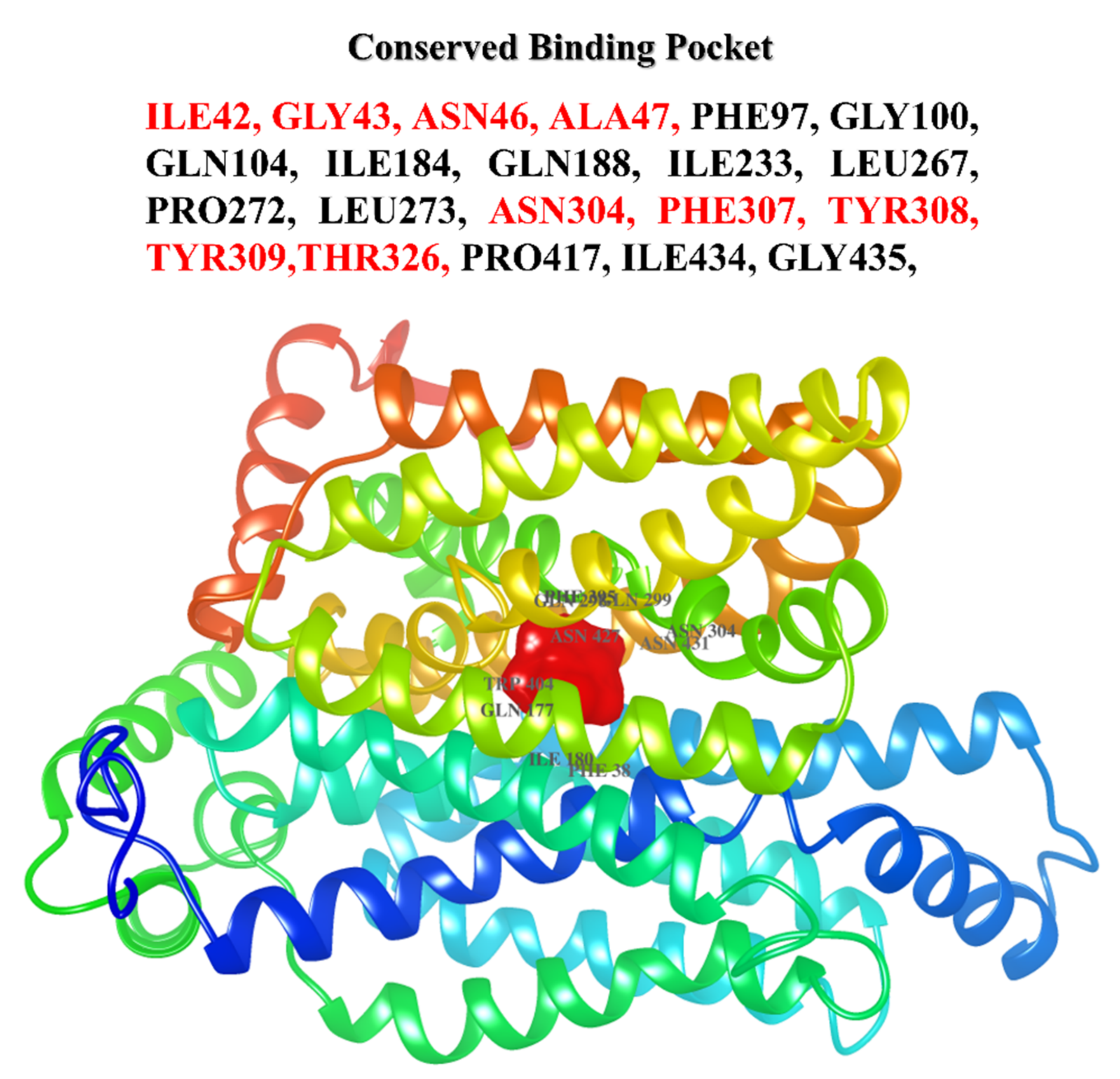
2.4. Binding Site Prediction
2.5. Virtual Screening and Molecular Docking Analysis
2.6. Physicochemical and ADMET Properties
2.7. Prediction of PAINS by Promiscuity Assessments
3. Results
3.1. Database Search, Comparative Phylogeny, and Physiochemical Properties Prediction
3.2. Structural Modeling, In Silico Characterization, and Model Validation
3.3. Principal Cavity Prediction
3.4. Molecular Docking Results
3.5. Physicochemical and ADMET Properties
3.6. PAINS Prediction
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Poznyak, A.; Grechko, A.V.; Poggio, P.; Myasoedova, V.A.; Alfieri, V.; Orekhov, A.N. The diabetes mellitus–atherosclerosis connection: The role of lipid and glucose metabolism and chronic inflammation. Int. J. Mol. Sci. 2020, 21, 1835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halim, M.; Halim, A. The effects of inflammation, aging, and oxidative stress on the pathogenesis of diabetes mellitus (type 2 diabetes). Diabetes Metab. Syndr. Clin. Res. Rev. 2019, 13, 1165–1172. [Google Scholar] [CrossRef] [PubMed]
- Pratley, R.; Weyer, C. The role of impaired early insulin secretion in the pathogenesis of type II diabetes mellitus. Diabetologia 2001, 44, 929–945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozougwu, J.C.; Obimba, K.C.; Belonwu, C.D.; Unakalamba, C.B. The pathogenesis and pathophysiology of type 1 and type 2 diabetes mellitus. J. Physiol. Pathophysiol. 2013, 4, 46–57. [Google Scholar] [CrossRef] [Green Version]
- Cruz, N.G.; Sousa, L.P.; Sousa, M.O.; Pietrani, N.T.; Fernandes, A.P.; Gomes, K.B. The linkage between inflammation and Type 2 diabetes mellitus. Diabetes Res. Clin. Pract. 2013, 99, 85–92. [Google Scholar] [CrossRef]
- Esser, N.; Legrand-Poels, S.; Piette, J.; Scheen, A.J.; Paquot, N. Inflammation as a link between obesity, metabolic syndrome and type 2 diabetes. Diabetes Res. Clin. Pract. 2014, 105, 141–150. [Google Scholar] [CrossRef] [Green Version]
- Akash, M.S.H.; Rehman, K.; Liaqat, A. Tumor necrosis factor-alpha: Role in development of insulin resistance and pathogenesis of type 2 diabetes mellitus. J. Cell. Biochem. 2018, 119, 105–110. [Google Scholar] [CrossRef]
- SLC2A4 Solute Carrier Family 2 Member 4 [Homo Sapiens (Human)]. Available online: https://www.ncbi.nlm.nih.gov/gene/6517 (accessed on 5 February 2022).
