Two Complete Mitochondrial Genomes of Mileewinae (Hemiptera: Cicadellidae) and a Phylogenetic Analysis



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sample Collection and DNA Extraction
2.2. Mitogenomes Sequencing, Assembly and Annotation
2.3. Sequence Analysis
2.4. Phylogenetic Analyses
3. Results and Discussion
3.1. Mitogenome Organization and Base Composition
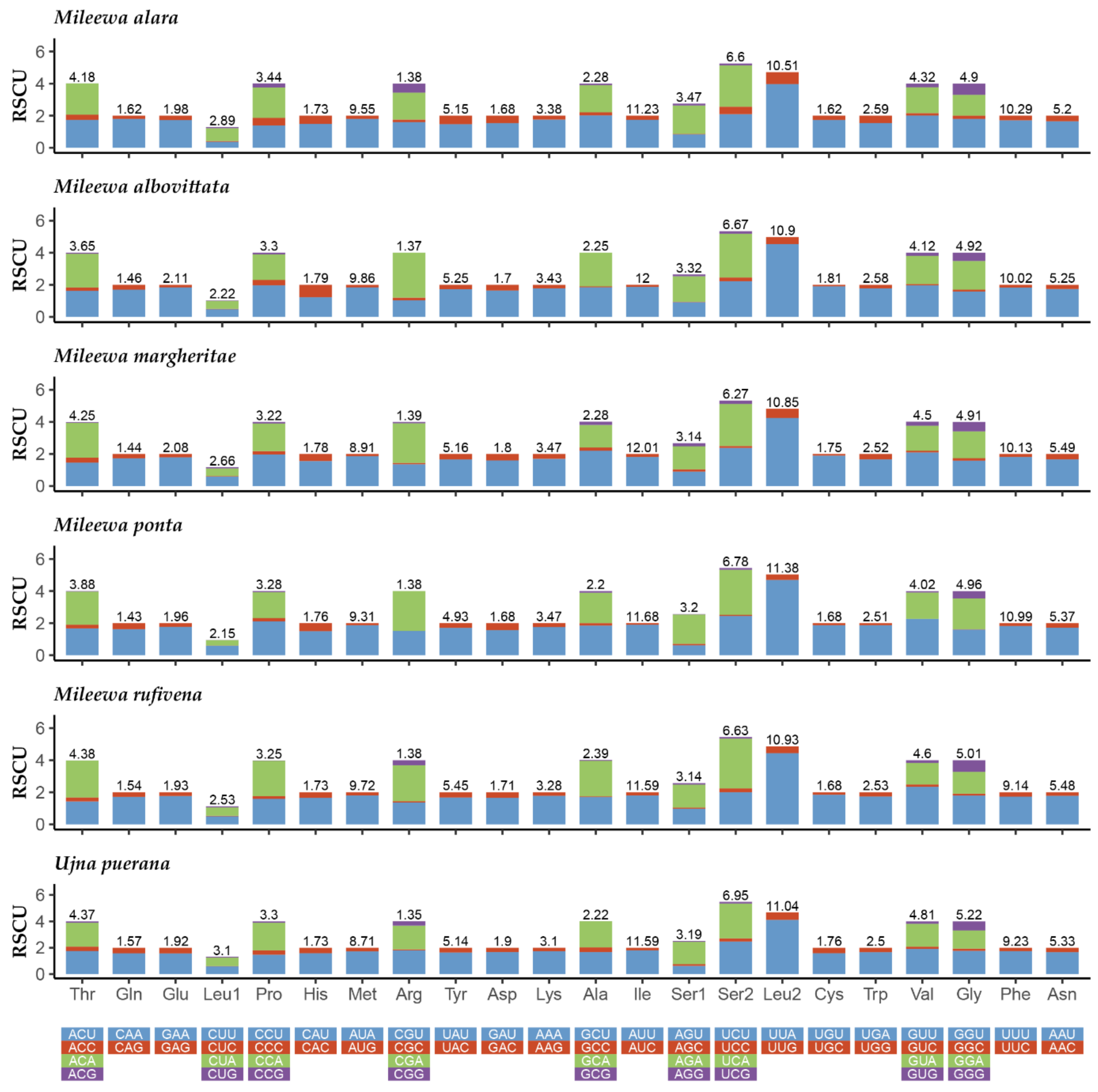
3.2. Protein-Coding Genes and Codon Usage
3.3. Gene Overlaps and Intergenic Spacers
3.4. Transfer and Ribosomal RNA Genes
3.5. Control Region
3.6. Nucleotide Diversity and Evolutionary Rate Analysis
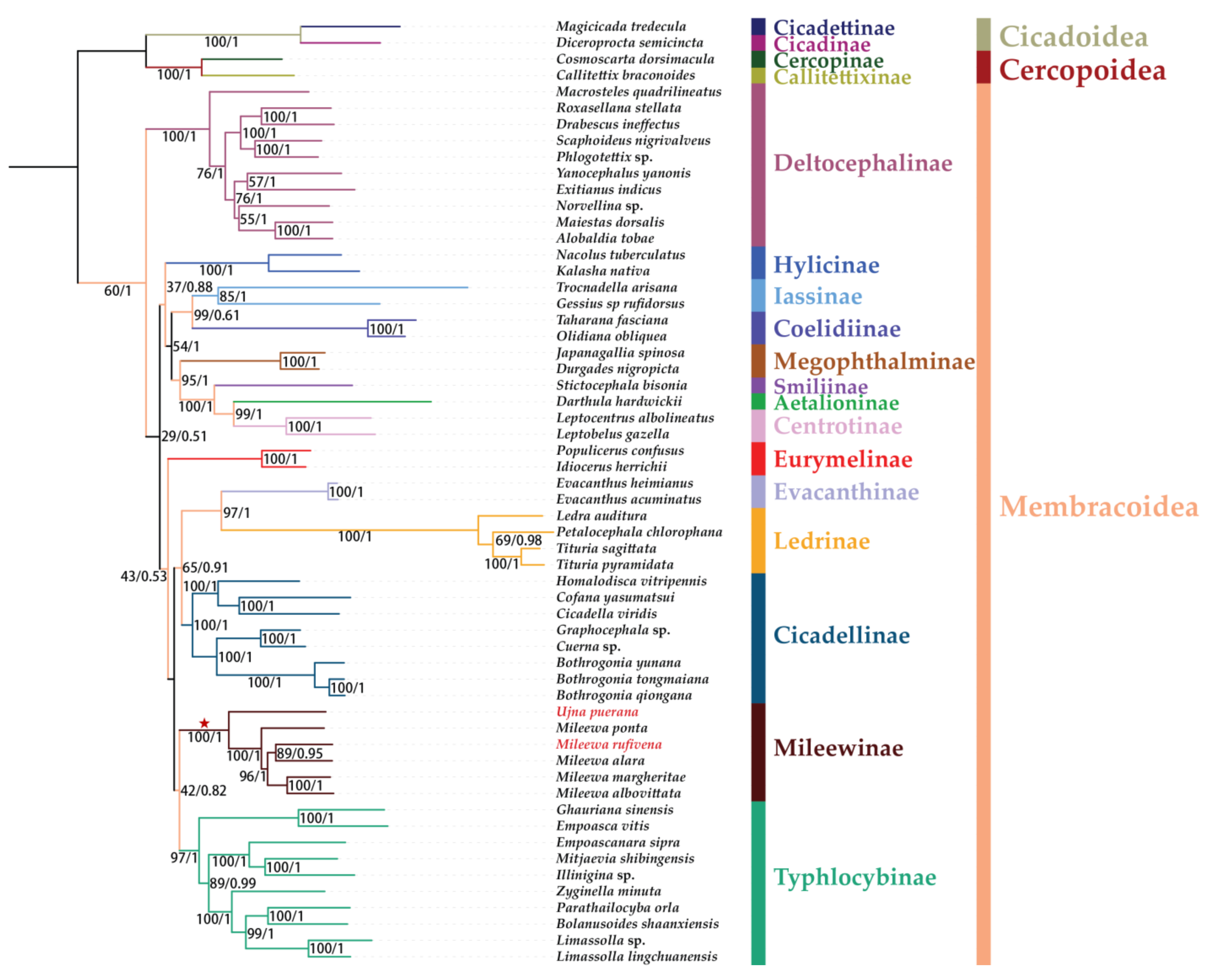
3.7. Phylogenetic Relationships
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Wallace, D.C. Structure and evolution of organelle genomes. Microbiol. Rev. 1982, 46, 208–240. [Google Scholar] [CrossRef]
- Boore, J.L. Animal mitochondrial genomes. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [Green Version]
- Abascal, F.; Posada, D.; Knight, R.D.; Zardoya, R. Parallel evolution of the genetic code in arthropod mitochondrial genomes. PLoS Biol. 2006, 4, e127. [Google Scholar] [CrossRef] [Green Version]
- Cameron, S.L. Insect mitochondrial genomics: Implications for evolution and phylogeny. Annu. Rev. Entomol. 2014, 59, 95–117. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wilson, A.C.; Cann, R.L.; Carr, S.M.; George, M.; Gyllensten, U.B.; Helm-Bychowski, K.M.; Higuchi, R.G.; Palumbi, S.R.; Prager, E.M.; Sage, R.D.; et al. Mitochondrial DNA and two perspectives on evolutionary genetics. Biol. J. Linn. Soc. 1985, 26, 375–400. [Google Scholar] [CrossRef]
- Yang, M.F.; Meng, Z.H.; He, Q.; Dietrich, C.H. Illustrated checklist of mileewine leafhoppers (Hemiptera: Cicadellidae: Mileewinae) of China, with descriptions of four new species. Zootaxa 2014, 3881, 175–189. [Google Scholar] [CrossRef] [Green Version]
- Dietrich, C.H. Tungurahualini, a new tribe of Neotropical leafhoppers, with notes on the subfamily Mileewinae (Hemiptera, Cicadellidae). Zookeys 2011, 124, 19–39. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Krishnankutty, S.M.; Dietrich, C.H. Review of Mileewine leafhoppers (Hemiptera: Cicadellidae: Mileewinae) in Madagascar, with description of seven new species. Ann. Entomol. Soc. Am. 2011, 104, 636–648. [Google Scholar] [CrossRef]
- Evans, J.W. A natural classification of leaf-hoppers (Jassoidea, Homoptera) Part 3. Jassidae. Trans. R. Entomol. Soc. Lond. 1947, 98, 105–271. [Google Scholar] [CrossRef]
