
Comparative Mitogenomic Analysis of Heptageniid Mayflies (Insecta: Ephemeroptera): Conserved Intergenic Spacer and tRNA Gene Duplication

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Mitogenome Sequencing and Assembly
2.3. Gene Annotation and Bioinformatic Analysis
2.4. Phylogenetic Analysis
3. Results and Discussion
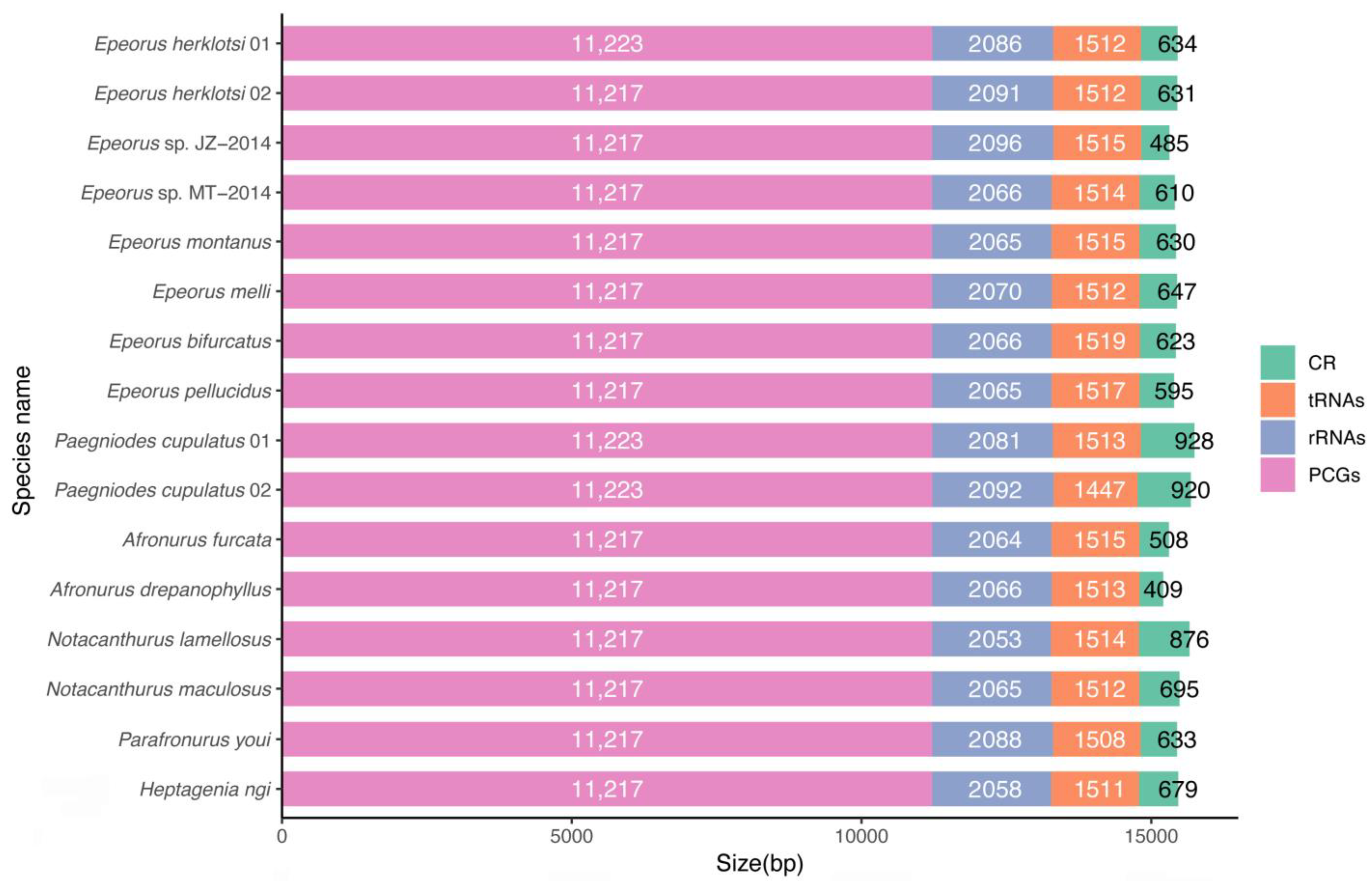
3.1. Genomic Organization and Composition
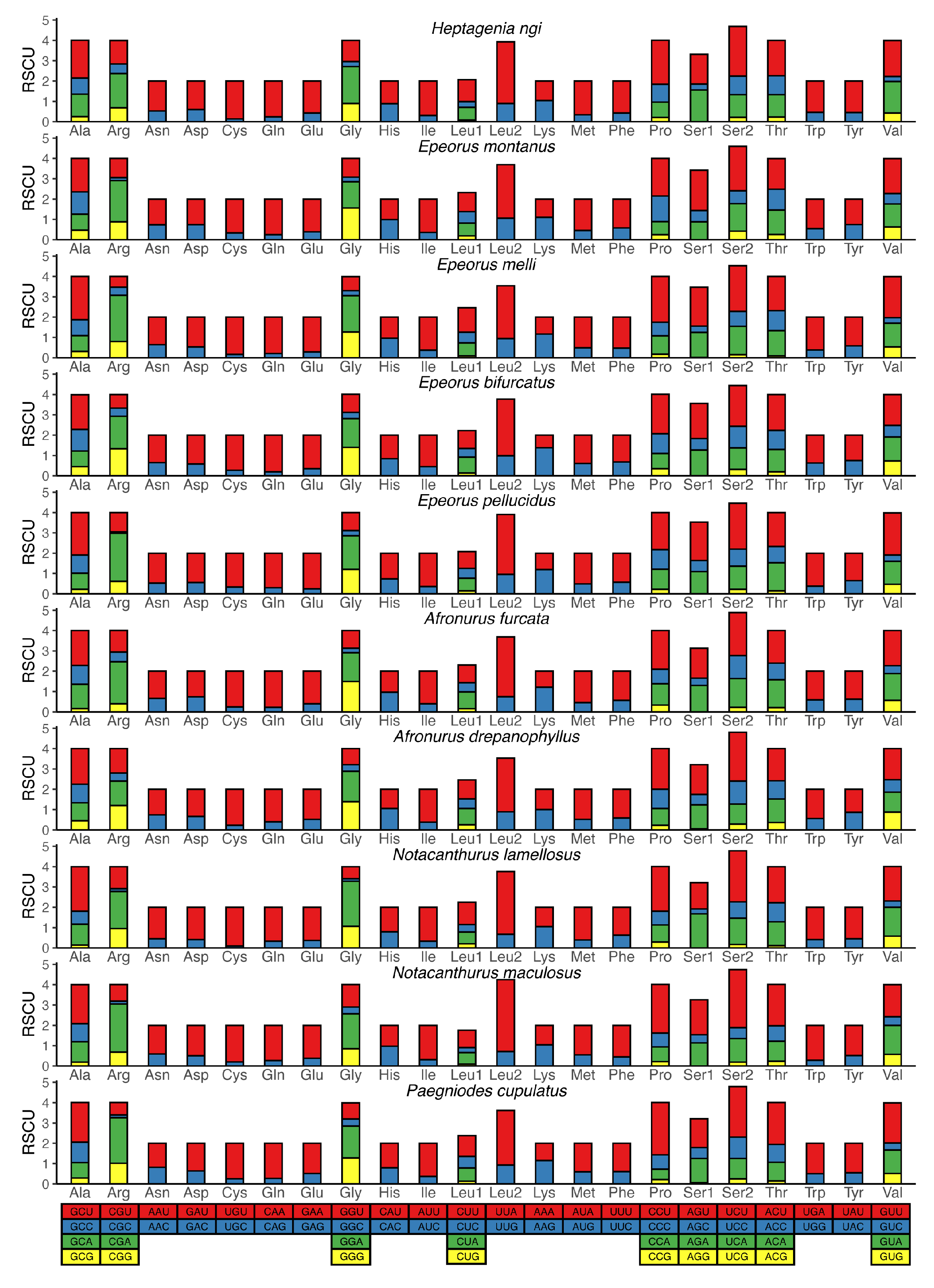
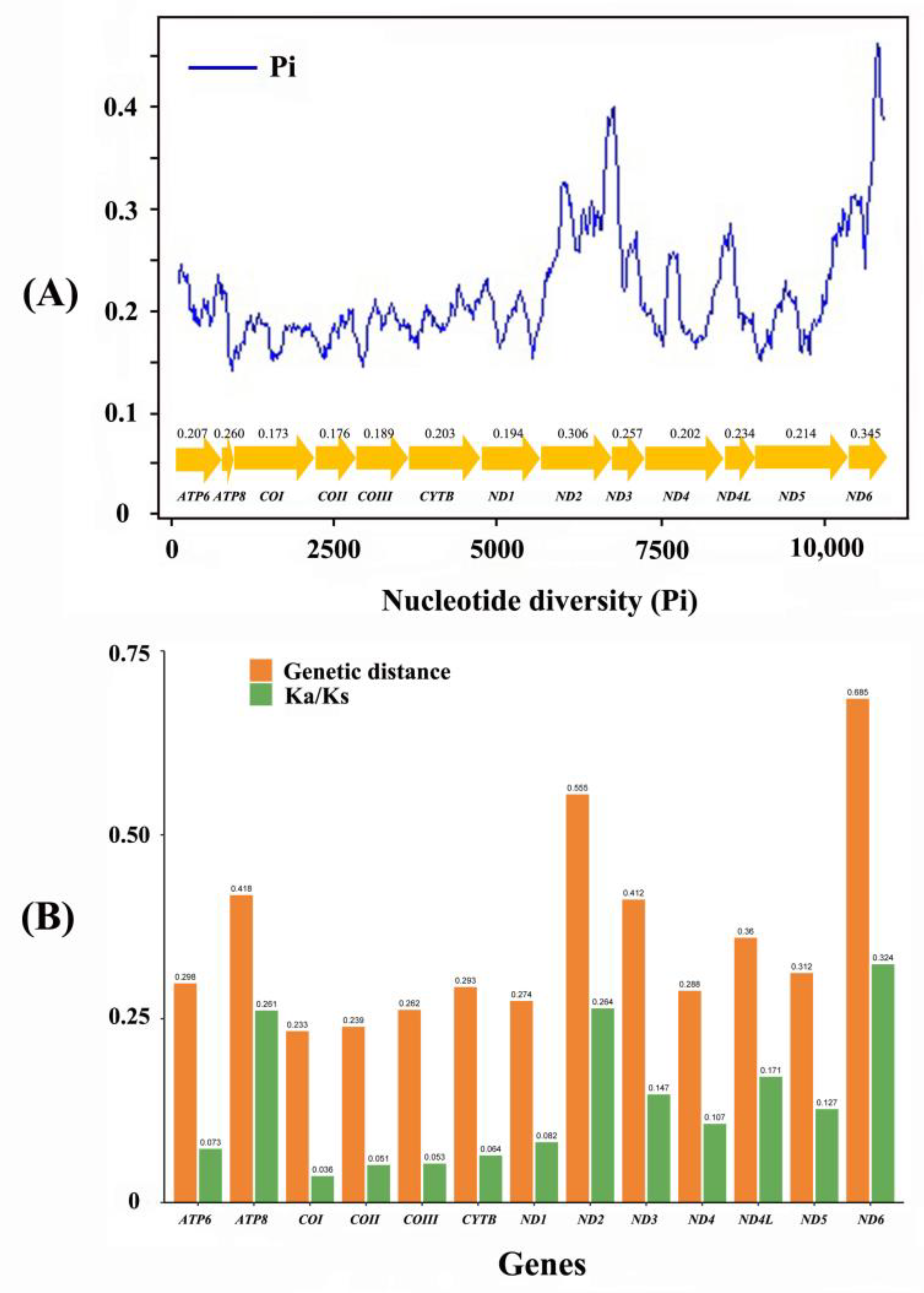
3.2. Protein-Coding Genes
3.3. Ribosomal and Transfer RNAs
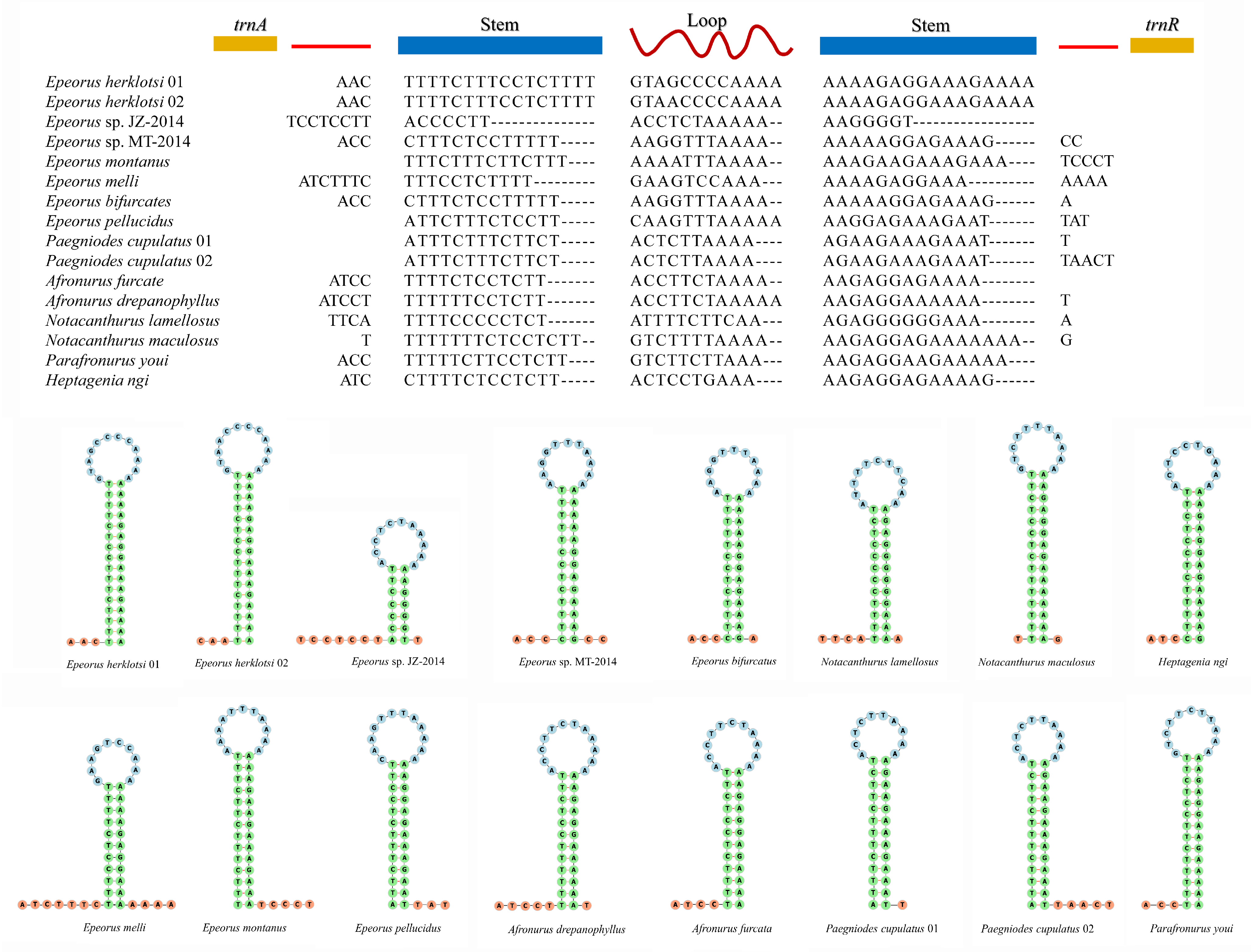
3.4. Non-Coding Regions
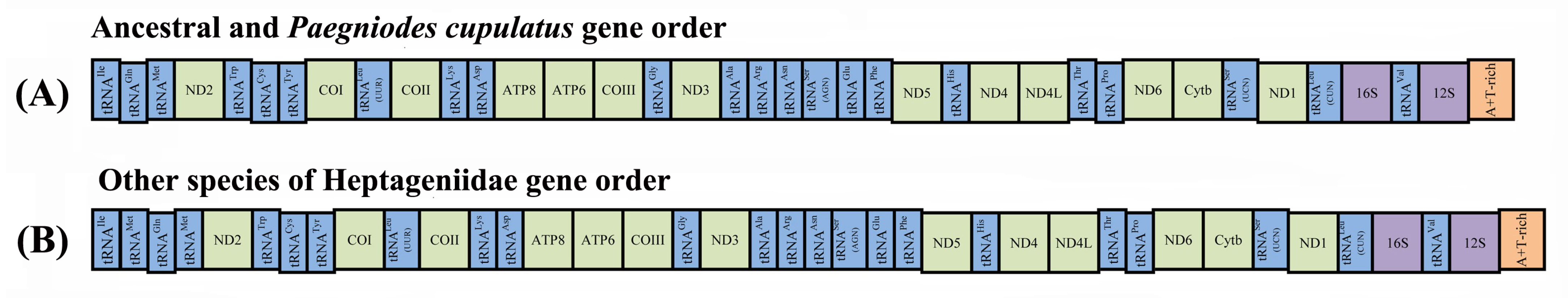
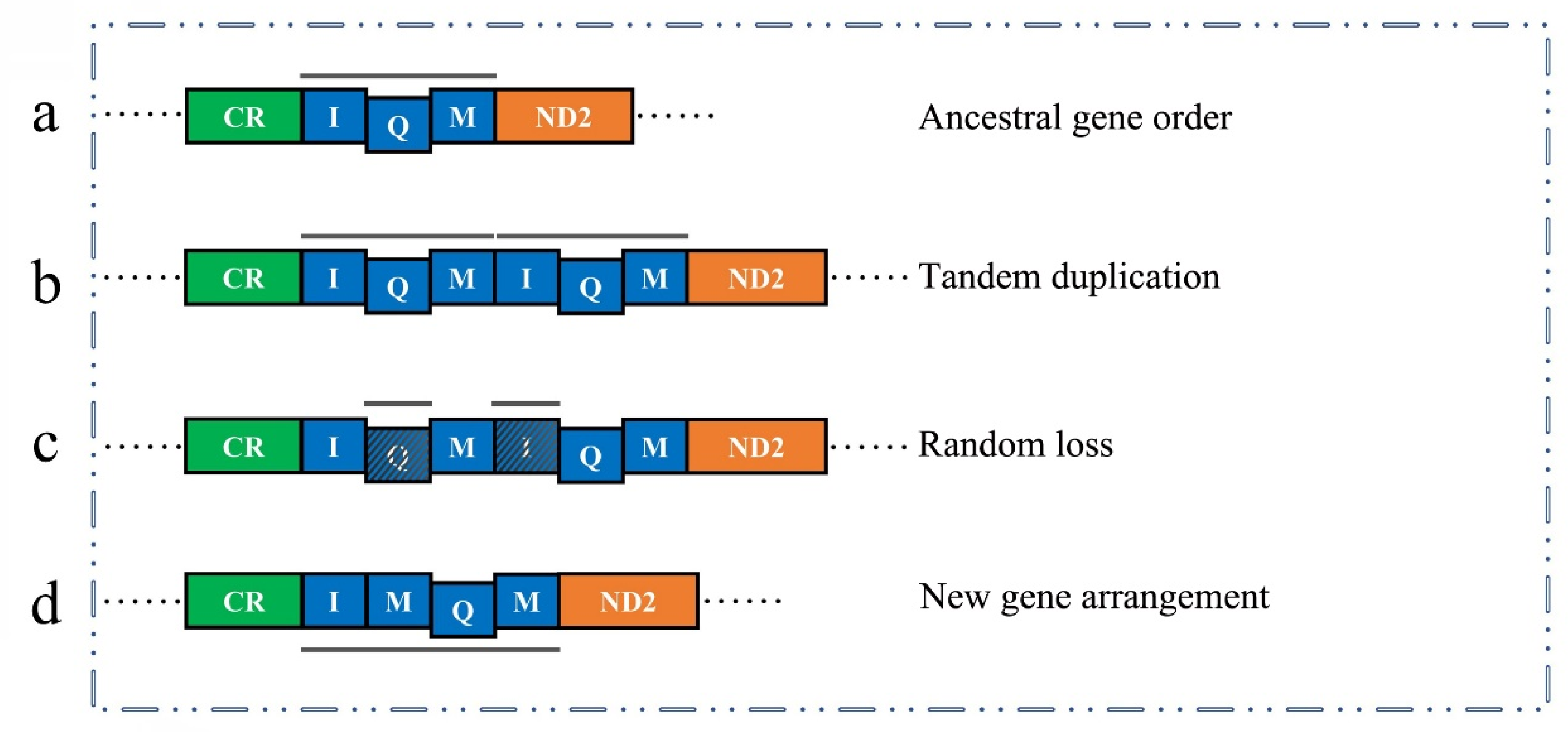
3.5. Gene Arrangement
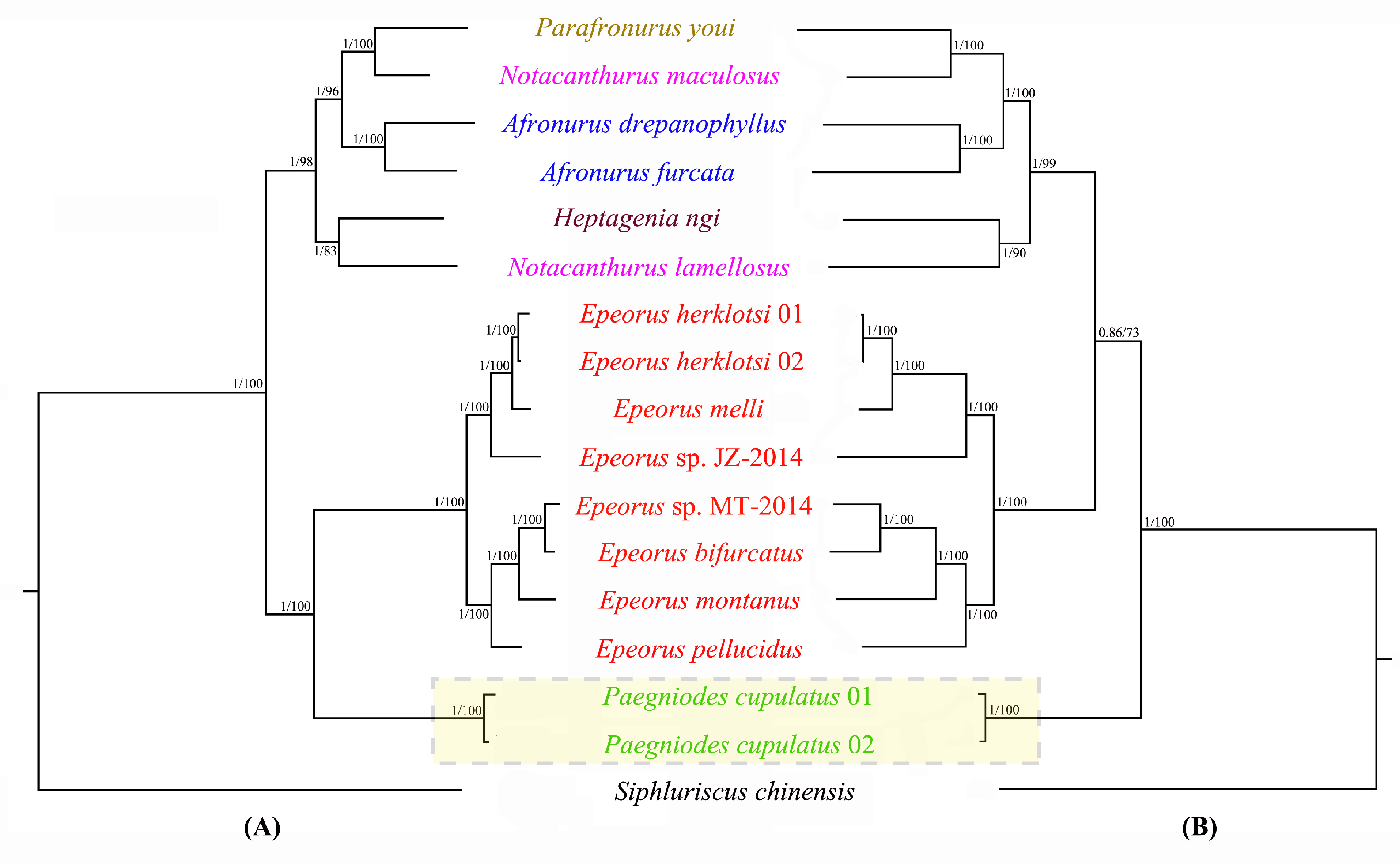
3.6. Phylogenetic Analyses
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wolstenholme, D.R. Animal mitochondrial DNA: Structure and evolution. Int. Rev. Cytol. 1992, 141, 173–216. [Google Scholar] [PubMed]
- Moritz, C.; Dowling, T.E.; Brown, W.M. Evolution of animal mitochondrial DNA: Relevance for population biology and systematics. Ann. Rev. Ecol. Syst. 1987, 18, 173–216. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Avise, J.C.; Arnold, J.; Ball, R.M.; Bermingham, E.; Lamb, T.; Neigel, J.E.; Reeb, C.A.; Saunders, N.C. Intraspecific phylogeography: The mitochondrial DNA bridge between population genetics and systematics. Ann. Rev. Ecol. Syst. 1987, 18, 489–522. [Google Scholar] [CrossRef]
- Zhou, N.; Dong, Y.; Qiao, P.; Yang, Z. Complete Mitogenomic Structure and Phylogenetic Implications of the Genus Ostrinia (Lepidoptera: Crambidae). Insects 2020, 11, 232. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Song, H.; Amédégnato, C.; Cigliano, M.M.; Desutter-Grandcolas, L.; Heads, S.W.; Huang, Y.; Otte, D.; Whiting, M.F. 300 million years of diversification: Elucidating the patterns of orthopteran evolution based on comprehensive taxon and gene sampling. Cladistics 2015, 31, 621–651. [Google Scholar] [CrossRef]
- Du, C.; Zhang, L.; Lu, T.; Ma, J.; Zeng, C.; Yue, B.; Zhang, X. Mitochondrial genomes of blister beetles (Coleoptera, Meloidae) and two large intergenic spacers in Hycleus genera. BMC Genom. 2017, 18, 698. [Google Scholar] [CrossRef]