- Bodhini, D.; Radha, V.; Ghosh, S.; Majumder, P.P.; Rao, M.S.; Mohan, V. GLUT4 gene polymorphisms and their association with type 2 diabetes in south Indians. Diabetes Technol. Ther. 2011, 13, 913–920. [Google Scholar] [CrossRef]
- Guo, T.; Bai, Y.; Cheng, X.; Han, H.; Du, H.; Hu, Y.; Jia, S.; Xing, X.; Ji, J. Insulin gene enhancer protein 1 mediates glycolysis and tumorigenesis of gastric cancer through regulating glucose transporter 4. Cancer Commun. 2021, 41, 258–272. [Google Scholar] [CrossRef]
- Stein, W.D.; Litman, T. Channels, Carriers, and Pumps: An Introduction to Membrane Transport, 2nd ed; Elsevier: Amsterdam, The Netherlands, 2014; ISBN 9780124165830. [Google Scholar]
- Turner, N.; Zeng, X.Y.; Osborne, B.; Rogers, S.; Ye, J.M. Repurposing drugs to target the diabetes epidemic. Trends Pharmacol. Sci. 2016, 37, 379–389. [Google Scholar] [CrossRef]
- Zhu, S.; Bai, Q.; Li, L.; Xu, T. Drug repositioning in drug discovery of T2DM and repositioning potential of antidiabetic agents. Comput. Struct. Biotechnol. J. 2022, 20, 2839–2847. [Google Scholar] [CrossRef]
- Kour, J.; Singh, K. Virtual screening using the ligand ZINC database for novel lipoxygenase-3 inhibitors. Bioinformation 2013, 9, 583–587. [Google Scholar] [CrossRef]
- Forni, C.; Facchiano, F.; Bartoli, M.; Pieretti, S.; Facchiano, A.; D’Arcangelo, D.; Norelli, S.; Valle, G.; Nisini, R.; Beninati, S. Beneficial role of phytochemicals on oxidative stress and age-related diseases. Biomed Res. Int. 2019, 2019, 8748253. [Google Scholar] [CrossRef] [Green Version]
- Arora, I.; Sharma, M.; Tollefsbol, T.O. Combinatorial epigenetics impact of polyphenols and phytochemicals in cancer prevention and therapy. Int. J. Mol. Sci. 2019, 20, 4567. [Google Scholar] [CrossRef] [Green Version]
- Benson, D.; Lipman, D.J.; Ostell, J. GenBank. Nucleic Acids Res. 1993, 21, 2963–2965. [Google Scholar] [CrossRef]
- Boutet, E.; Lieberherr, D.; Tognolli, M.; Schneider, M.; Bairoch, A. Uniprotkb/Swiss-Prot. In Plant Bioinformatics; Springer: Berlin/Heidelberg, Germany, 2007; pp. 89–112. [Google Scholar]
- Madden, T. The BLAST sequence analysis tool. In NCBI Handbook; National Center for Biotechnology Information: Bethesda, MD, USA, 2013; Volume 2, pp. 425–436. [Google Scholar]
- Sievers, F.; Higgins, D.G. Clustal omega. Curr. Protoc. Bioinforma. 2014, 48, 3–13. [Google Scholar] [CrossRef]
- Bailey, T.L.; Johnson, J.; Grant, C.E.; Noble, W.S. The MEME Suite. Nucleic Acids Res. 2015, 43, W39–W49. [Google Scholar] [CrossRef] [Green Version]