- Young, D.A. Western Hemisphere Mileewanini (Homoptera, Cicadellidae). Zool. Beiträge 1965, 11, 369–380. [Google Scholar]
- Mahmood, S.H. A study of the Typhlocybine genera of the Oriental Region (Thailand, the Philippines and adjoining areas). Pac. Insects Monogr. 1967, 12, 1–52. [Google Scholar]
- Young, D.A. Taxonomic study of the Cicadellinae (Homoptera: Cicadellidae). Part 1. Proconiini. Bull. United States Natl. Mus. 1968, 261, 1–287. [Google Scholar] [CrossRef]
- Dietrich, C.H.; Dmitriev, D.A.; Rakitov, R.A.; Takiya, D.M.; Zahniser, J.N. Phylogeny of Cicadellidae (Cicadomorpha: Membracoidea) based on combined morphological and 28S rDNA sequence data. In Abstracts of Talks and Posters, Proceedings of the 12th International Auchenorrhyncha Congress, Berkeley, CA, USA, 8–12 August 2005; Purcell, A., Ed.; University of California: Berkeley, CA, USA, 2005; pp. S13–S14. [Google Scholar]
- He, H.L.; Li, H.X.; Yang, M.F. Complete mitochondrial genome sequence of Mileewa albovittata (Hemiptera: Cicadellidae: Mileewinae). Mitochondrial DNA Part B Resour. 2019, 4, 740–741. [Google Scholar] [CrossRef] [Green Version]
- He, H.L.; Yang, M.F. Characterization and phylogenetic analysis of the mitochondrial genome of Mileewa ponta (Hemiptera: Cicadellidae: Mileewinae). Mitochondrial DNA B Resour. 2020, 5, 2976–2977. [Google Scholar] [CrossRef]
- He, H.L.; Yang, M.F. The mitogenome of Mileewa margheritae (Hemiptera: Cicadellidae: Mileewinae). Mitochondrial DNA B Resour. 2020, 5, 3163–3164. [Google Scholar] [CrossRef]
- He, H.L.; Yang, M.F. Characterization of the leafhopper mitogenome of Mileewa alara (Hemiptera: Cicadellidae: Mileewinae) and its phylogenetic analysis. Mitochondrial DNA B Resour. 2021, 6, 1265–1266. [Google Scholar] [CrossRef]
- Yang, M.F.; Meng, Z.H.; Li, Z.Z. Hemiptera: Cicadellidae (II): Cicadellinae; Fauna Sinica: Insecta; Science Press: Beijing, China, 2017; Volume 67, pp. 1–637. [Google Scholar]
- Hahn, C.; Bachmann, L.; Chevreux, B. Reconstructing mitochondrial genomes directly from genomic next-generation sequencing reads--a baiting and iterative mapping approach. Nucleic Acids Res. 2013, 41, e129. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bernt, M.; Donath, A.; Juhling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Putz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo metazoan mitochondrial genome annotation. Mol. Phylogenetics Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Grant, J.R.; Stothard, P. The CGView Server: A comparative genomics tool for circular genomes. Nucleic Acids Res. 2008, 36, W181–W184. [Google Scholar] [CrossRef] [PubMed]