- Sun, Z.; Liu, Y.; Wilson, J.J.; Chen, Z.; Song, F.; Cai, W.Z.; Li, H. Mitochondrial genome of Phalantus geniculatus (Hemiptera: Reduviidae): trnT duplication and phylogenetic implications. Int. J. Biol. Macromol. 2019, 129, 110–115. [Google Scholar] [CrossRef]
- Cao, J.J.; Wang, Y.; Li, W.H. Comparative mitogenomic analysis of species in the subfamily Amphinemurinae (Plecoptera: Nemouridae) reveal conserved mitochondrial genome organization. Int. J. Biol. Macromol. 2019, 138, 292–301. [Google Scholar] [CrossRef]
- Bauernfeind, E.; Soldán, T. The Mayflies of Europe (Ephemeroptera); Apollo Books: Ollerup, Denmark, 2012; p. 781. [Google Scholar]
- Li, D.; Qin, J.C.; Zhou, C.F. The phylogeny of Ephemeroptera in Pterygota revealed by the mitochondrial genome of Siphluriscus chinensis (Hexapoda: Insecta). Gene 2014, 545, 132–140. [Google Scholar] [CrossRef] [PubMed]
- Gao, X.Y.; Zhang, S.S.; Zhang, L.P.; Yu, D.N.; Zhang, J.Y.; Cheng, H.Y. The complete mitochondrial genome of Epeorus herklotsi (Ephemeroptera: Heptageniidae) and its phylogeny. Mitochondrial DNA B 2018, 3, 303–304. [Google Scholar] [CrossRef] [Green Version]
- Li, R.; Zhang, W.; Ma, Z.X.; Zhou, C.F. Novel gene rearrangement pattern in the mitochondrial genomes of Torleya mikhaili and Cincticostella fusca (Ephemeroptera: Ephemerellidae). Int. J. Biol. Macromol. 2020, 165, 3106–3114. [Google Scholar] [CrossRef]
- Zhang, J.Y.; Zhou, C.F.; Gai, Y.H.; Song, D.X.; Zhou, K.Y. The complete mitochondrial genome of Parafronurus youi (Insecta: Ephemeroptera) and phylogenetic position of the Ephemeroptera. Gene 2008, 424, 18–24. [Google Scholar] [CrossRef] [PubMed]
- Zhou, D.; Wang, Y.Y.; Sun, J.Z.; Han, Y.K.; Zhou, C.F. The complete mitochondrial genome of Paegniodes cupulatus (Ephemeroptera: Heptageniidae). Mitochondrial DNA A 2016, 27, 925–926. [Google Scholar] [CrossRef] [PubMed]
- Webb, J.M.; McCafferty, W.P. Heptageniidae of the World. Part II: Key to the Genera. Can. J. Arthrop. Ident. 2008, 7, 1–55. [Google Scholar]