- Webb, B.; Sali, A. Comparative Protein Structure Modeling Using Modeller. In Current Protocols in Bioinformatics 54; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2016; pp. 5.6.1–5.6.37. [Google Scholar]
- Zhang, Y.; Zhang, T.J.; Tu, S.; Zhang, Z.H.; Meng, F.H. Identification of novel Src inhibitors: Pharmacophore-based virtual screening, molecular docking and molecular dynamics simulations. Molecules 2020, 25, 4094. [Google Scholar] [CrossRef]
- Chemical Computing Group. Molecular Operating Environment (MOE), Version 2011.10; Chemical Computing Group Inc: Montreal, QC, Canada, 2011. [Google Scholar]
- Cheng, F.; Li, W.; Zhou, Y.; Shen, J.; Wu, Z.; Liu, G.; Lee, P.W.; Tang, Y. admetSAR: A comprehensive source and free tool for assessment of chemical ADMET properties. J. Chem. Inf. Model. 2012, 52, 3099–3105. [Google Scholar] [CrossRef]
- Available online: https://preadmet.webservice.bmdrc.org/adme-prediction/ (accessed on 5 February 2022).
- Magalhães, P.R.; Reis, P.B.; Vila-Viçosa, D.; Machuqueiro, M.; Victor, B.L. Identification of Pan-Assay INterference compoundS (PAINS) Using an MD-Based Protocol. Comput. Des. Membr. Proteins 2021, 2315, 263–271. [Google Scholar]
- Stork, C.; Wagner, J.; Friedrich, N.O.; Kops, C.B.; Sicho, M.; Kirchmair, J. Hit Dexter: A Machine-Learning Model for the Prediction of Frequent Hitters. Chem. Med. Chem. 2018, 13, 564–571. [Google Scholar] [CrossRef] [PubMed]
- James, J.P.; Sasidharan, P.; Mandal, S.P.; Dixit, S.R. Virtual Screening of Alkaloids and Flavonoids as Acetylcholinesterase and MAO-B Inhibitors by Molecular Docking and Dynamic Simulation Studies. Polycycl. Aromat. Compd. 2022, 1–25. [Google Scholar] [CrossRef]
- Daina, A.; Michielin, O.; Zoete, V. SwissADME: A free web tool to evaluate pharmacokinetics, drug-likeness and medicinal chemistry friendliness of small molecules. Sci. Rep. 2017, 7, 42717. [Google Scholar] [CrossRef] [Green Version]
- Elksnis, A.; Martinell, M.; Eriksson, O.; Espes, D. Heterogeneity of metabolic defects in type 2 diabetes and its relation to reactive oxygen species and alterations in beta-cell mass. Front. Physiol. 2019, 10, 107. [Google Scholar] [CrossRef] [Green Version]
- Alam, F.; Islam, M.A.; Khalil, M.I.; Gan, S.H. Metabolic Control of Type 2 Diabetes by Targeting the GLUT4 Glucose Transporter: Intervention Approaches. Curr. Pharm. Des. 2016, 22, 3034–3049. [Google Scholar] [CrossRef]