- Zhang, D.; Gao, F.; Jakovlic, I.; Zhou, H.; Zhang, J.; Li, W.X.; Wang, G.T. PhyloSuite: An integrated and scalable desktop platform for streamlined molecular sequence data management and evolutionary phylogenetics studies. Mol. Ecol. Resour. 2019, 20, 348–355. [Google Scholar] [CrossRef]
- Perna, N.T.; Kocher, T.D. Patterns of nucleotide composition at fourfold degenerate sites of animal mitochondrial genomes. J. Mol. Evol. 1995, 41, 353–358. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Rozas, J.; Ferrer-Mata, A.; Sanchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sanchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large data sets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Du, Z.; Hasegawa, H.; Cooley, J.R.; Simon, C.; Yoshimura, J.; Cai, W.; Sota, T.; Li, H. Mitochondrial genomics reveals shared phylogeographic patterns and demographic history among three periodical cicada species groups. Mol. Biol. Evol. 2019, 36, 1187–1200. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Bu, C.; Wipfler, B.; Liang, A. Comparative analysis of the mitochondrial genomes of Callitettixini spittlebugs (Hemiptera: Cercopidae) confirms the overall high evolutionary speed of the AT-rich region but reveals the presence of short conservative elements at the tribal level. PLoS ONE 2014, 9, e109140. [Google Scholar] [CrossRef] [Green Version]
- Liang, A.P.; Gao, J.; Zhao, X. Characterization of the complete mitochondrial genome of the treehopper Darthula hardwickii (Hemiptera: Aetalionidae). Mitochondrial DNA Part A 2016, 27, 3291–3292. [Google Scholar] [CrossRef]
- Hu, K.; Yuan, F.; Dietrich, C.H.; Yuan, X.Q. Structural features and phylogenetic implications of four new mitogenomes of Centrotinae (Hemiptera: Membracidae). Int. J. Biol. Macromol. 2019, 139, 1018–1027. [Google Scholar] [CrossRef] [PubMed]
- Yu, R.; Feng, L.; Yuan, X. Complete mitochondrial genome sequence of the global invasive species Stictocephala bisonia (Hemiptera: Membracidae: Smiliinae). Mitochondrial DNA Part B Resour. 2021, 6, 1601–1602. [Google Scholar] [CrossRef]
- Zhong, L.K.; Yang, M.F.; Yu, X.F. The mitochondrial genome of Cofana yasumatsui (Hemiptera: Cicadellidae: Cicadellinae). Mitochondrial DNA B Resour. 2020, 5, 1075–1076. [Google Scholar] [CrossRef] [Green Version]
- Song, N.; Cai, W.; Li, H. Insufficient power of mitogenomic data in resolving the auchenorrhynchan monophyly. Zool. J. Linn. Soc. 2008, 183, 776–790. [Google Scholar] [CrossRef]