- Webb, J.M.; McCafferty, W.P. Contributions to the larvae of North American Nixe (Ephemeroptera: Heptageniidae), with the description of N. dorothae sp. nov. from southern Indiana. Zootaxa 2011, 3065, 27–37. [Google Scholar] [CrossRef]
- Ball, S.L.; Hebert, P.D.N.; Burian, S.K.; Webb, J.M. Biological identifications of mayflies (Ephemeroptera) using DNA barcodes. J. N. Am. Benthol. Soc. 2005, 24, 508–524. [Google Scholar] [CrossRef]
- Barber-James, H.M.; Gattolliat, J.L.; Sartori, M.; Hubbard, M.D. Global diversity of mayflies (Ephemeroptera, Insecta) in freshwater. Hydrobiologia 2008, 595, 339–350. [Google Scholar] [CrossRef]
- Tshernova, O.A. The generic composition of the mayflies of the family Heptageniidae (Ephemeroptera) in the Holarctic and Oriental region. Entomol. Obozr. 1974, 53, 801–814. [Google Scholar]
- Tomka, I.; Zurwerra, A. Key to the genera of the Heptageniidae (Ephemeroptera) of the Holarctic, Oriental and Ethiopian region. Entomol. Berich. Luz. 1985, 14, 113–126. [Google Scholar]
- Yanai, Z.; Sartori, M.; Dor, R.; Dorchin, N. Molecular phylogeny and morphological analysis resolve a long-standing controversy over generic concepts in Ecdyonurinae mayflies (Ephemeroptera: Heptageniidae). Syst. Entomol. 2017, 42, 182–193. [Google Scholar] [CrossRef]
- Polášek, M.; Godunko, R.J.; Rutschmann, S.; Svitok, M.; Novikmec, M.; Zahrádková, S. Integrative taxonomy of genus Electrogena (Ephemeroptera: Heptageniidae): The role of innovative morphological analyses for species delimitation. Arthropod Syst. Phylogeny 2018, 76, 449–462. [Google Scholar]
- Gattolliat, J.L.; Monaghan, M.T.; Sartori, M.; Elouard, J.M.; Vogler, A.P. A molecular analysis of the Afrotropical Baetidae. In International Advances in the Ecology, Zoogeography and Systematics of Mayflies and Stoneflies; University of California Press: Berkeley, CA, USA, 2008; pp. 219–232. [Google Scholar]
- Ogden, T.H.; Osborne, J.T.; Jacobus, L.M.; Whiting, M.F. Combined molecular and morphological phylogeny of Ephemerellinae (Ephemerellidae: Ephemeroptera), with remarks about classification. Zootaxa 2009, 1991, 28–42. [Google Scholar] [CrossRef]