- Chellappan, D.K.; Yap, W.S.; Nurfatiah, B.A.S.; Gupta, G.; Dua, K. Current therapies and targets for type 2 diabetes mellitus. Panminerva Med. 2018, 60, 117–131. [Google Scholar] [CrossRef]
- Basu, A.; Sarkar, A.; Maulik, U. Molecular docking study of potential phytochemicals and their effects on the complex of SARS-CoV2 spike protein and human ACE2. Sci. Rep. 2020, 10, 17699. [Google Scholar] [CrossRef]
- James, J.P.; Kumar, P.; Kumar, A.; Bhat, K.I.; Shastry, C.S. In Silico Anticancer Evaluation, Molecular Docking and Pharmacophore Modeling of Flavonoids against Various Cancer Targets. Lett. Drug Des. Discov. 2020, 17, 1485–1501. [Google Scholar] [CrossRef]
- Kodical, D.D.; James, J.P.; Deepthi, K.; Kumar, P.; Cyriac, C.; Gopika, K.V. ADMET, Molecular docking studies and binding energy calculations of Pyrimidine-2-Thiol Derivatives as Cox Inhibitors. Res. J. Pharm. Technol. 2020, 13, 4200–4206. [Google Scholar] [CrossRef]
- Mittal, L.; Kumari, A.; Srivastava, M.; Singh, M.; Asthana, S. Identification of potential molecules against COVID-19 main protease through structure-guided virtual screening approach. J. Biomol. Struct. Dyn. 2020, 39, 3662–3680. [Google Scholar] [CrossRef]
- James, J.P.; Devaraji, V.; Sasidharan, P.; Pavan, T.S. Pharmacophore Modeling, 3D QSAR, Molecular Dynamics Studies and Virtual Screening on Pyrazolopyrimidines as anti-Breast Cancer Agents. Polycycl. Aromat. Compd. 2022, 4, 1–8. [Google Scholar] [CrossRef]
- Eswar, N.; Eramian, D.; Webb, B.; Shen, M.-Y.; Sali, A. Protein structure modeling with MODELLER. In Structural Proteomics; Springer: Berlin/Heidelberg, Germany, 2008; pp. 145–159. [Google Scholar]
- Yang, J.; Yan, R.; Roy, A.; Xu, D.; Poisson, J.; Zhang, Y. The I-TASSER Suite: Protein structure and function prediction. Nat. Methods 2015, 12, 7–8. [Google Scholar] [CrossRef] [Green Version]
- Gopalakrishnan, K.; Sowmiya, G.; Sheik, S.S.; Sekar, K. Ramachandran plot on the web (2.0). Protein Pept. Lett. 2007, 14, 669–671. [Google Scholar] [CrossRef]
- Siddiqui, S.; Upadhyay, S.; Ahmad, R.; Gupta, A.; Srivastava, A.; Trivedi, A.; Husain, I.; Ahmad, B.; Ahamed, M.; Khan, M.A. Virtual screening of phytoconstituents from miracle herb nigella sativa targeting nucleocapsid protein and papain-like protease of SARS-CoV-2 for COVID-19 treatment. J. Biomol. Struct. Dyn. 2020, 40, 3928–3948. [Google Scholar] [CrossRef]