- Wang, X.; Wang, J.; Dai, R.H. Mitogenomics of five Olidiana leafhoppers (Hemiptera: Cicadellidae: Coelidiinae) and their phylogenetic implications. PeerJ 2021, 9, e11086. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Li, H.; Dai, R. Complete mitochondrial genome of Taharana fasciana (Insecta, Hemiptera: Cicadellidae) and comparison with other Cicadellidae insects. Genetica 2017, 145, 593–602. [Google Scholar] [CrossRef] [PubMed]
- Song, N.; Cai, W.; Li, H. Deep-level phylogeny of Cicadomorpha inferred from mitochondrial genomes sequenced by NGS. Sci. Rep. 2017, 7, 11. [Google Scholar] [CrossRef] [Green Version]
- Xu, D.; Yu, T.; Zhang, Y. Characterization of the complete mitochondrial genome of Drabescus ineffectus and Roxasellana stellata (Hemiptera: Cicadellidae: Deltocephalinae: Drabescini) and their phylogenetic implications. Insects 2020, 11, 534. [Google Scholar] [CrossRef]
- Mao, M.; Yang, X.; Bennett, G. The complete mitochondrial genome of Macrosteles quadrilineatus (Hemiptera: Cicadellidae). Mitochondrial DNA B Resour. 2017, 2, 173–175. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Zhang, C.; Dietrich, C.H.; Zhang, Y.; Dai, W. Characterization of the complete mitochondrial genomes of Maiestas dorsalis and Japananus hyalinus (Hemiptera: Cicadellidae) and comparison with other Membracoidea. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Du, Y.; Dai, W.; Dietrich, C.H. Mitochondrial genomic variation and phylogenetic relationships of three groups in the genus Scaphoideus (Hemiptera: Cicadellidae: Deltocephalinae). Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Shan, L.C.Y.; Di, X.C.; Luo, H.; Zhang, B. The complete mitochondrial genome of the leafhopper Idiocerus herrichii (Hemiptera: Cicadellidae: Idiocerinae). Mitochondrial DNA Part B Resour. 2020, 5, 1465–1466. [Google Scholar] [CrossRef] [Green Version]
- Yuan, Z.; Yang, X.; Li, C.; Song, Y. The complete mitochondrial genome of the leafhopper Evacanthus acuminatus (Hemiptera: Cicadellidae: Evacanthinae). Mitochondrial DNA B Resour. 2019, 4, 3866–3867. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.J.; Yang, M.F.; Dai, R.H.; Li, H. Complete mitochondrial genome of Evacanthus heimianus (Hemiptera: Cicadellidae: Evacanthinae) from China. Mitochondrial DNA Part B Resour. 2018, 4, 284–285. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Huang, W.; Zhang, Y. The complete mitochondrial genome of four Hylicinae (Hemiptera: Cicadellidae): Structural features and phylogenetic implications. Insects 2020, 11, 869. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Wu, Y.; Dai, R.; Yang, M. Comparative mitogenomes of six species in the subfamily Iassinae (Hemiptera: Cicadellidae) and phylogenetic analysis. Int. J. Biol. Macromol. 2020, 149, 1294–1303. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.J.; Li, D.F.; Li, H.; Yang, M.F.; Dai, R.H. Structural and phylogenetic implications of the complete mitochondrial genome of Ledra auditura. Sci. Rep. 2019, 9, 11. [Google Scholar] [CrossRef]
- Huang, W.; Zhang, Y. Characterization of two complete mitochondrial genomes of Ledrinae (Hemiptera: Cicadellidae) and phylogenetic analysis. Insects 2020, 11, 609. [Google Scholar] [CrossRef]
- Wang, J.; Dai, R.; Li, H.; Zhan, H. Characterization of the complete mitochondrial genome of Japanagallia spinosa and Durgades nigropicta (Hemiptera: Cicadellidae: Megophthalminae). Biochem. Syst. Ecol. 2017, 74, 33–41. [Google Scholar] [CrossRef]
- Zhou, N.; Wang, M.; Cui, L.; Chen, X.; Han, B. Complete mitochondrial genome of Empoasca vitis (Hemiptera: Cicadellidae). Mitochondrial DNA Part A 2016, 27, 1052–1053. [Google Scholar] [CrossRef]
- Tan, C.; Chen, X.X.; Li, C.; Song, Y.H. The complete mitochondrial genome of Empoascanara sipra (Hemiptera:Cicadellidae: Typhlocybinae) with phylogenetic consideration. Mitochondrial DNA Part B Resour. 2020, 5, 260–261. [Google Scholar] [CrossRef] [Green Version]
- Shi, R.; Yu, X.F.; Yang, M.F. Complete mitochondrial genome of Ghauriana sinensis (Hemiptera: Cicadellidae: Typhlocybinae). Mitochondrial DNA Part B Resour. 2020, 5, 1367–1368. [Google Scholar] [CrossRef] [Green Version]
- Yuan, X.; Xiong, K.; Li, C.; Song, Y. The complete mitochondrial genome of Limassolla lingchuanensis (Hemiptera: Cicadellidae: Typhlocybinae). Mitochondrial DNA Part B Resour. 2020, 5, 229–230. [Google Scholar] [CrossRef] [Green Version]
- Zhou, X.; Dietrich, C.H.; Huang, M. Characterization of the complete mitochondrial genomes of two species with preliminary investigation on phylogenetic status of Zyginellini (Hemiptera: Cicadellidae: Typhlocybinae). Insects 2020, 11, 684. [Google Scholar] [CrossRef] [PubMed]
- Jiang, J.; Yuan, X.; Yuan, Z.; Song, Y. The complete mitochondrial genome of Parathailocyba orla (Hemiptera: Cicadellidae: Typhlocybinae). Mitochondrial DNA Part B Resour. 2020, 5, 1981–1982. [Google Scholar] [CrossRef]