- Meng, G.; Li, Y.; Yang, C.; Liu, S. MitoZ: A toolkit for mitochondrial genome assembly, annotation and visualization. Nucleic Acids Res. 2019, 47, e63. [Google Scholar] [CrossRef] [PubMed]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE Online: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef]
- Laslett, D.; Canbäck, B. ARWEN, a program to detect tRNA genes in metazoan mitochondrial nucleotide sequences. Bioinformatics 2008, 24, 172–175. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular evolutionary genetics analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Alejandro, S.G. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [Green Version]
- Zhang, D.; Gao, F.; Jakovlic, I.; Zhou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Res. 2020, 20, 348–355. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Calcott, B.; Ho, S.Y.; Guindon, S. PartitionFinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ronquist, F.; Teslenko, M.; Van Der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.A.; Wayne, P.; Terri, S. The CIPRES science gateway: A community resource for phylogenetic analyses. In Proceedings of the 2011 TeraGrid Conference: Extreme Digital Discovery, Salt Lake City, UT, USA, 18–21July 2011; pp. 1–8. [Google Scholar]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis and post-analysis of large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef] [PubMed]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef]
- Zhang, R.Y.; Li, J.; Geng, S.; Yang, J.; Zhang, X.; An, Y.X.; Li, C.; Cui, H.R.; Li, X.Y.; Wang, Y.Y. The first mitochondrial genome for Phaudidae (Lepidoptera) with phylogenetic analyses of Zygaenoidea. Int. J. Biol. Macromol. 2020, 149, 951–961. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.Y.; Liu, F.F.; Chiba, H.; Yuan, X.Q. The mitochondrial genomes of three skippers: Insights into the evolution of the family Hesperiidae (Lepidoptera). Genomics 2020, 112, 432–441. [Google Scholar] [CrossRef] [PubMed]
