
Mitochondrial Genomes of Hestina persimilis and Hestinalis nama (Lepidoptera, Nymphalidae): Genome Description and Phylogenetic Implications

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling and DNA Sequencing
2.2. Annotation and Analysis of Mitochondrial DNA
2.3. Phylogenetic Analysis
3. Results and Discussion
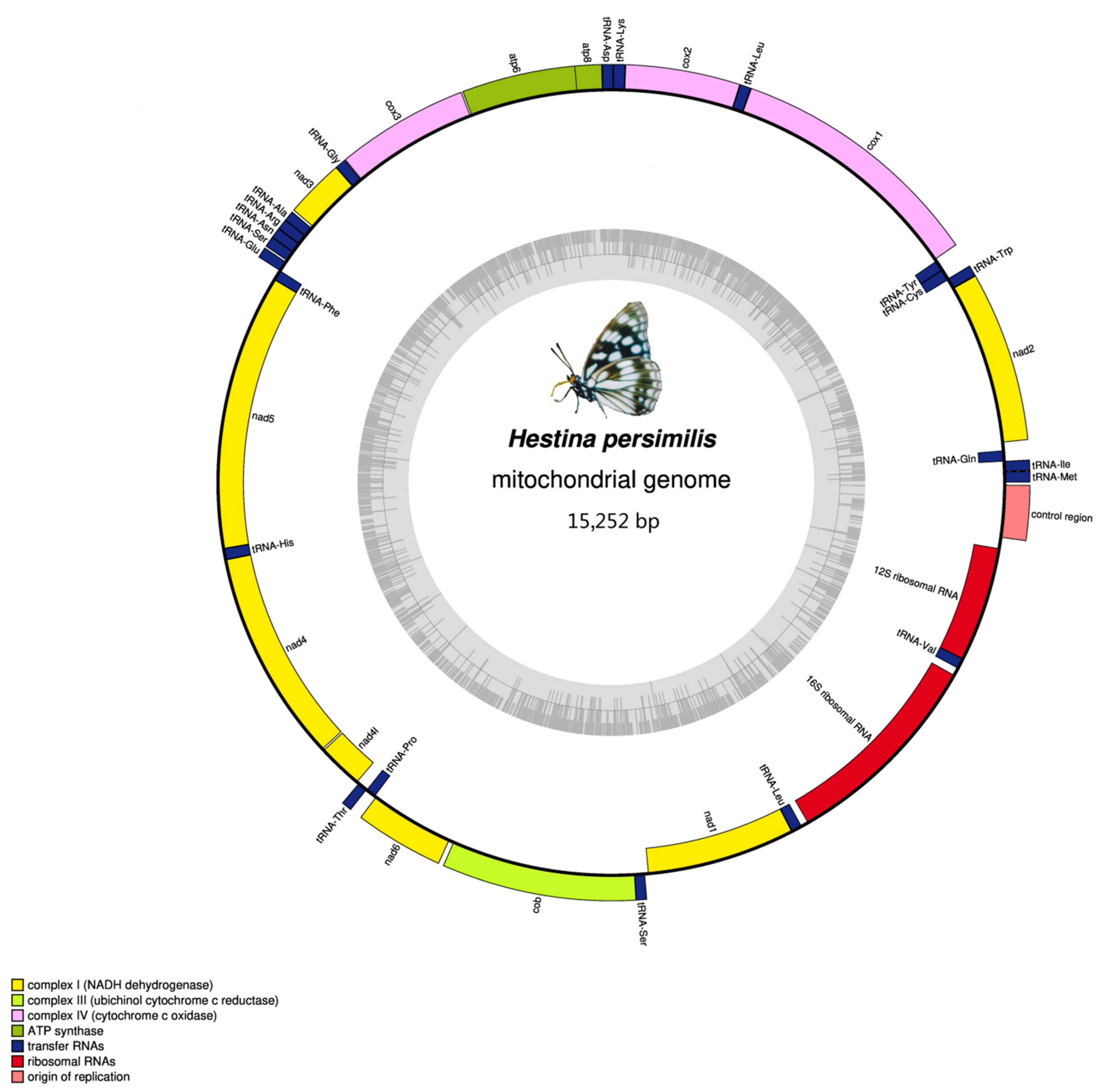
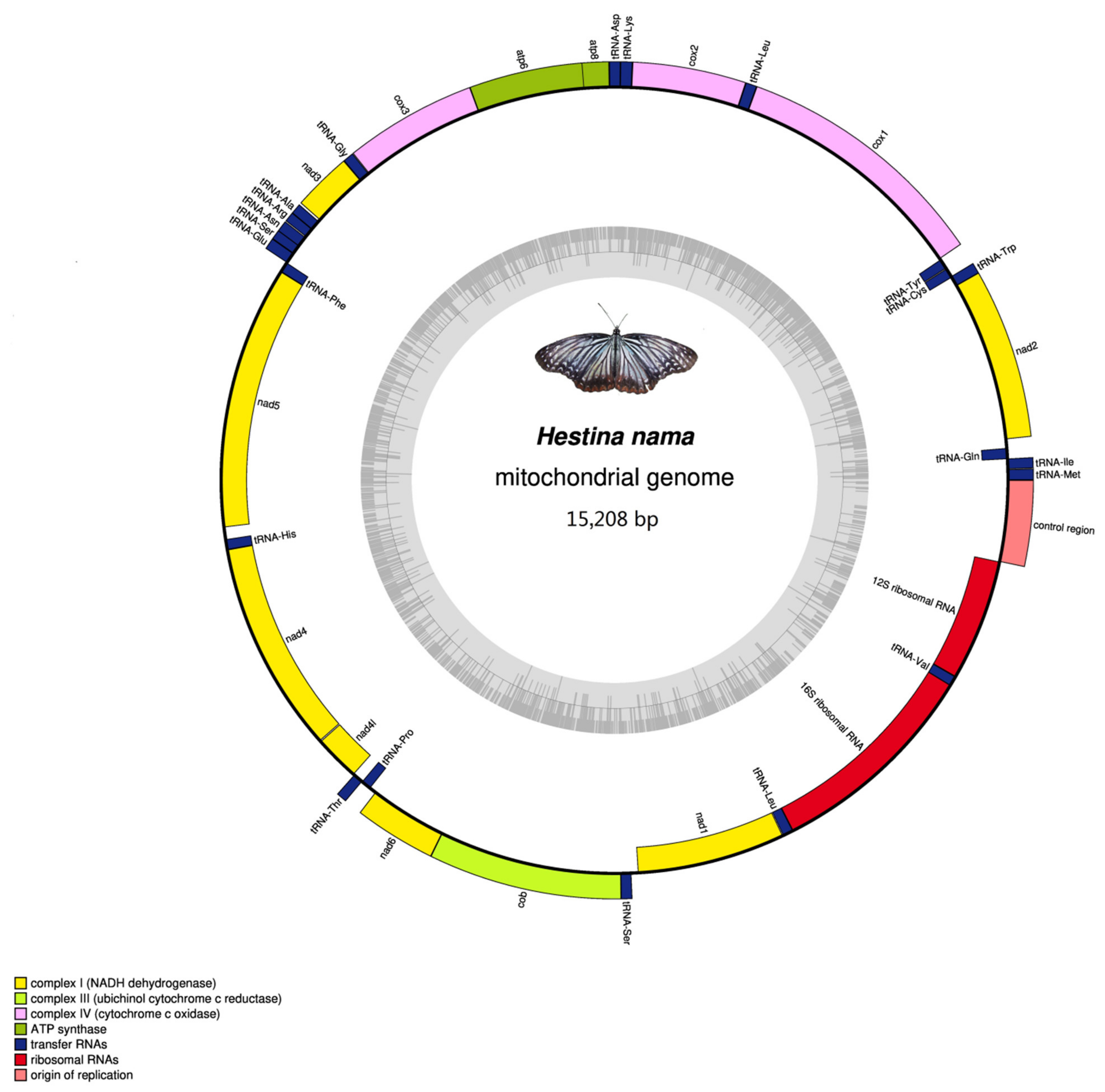
3.1. Mitogenomes Organization
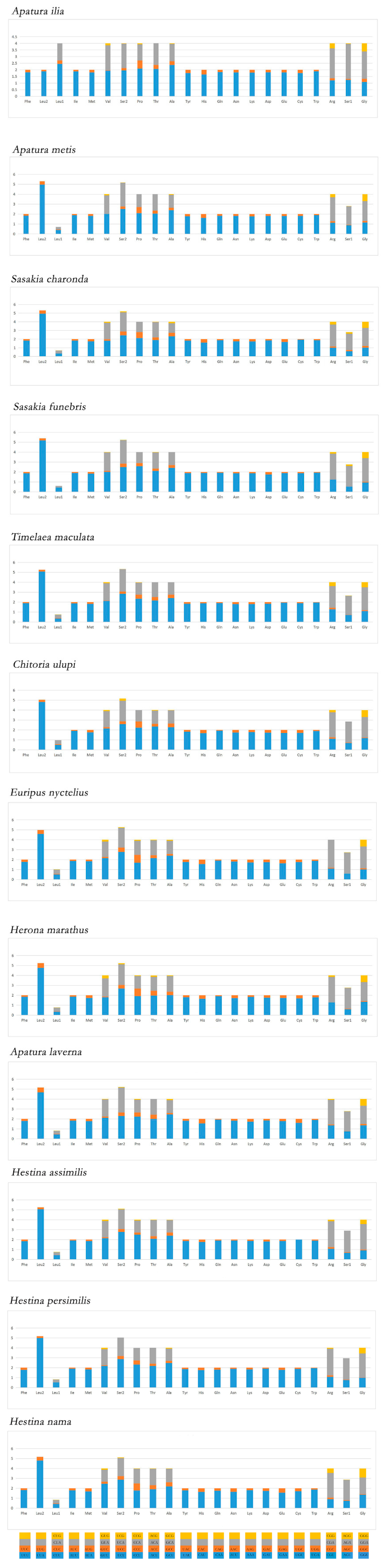
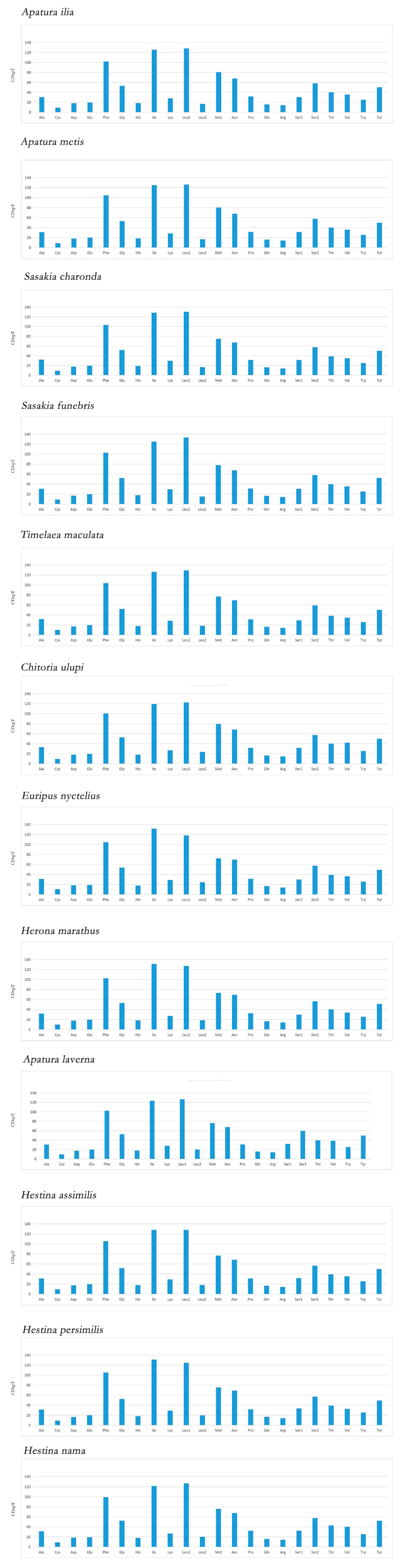