- Guan, L.; Yang, H.; Cai, Y.; Sun, L.; Di, P.; Li, W.; Liu, G.; Tang, Y. ADMET-score—A comprehensive scoring function for evaluation of chemical drug-likeness. Medchemcomm 2018, 10, 148–157. [Google Scholar] [CrossRef]
- Domínguez-Villa, F.X.; Durán-Iturbide, N.A.; Ávila-Zárraga, J.G. Synthesis, molecular docking, and in silico ADME/Tox profiling studies of new 1-aryl-5-(3-azidopropyl) indol-4-ones: Potential inhibitors of SARS CoV-2 main protease. Bioorganic Chem. 2021, 106, 104497. [Google Scholar] [CrossRef]
- Ekins, S.; Nikolsky, Y.; Nikolskaya, T. Techniques: Application of systems biology to absorption, distribution, metabolism, excretion and toxicity. Trends Pharmacol. Sci. 2005, 26, 202–209. [Google Scholar] [CrossRef]
- Morris, G.M.; Lim-Wilby, M. Molecular docking. In Molecular Modeling of Proteins; Springer: Berlin/Heidelberg, Germany, 2008; pp. 365–382. [Google Scholar]
- Lipinski, C.A.; Lombardo, F.; Dominy, B.W.; Feeney, P.J. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv. Drug Del. Rev. 2001, 46, 3–26. [Google Scholar] [CrossRef]
- Zhao, Y.H.; Le, J.; Abraham, M.H.; Hersey, A.; Eddershaw, P.J.; Luscombe, C.N.; Butina, D.; Beck, G.; Sherborne, B.; Cooper, I.; et al. Evaluation of human intestinal absorption data and subsequent derivation of a quantitative structure-activity relationship (QSAR) with the Abraham descriptors. J. Pharm. Sci. 2001, 90, 749–784. [Google Scholar] [CrossRef] [Green Version]
- Ma, X.; Chen, C.; Yang, J. Predictive model of blood-brain barrier penetration of organic compounds. Acta Pharm. Sin. 2005, 26, 500–512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Godin, D.V. Pharmacokinetics: Disposition and metabolism of drugs. In Principles of Pharmacology; Munson, P.L., Ed.; Chapman & Hall: New York, NY, USA, 1995. [Google Scholar]
- Yamashita, S.; Furubayashi, T.; Kataoka, M.; Sakane, T.; Sezaki, H.; Tokuda, H. Optimized conditions for prediction of intestinal drug permeability using Caco-2 cells. Eur. J. Pharm. 2000, 10, 195–204. [Google Scholar] [CrossRef] [PubMed]
- Irvine, J.D.; Takahashi, L.; Lockhart, K.; Cheong, J.; Tolan, J.W.; Selick, H.E.; Grove, J.R. MDCK (Madin-Darby canine kidney) cells: A tool for membrane permeability screening. J. Pharm. Sci. 1999, 88, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Doak, B.C.; Over, B.; Giordanetto, F.; Kihlberg, J. Oral druggable space beyond the rule of 5: Insights from drugs and clinical candidates. Chem. Biol. 2014, 21, 1115–1142. [Google Scholar] [CrossRef] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Ligand | Canonical_SMILES |