- Jia, W.Z.; Yan, H.B.; Guo, A.J.; Zhu, X.Q.; Wang, Y.C.; Shi, W.G.; Chen, H.T.; Fang, Z.; Zhang, S.H.; Fu, B.Q. Complete mitochondrial genomes of Taenia multiceps, T. hydatigena and T. pisiformis: Additional molecular markers for a tapeworm genus of human and animal health significance. BMC Genom. 2010, 11, 447. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, T.; He, B.; Li, K.; Liang, A. Comparative analysis of the mitochondrial genomes of oriental spittlebug trible Cosmoscartini: Insights into the relationships among closely related taxa. BMC Genom. 2018, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Hu, K.; Yuan, F.; Dietrich, C.H.; Yuan, X.Q. Structural features and phylogenetic implications of four new mitogenomes of Centrotinae (Hemiptera: Membracidae). Int. J. Biol. Macromol. 2019, 139, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Clayton, D.A. Replication and transcription of vertebrate mitochondrial DNA. Annu. Rev. Cell Biol. 1991, 7, 453–478. [Google Scholar] [CrossRef]
- Fernandez-Silva, P.; Enriquez, J.A.; Montoya, J. Replication and transcription of mammalian mitochondrial DNA. Exp. Physiol. 2003, 88, 41–56. [Google Scholar] [CrossRef] [PubMed]
- Ruan, H.T.; Li, M.; Li, Z.H.; Huang, J.J.; Chen, W.Y.; Sun, J.J.; Liu, L.; Zou, K.S. Comparative analysis of complete mitochondrial genomes of three Gerres fishes (Perciformes: Gerreidae) and primary exploration of their evolution history. Int. J. Mol. Sci. 2020, 21, 1874. [Google Scholar] [CrossRef] [Green Version]
- Yu, X.L.; Tan, W.; Zhang, H.Y.; Jiang, W.L.; Gao, H.; Wang, W.X.; Gao, H.; Wang, W.X.; Liu, Y.X.; Wang, Y.; et al. Characterization of the complete mitochondrial genome of Harpalus sinicus and its implications for phylogenetic analyses. Genes 2019, 10, 724. [Google Scholar] [CrossRef] [Green Version]
- Gong, L.; Liu, L.Q.; Guo, B.Y.; Ye, Y.Y.; Lü, Z.M. The complete mitochondrial genome characterization of Thunnus obesus (Scombriformes: Scombridae) and phylogenetic analyses of Thunnus. Conserv. Genet. Res. 2017, 9, 379–383. [Google Scholar] [CrossRef]
- Seligmann, H.; Krishnan, N.M.; Rao, B.J. Possible multiple origins of replication in primate mitochondria: Alternative role of tRNA sequences. J. Theor. Biol. 2006, 241, 321–332. [Google Scholar] [CrossRef]
- Seligmann, H.; Labra, A. The relation between hairpin formation by mitochondrial WANCY tRNAs and the occurrence of the light strand replication origin in Lepidosauria. Gene 2014, 542, 248–257. [Google Scholar] [CrossRef]