- Han, C.; Yan, B.; Yu, X.; Yang, M. Complete mitochondrial genome of Zyginella minuta (Cicadellidae: Typhlocybinae: Zyginellini) from China, with its phylogenetic analysis. Mitochondrial DNA Part B Resour. 2020, 5, 2792–2793. [Google Scholar] [CrossRef]
- Talavera, G.; Castresana, J. Improvement of phylogenies after removing divergent and ambiguously aligned blocks from protein sequence alignments. Syst. Biol. 2007, 56, 564–577. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular and morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; van der Mark, P.; Ayres, D.L.; Darling, A.; Hohna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior summarization in Bayesian phylogenetics using tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.A.; Pfeiffer, W.; Schwartz, T. Creating the CIPRES Science Gateway for inference of large phylogenetic trees. In Proceedings of the Gateway Computing Environments Workshop (GCE), New Orleans, LA, USA, 14 November 2010; Institute of Electrical and Electronics Engineers (IEEE): New Orleans, LA, USA, 2010; pp. 1–8. [Google Scholar]
- Boore, J.L.; Lavrov, D.V.; Brown, W.M. Gene translocation links insects and crustaceans. Nature 1998, 392, 667–668. [Google Scholar] [CrossRef]
- Ojala, D.; Montoya, J.; Attardi, G. tRNA punctuation model of RNA processing in human mitochondria. Nature 1981, 290, 470–474. [Google Scholar] [CrossRef] [PubMed]
- Doublet, V.; Ubrig, E.; Alioua, A.; Bouchon, D.; Marcade, I.; Marechal-Drouard, L. Large gene overlaps and tRNA processing in the compact mitochondrial genome of the crustacean Armadillidium vulgare. RNA Biol. 2015, 12, 1159–1168. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garey, J.R.W.; Wolstenholme, D.R. Platyhelminth mitochondrial DNA: Evidence for early evolutionary origin of a tRNAserAGN that contains a dihydrouridine arm replacement loop, and of serine-specifying AGA and AGG codons. J. Mol. Evol. 1989, 28, 374–387. [Google Scholar] [CrossRef]
- Du, Y.; Dietrich, C.H.; Dai, W. Complete mitochondrial genome of Macrosteles quadrimaculatus (Matsumura) (Hemiptera: Cicadellidae: Deltocephalinae) with a shared tRNA rearrangement and its phylogenetic implications. Int. J. Biol. Macromol. 2019, 122, 1027–1034. [Google Scholar] [CrossRef] [PubMed]
- Du, Y.; Liang, Z.; Dietrich, C.H.; Dai, W. Comparative analysis of mitochondrial genomes of Nirvanini and Evacanthini (Hemiptera: Cicadellidae) reveals an explicit evolutionary relationship. Genomics 2021, 113, 1378–1385. [Google Scholar] [CrossRef] [PubMed]