|---|---|
| ZINC000001576020 | C[C@@H](NC(=O)[C@H]1C[C@H](O)CN1C(=O)CN)C(=O)O |
| ZINC000001643171 | CN(/N=C/C=N/N(C)C1=NCCN1)C1=NCCN1 |
| ZINC000001704450 | CC(C)C[C@H](N)C(=O)O[C@H](N)C(=O)NCCN(C)C |
| ZINC000017064359 | Cn1c(=O)c2[nH]c(-[n+]3cccc(CO)c3)nc2n(C)c1=O |
| ZINC000216155214 | CN1CCN(C2=C3C(=O)NON3[C@@H]([N+](=O)[O-])C=C2)CC1 |
| ZINC000618254662 | O=C(O)CNC(=O)CN/C=C1\SC(=S)NC1=O |
| Ligand | Molecular Weight (g/Mol) | Octanol−Water Partition Coefficient (LogP) | H-Donor | H-Acceptor | Lipinski Violation | Toxicity |
|---|---|---|---|---|---|---|
| ZINC000001576020 | 259.262 | −2.5038 | 4 | 5 | 0 | Non-toxic |
| ZINC000001643171 | 250.31 | −1.26 | 2 | 8 | 0 | Non-toxic |
| ZINC000001704450 | 274.365 | −1.1344 | 3 | 6 | 0 | Non-toxic |
| ZINC000017064359 | 288.287 | −1.2707 | 2 | 6 | 0 | Non-toxic |
| ZINC000216155214 | 281.272 | −1.1036 | 1 | 7 | 0 | Non-toxic |
| ZINC000618254662 | 275.311 | −1.2339 | 4 | 6 | 0 | Non-toxic |
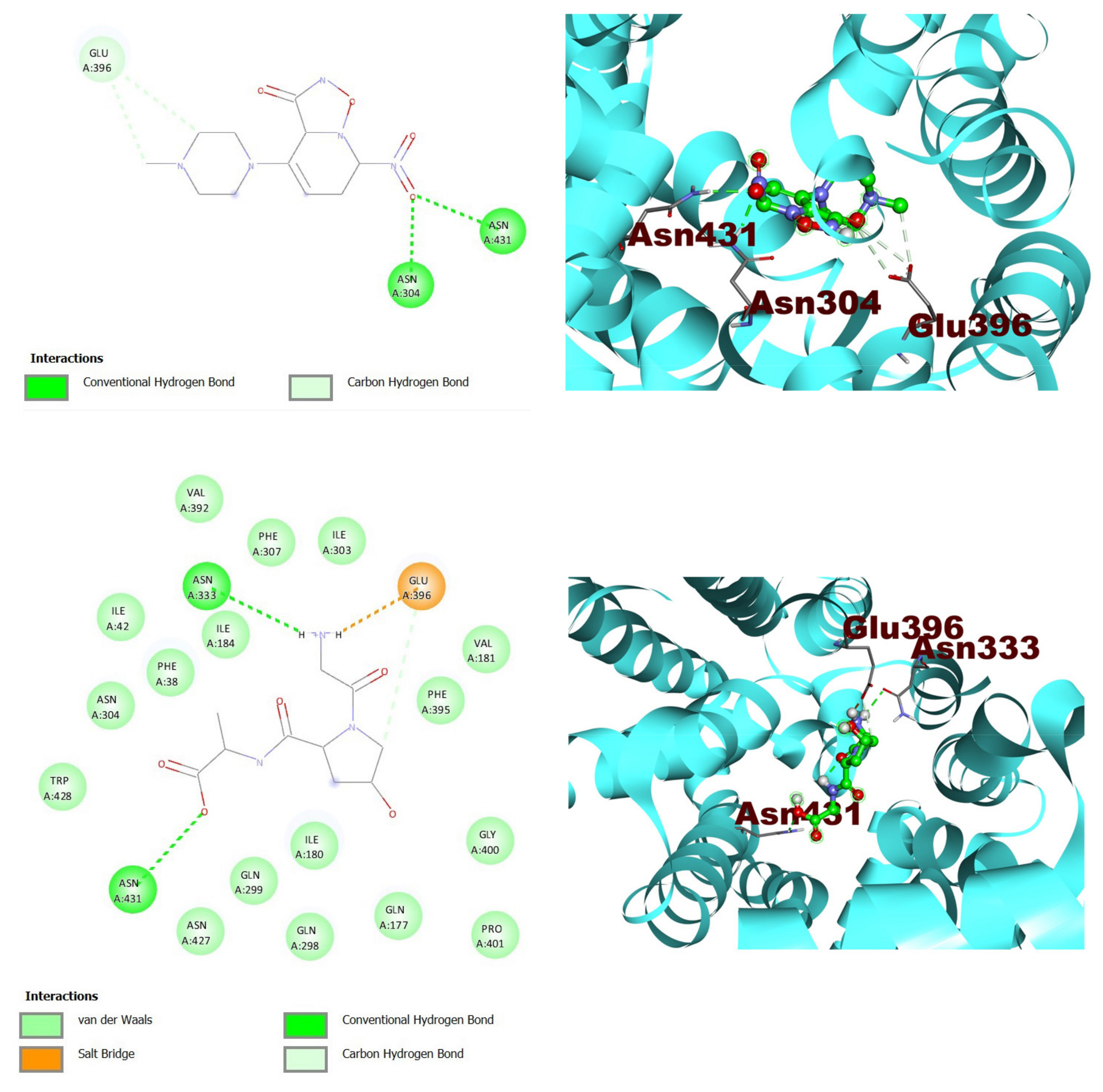
| Ligand | S-Score (Kcal/Mol) | RMSD (Å) | Interacting Residues |
|---|---|---|---|
| ZINC000216155214 | −6.5 | 0.0 | ASN304, GLU396, ASN431 |
| ZINC000001576020 | −5.9 | 1.1 | ASN333, GLU396, ASN431 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Aldahish, A.; Balaji, P.; Vasudevan, R.; Kandasamy, G.; James, J.P.; Prabahar, K. Elucidating the Potential Inhibitor against Type 2 Diabetes Mellitus Associated Gene of GLUT4. J. Pers. Med. 2023, 13, 660. https://doi.org/10.3390/jpm13040660
Aldahish A, Balaji P, Vasudevan R, Kandasamy G, James JP, Prabahar K. Elucidating the Potential Inhibitor against Type 2 Diabetes Mellitus Associated Gene of GLUT4. Journal of Personalized Medicine. 2023; 13(4):660. https://doi.org/10.3390/jpm13040660
Chicago/Turabian StyleAldahish, Afaf, Prasanalakshmi Balaji, Rajalakshimi Vasudevan, Geetha Kandasamy, Jainey P. James, and Kousalya Prabahar. 2023. "Elucidating the Potential Inhibitor against Type 2 Diabetes Mellitus Associated Gene of GLUT4" Journal of Personalized Medicine 13, no. 4: 660. https://doi.org/10.3390/jpm13040660
APA StyleAldahish, A., Balaji, P., Vasudevan, R., Kandasamy, G., James, J. P., & Prabahar, K. (2023). Elucidating the Potential Inhibitor against Type 2 Diabetes Mellitus Associated Gene of GLUT4. Journal of Personalized Medicine, 13(4), 660. https://doi.org/10.3390/jpm13040660