- Dowton, M.; Campbell, N.J. Intramitochondrial recombination–is it why some mitochondrial genes sleep around? Trends Ecol. Evol. 2001, 16, 269–271. [Google Scholar] [CrossRef]
- Mao, M.; Gibson, T.; Dowton, M. Evolutionary dynamics of the mitochondrial genome in the Evaniomorpha (Hymenoptera)—a group with an intermediate rate of gene rearrangement. Genome Biol. Evol. 2014, 6, 1862–1874. [Google Scholar] [CrossRef] [PubMed]
- Gong, L.; Lu, X.T.; Luo, H.R.; Zhang, Y.; Shi, W.; Liu, L.Q.; Lü, Z.M.; Liu, B.J.; Jiang, L.H. Novel gene rearrangement pattern in Cynoglossus melampetalus mitochondrial genome: New gene order in genus Cynoglossus (Pleuronectiformes: Cynoglossidae). Int. J. Biol. Macromol. 2020, 149, 1232–1240. [Google Scholar] [CrossRef]
- Liu, Y.; Li, H.; Song, F.; Zhao, Y.; Wilson, J.J.; Cai, W.Z. Higher-level phylogeny and evolutionary history of Pentatomomorpha (Hemiptera: Heteroptera) inferred from mitochondrial genome sequences. Syst. Entomol. 2019, 44, 810–819. [Google Scholar] [CrossRef]
- Feng, Z.; Wu, Y.; Yang, C.; Gu, X.; Wilson, J.J.; Li, H.; Cai, W.Z.; Yang, H.L.; Song, F. Evolution of tRNA gene rearrangement in the mitochondrial genome of ichneumonoid wasps (Hymenoptera: Ichneumonoidea). Int. J. Biol. Macromol. 2020, 164, 540–547. [Google Scholar] [CrossRef] [PubMed]
- Ma, Z.X.; Han, N.; Zhang, W.; Zhou, C.F. Position and definition of the genus Paegniodes Eaton, 1881 based on redescription on the type species Paegniodes cupulatus (Eaton, 1871) (Ephemeroptera: Heptageniidae). Aquat. Insect. 2018, 39, 362–374. [Google Scholar] [CrossRef]
- Zhang, W.; Zhang, M.; Han, N.; Zhou, C.F. Two new species of the genus Notacanthurus from China (Ephemeroptera: Heptageniidae, Ecdyonurinae). Zootaxa 2020, 4802, 335–348. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Subfamily | Species | Size (bp) | GenBank # |
|---|---|---|---|
| Rhithrogeninae | Epeorus herklotsi 01 | 15,502 | MG870104 |
| Epeorus herklotsi 02 | 15,499 | MH752075 | |
| Epeorus sp. JZ-2014 | 15,338 | KJ493406 | |
| Epeorus sp. MT-2014 | 15,456 | KM244708 | |
| Epeorus montanus | 15,472 | This study | |