- Hebert, P.D.; Cywinska, A.; Ball, S.L.; de Waard, J.R. Biological identifications through DNA barcodes. Proc. Biol. Sci. 2003, 270, 313–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Yuan, Z.; Li, C.; Dietrich, C.H.; Song, Y. Structural features and phylogenetic implications of Cicadellidae subfamily and two new mitogenomes leafhoppers. PLoS ONE 2021, 16, e0251207. [Google Scholar] [CrossRef]
- Dietrich, C.H.; Rakitov, R.A.; Holmes, J.L.; Black, W.C.T. Phylogeny of the major lineages of Membracoidea (Insecta: Hemiptera: Cicadomorpha) based on 28S rDNA sequences. Mol. Phylogenetics Evol. 2001, 18, 293–305. [Google Scholar] [CrossRef] [PubMed]
- Dietrich, C.H. The role of grasslands in the diversification of leafhoppers (Homoptera: Cicadellidae): A phylogenetic perspective. In Proceedings of the Fifteenth North American Prairie Conference; Natural Areas Association: Bend, OR, USA, 1999; pp. 44–49. [Google Scholar]











{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Superfamily | Family | Subfamily | Species | Accession Number | Reference |
|---|---|---|---|---|---|
| Cicadoidea | Cicadidae | Cicadinae | Diceroprocta semicincta | KM000131 | Unpublished |
| Cicadettinae | Magicicada tredecula | MH937705 | [27] | ||
| Cercopoidea | Cercopidae | Callitettixinae | Callitettix braconoides | NC_025497 | [28] |
| Cercopinae | Cosmoscarta dorsimacula | NC_040115 | Unpublished | ||
| Membracoidea | Aetalionidae | Aetalioninae | Darthula hardwickii | NC_026699 | [29] |
| Membracidae | Centrotinae | Leptobelus gazella | JF801955 | [30] | |
| Leptocentrus albolineatus | NC_044707 | [30] | |||
| Smiliinae | Stictocephala bisonia | MW342606 | [31] | ||
| Cicadellidae | Cicadellinae | Bothrogonia qiongana | NC_049894 | Unpublished | |
| Bothrogonia tongmaiana | NC_049895 | Unpublished | |||
| Bothrogonia yunana | NC_049896 | Unpublished | |||
| Cicadella viridis | KY752061 | Unpublished | |||
| Cofana yasumatsui | NC_049087 | [32] | |||
| Cuerna sp. | KX437741 | [33] | |||
| Graphocephala sp. | KX437740 | [33] | |||
| Homalodisca vitripennis | NC_006899 | Unpublished | |||
| Coelidiinae | Olidiana obliquea | MN780583 | [34] | ||
| Taharana fasciana | NC_036015 | [35] | |||
| Deltocephalinae | Alobaldia tobae | KY039116 | [36] | ||
| Drabescus ineffectus | NC_050258 | [37] | |||
| Exitianus indicus | KY039128 | [35] | |||
| Macrosteles quadrilineatus | NC_034781 | [38] | |||
| Maiestas dorsalis | NC_036296 | [39] | |||
| Norvellina sp. | KY039131 | [36] | |||
| Phlogotettix sp. | KY039135 | [36] | |||
| Roxasellana stellata | NC_050257 | [37] | |||
| Scaphoideus nigrivalveus | KY817244 | [40] | |||
| Yanocephalus yanonis | NC_036131 | [36] | |||
| Eurymelinae | Idiocerus herrichii | MN935487 | [41] | ||
| Populicerus confusus | NC_050982 | Unpublished | |||
| Evacanthinae | Evacanthus acuminatus | MK948205 | [42] | ||
| Evacanthus heimianus | MG813486 | [43] | |||
| Hylicinae | Kalasha nativa | MW218662 | [44] | ||
| Nacolus tuberculatus | MW218663 | [44] | |||
| Iassinae | Gessius rufidorsus | MN577633 | [45] | ||
| Trocnadella arisana | NC_036480 | [45] | |||
| Ledrinae | Ledra auditura | MK387845 | [46] | ||
| Petalocephala chlorophana | NC_051527 | [47] | |||
| Tituria pyramidata | NC_046701 | Unpublished | |||
| Tituria sagittata | NC_051528 | [47] | |||
| Megophthalminae | Durgades nigropicta | NC_035684 | [48] | ||
| Japanagallia spinosa | NC_035685 | [48] | |||
| Mileewinae | Mileewa alara | MW533151 | [17] | ||
| Mileewa albovittata | MK138358 | [14] | |||
| Mileewa margheritae | MT483998 | [16] | |||
| Mileewa ponta | MT497465 | [15] | |||
| Mileewa rufivena | MZ326689 | This study | |||
| Ujna puerana | MZ326688 | This study | |||
| Typhlocybinae | Bolanusoides shaanxiensis | MN661136 | Unpublished | ||
| Empoasca vitis | NC_024838 | [49] | |||
| Empoascanara sipra | NC_048516 | [50] | |||
| Ghauriana sinensis | MN699874 | [51] | |||
| Illinigina sp. | KY039129 | [36] | |||
| Limassolla lingchuanensis | NC_046037 | [52] | |||
| Limassolla sp. | MT683892 | [53] | |||
| Mitjaevia shibingensis | MT981879 | Unpublished | |||
| Parathailocyba orla | MN894531 | [54] | |||
| Zyginella minuta | NC_052876 | [55] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Yu, T.; Zhang, Y. Two Complete Mitochondrial Genomes of Mileewinae (Hemiptera: Cicadellidae) and a Phylogenetic Analysis. Insects 2021, 12, 668. https://doi.org/10.3390/insects12080668
Yu T, Zhang Y. Two Complete Mitochondrial Genomes of Mileewinae (Hemiptera: Cicadellidae) and a Phylogenetic Analysis. Insects. 2021; 12(8):668. https://doi.org/10.3390/insects12080668
Chicago/Turabian StyleYu, Tinghao, and Yalin Zhang. 2021. "Two Complete Mitochondrial Genomes of Mileewinae (Hemiptera: Cicadellidae) and a Phylogenetic Analysis" Insects 12, no. 8: 668. https://doi.org/10.3390/insects12080668
APA StyleYu, T., & Zhang, Y. (2021). Two Complete Mitochondrial Genomes of Mileewinae (Hemiptera: Cicadellidae) and a Phylogenetic Analysis. Insects, 12(8), 668. https://doi.org/10.3390/insects12080668