| Epeorus melli | 15,490 | This study | |
| Epeorus bifurcatus | 15,466 | This study | |
| Epeorus pellucidus | 15,435 | This study | |
| Paegniodes cupulatus 01 | 15,715 | HM004123 | |
| Paegniodes cupulatus 02 | 15721 | This study | |
| Ecdyonurinae | Parafronurus youi | 15,481 | EU349015 |
| Afronurus furcata | 15,334 | This study | |
| Afronurus drepanophyllus | 15,242 | This study | |
| Notacanthurus lamellosus | 15,693 | This study | |
| Notacanthurus maculosus | 15,524 | This study | |
| Heptageniinae | Heptagenia ngi | 15,495 | This study |
| Outgroup | |||
| Siphluriscidae | Siphluriscus chinensis | 16,616 | HQ875717 |
| Species | A+T (%) | AT-Skew | GC-Skew | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| All | PCGs | tRNAs | rRNAs | CR | All | PCGs | tRNAs | rRNAs | CR | All | PCGs | tRNAs | rRNAs | CR | |
| Epeorus herklotsi 01 | 65.66 | 65.10 | 65.74 | 65.77 | 74.45 | −0.002 | −0.196 | −0.018 | 0.007 | 0.017 | −0.245 | −0.033 | 0.131 | 0.311 | −0.086 |
| Epeorus herklotsi 02 | 65.71 | 65.15 | 65.87 | 65.61 | 74.80 | −0.004 | −0.197 | −0.020 | 0.007 | 0.008 | −0.243 | −0.031 | 0.140 | 0.305 | −0.082 |
| Epeorus sp. JZ-2014 | 64.60 | 64.03 | 64.62 | 66.03 | 72.78 | −0.002 | −0.201 | −0.021 | 0.012 | 0.042 | −0.209 | −0.026 | 0.123 | 0.270 | 0.000 |
| Epeorus sp. MT-2014 | 64.07 | 62.53 | 65.19 | 67.18 | 78.03 | −0.010 | −0.197 | −0.023 | 0.027 | 0.076 | −0.232 | −0.023 | 0.112 | 0.292 | −0.164 |
| Epeorus montanus | 64.86 | 63.52 | 64.75 | 67.31 | 78.89 | −0.019 | −0.205 | −0.019 | 0.026 | 0.082 | −0.210 | −0.021 | 0.105 | 0.262 | −0.128 |
| Epeorus melli | 65.73 | 65.28 | 65.01 | 65.70 | 74.34 | −0.004 | −0.199 | −0.019 | 0.004 | −0.040 | −0.245 | −0.022 | 0.127 | 0.313 | −0.084 |
| Epeorus bifurcatus | 65.01 | 63.72 | 65.64 | 67.28 | 78.01 | −0.006 | −0.194 | −0.021 | 0.026 | 0.082 | −0.245 | −0.023 | 0.111 | 0.281 | −0.255 |
| Epeorus pellucidus | 66.39 | 65.56 | 66.78 | 67.36 | 76.64 | −0.028 | −0.194 | −0.011 | 0.041 | 0.083 | −0.215 | −0.017 | 0.115 | 0.279 | −0.036 |
| Paegniodes cupulatus 01 | 65.59 | 65.54 | 65.50 | 66.41 | 63.15 | −0.009 | −0.209 | −0.007 | 0.020 | 0.154 | −0.202 | −0.033 | 0.138 | 0.313 | 0.099 |
| Paegniodes cupulatus 02 | 64.73 | 64.62 | 66.14 | 66.59 | 58.70 | −0.005 | −0.213 | −0.018 | 0.025 | 0.244 | −0.203 | −0.036 | 0.135 | 0.296 | −0.011 |
| Parafronurus youi | 66.38 | 67.04 | 65.38 | 66.19 | 57.03 | −0.016 | −0.199 | −0.020 | 0.017 | −0.003 | −0.220 | −0.018 | 0.111 | 0.309 | −0.022 |
| Afronurus furcata | 64.66 | 64.85 | 63.23 | 65.50 | 61.22 | 0.008 | −0.184 | −0.002 | −0.004 | 0.068 | −0.214 | −0.021 | 0.102 | 0.326 | 0.096 |
| Afronurus drepanophyllus | 63.68 | 63.26 | 64.31 | 64.67 | 66.26 | −0.021 | −0.204 | −0.026 | 0.021 | 0.063 | −0.176 | −0.025 | 0.115 | 0.299 | 0.087 |
| Notacanthurus lamellosus | 66.26 | 66.51 | 64.46 | 66.68 | 64.95 | −0.046 | −0.193 | −0.023 | 0.065 | −0.072 | −0.167 | −0.015 | 0.141 | 0.249 | −0.225 |
| Notacanthurus maculosus | 66.77 | 67.03 | 65.01 | 67.17 | 64.46 | −0.014 | −0.200 | −0.030 | 0.025 | 0.045 | −0.219 | −0.010 | 0.127 | 0.298 | −0.296 |
| Heptagenia ngi | 64.12 | 64.03 | 62.81 | 63.95 | 68.78 | −0.005 | −0.196 | −0.022 | −0.008 | −0.058 | −0.177 | 0.000 | 0.121 | 0.275 | −0.094 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Li, R.; Lei, Z.; Li, W.; Zhang, W.; Zhou, C. Comparative Mitogenomic Analysis of Heptageniid Mayflies (Insecta: Ephemeroptera): Conserved Intergenic Spacer and tRNA Gene Duplication. Insects 2021, 12, 170. https://doi.org/10.3390/insects12020170
Li R, Lei Z, Li W, Zhang W, Zhou C. Comparative Mitogenomic Analysis of Heptageniid Mayflies (Insecta: Ephemeroptera): Conserved Intergenic Spacer and tRNA Gene Duplication. Insects. 2021; 12(2):170. https://doi.org/10.3390/insects12020170
Chicago/Turabian StyleLi, Ran, Zhiming Lei, Wenjuan Li, Wei Zhang, and Changfa Zhou. 2021. "Comparative Mitogenomic Analysis of Heptageniid Mayflies (Insecta: Ephemeroptera): Conserved Intergenic Spacer and tRNA Gene Duplication" Insects 12, no. 2: 170. https://doi.org/10.3390/insects12020170
APA StyleLi, R., Lei, Z., Li, W., Zhang, W., & Zhou, C. (2021). Comparative Mitogenomic Analysis of Heptageniid Mayflies (Insecta: Ephemeroptera): Conserved Intergenic Spacer and tRNA Gene Duplication. Insects, 12(2), 170. https://doi.org/10.3390/insects12020170