3.2. Protein Coding Genes and Codon Usage
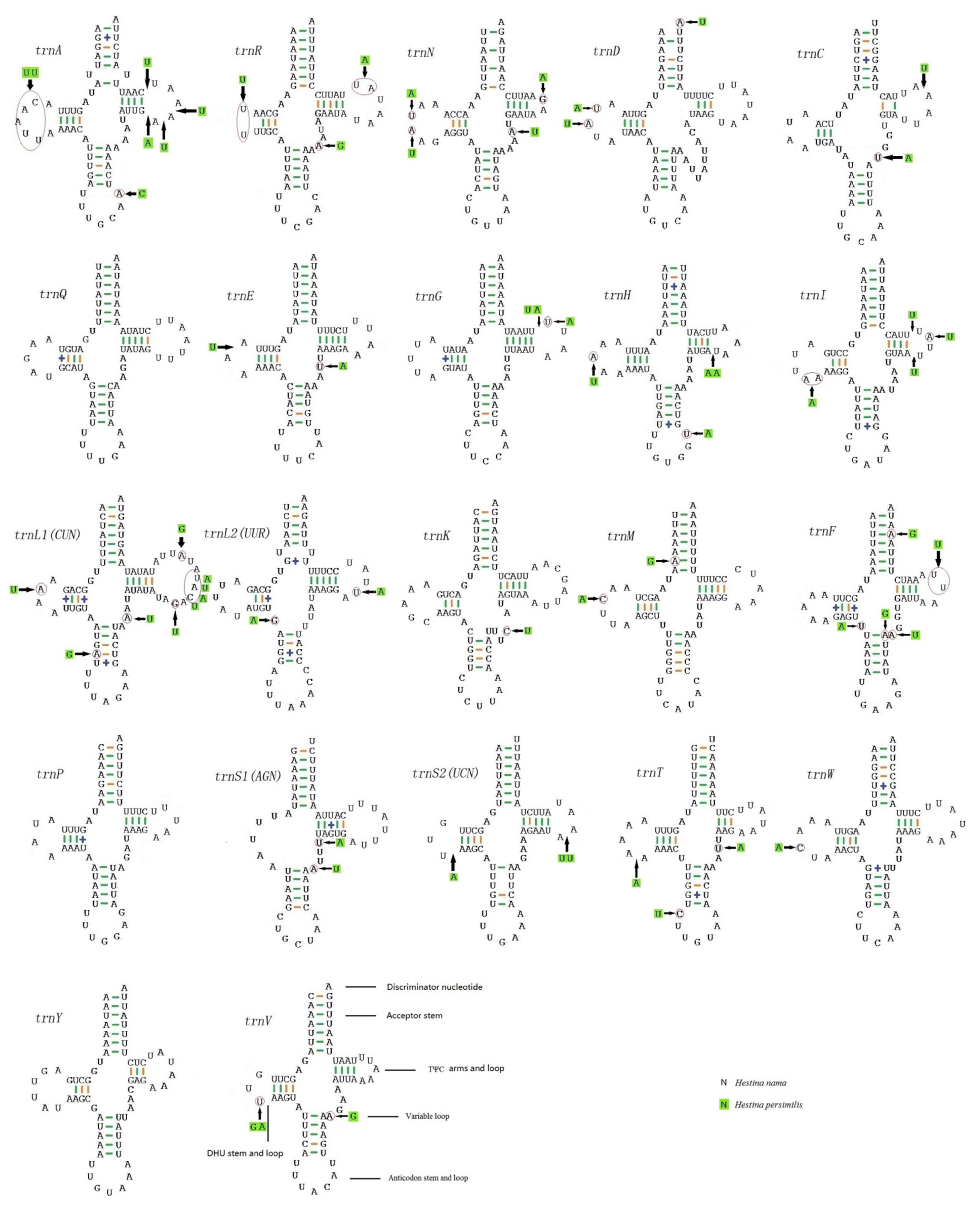
3.3. Transfer RNAs and Ribosomal RNAs
3.4. Intergenic and Overlapping Regions
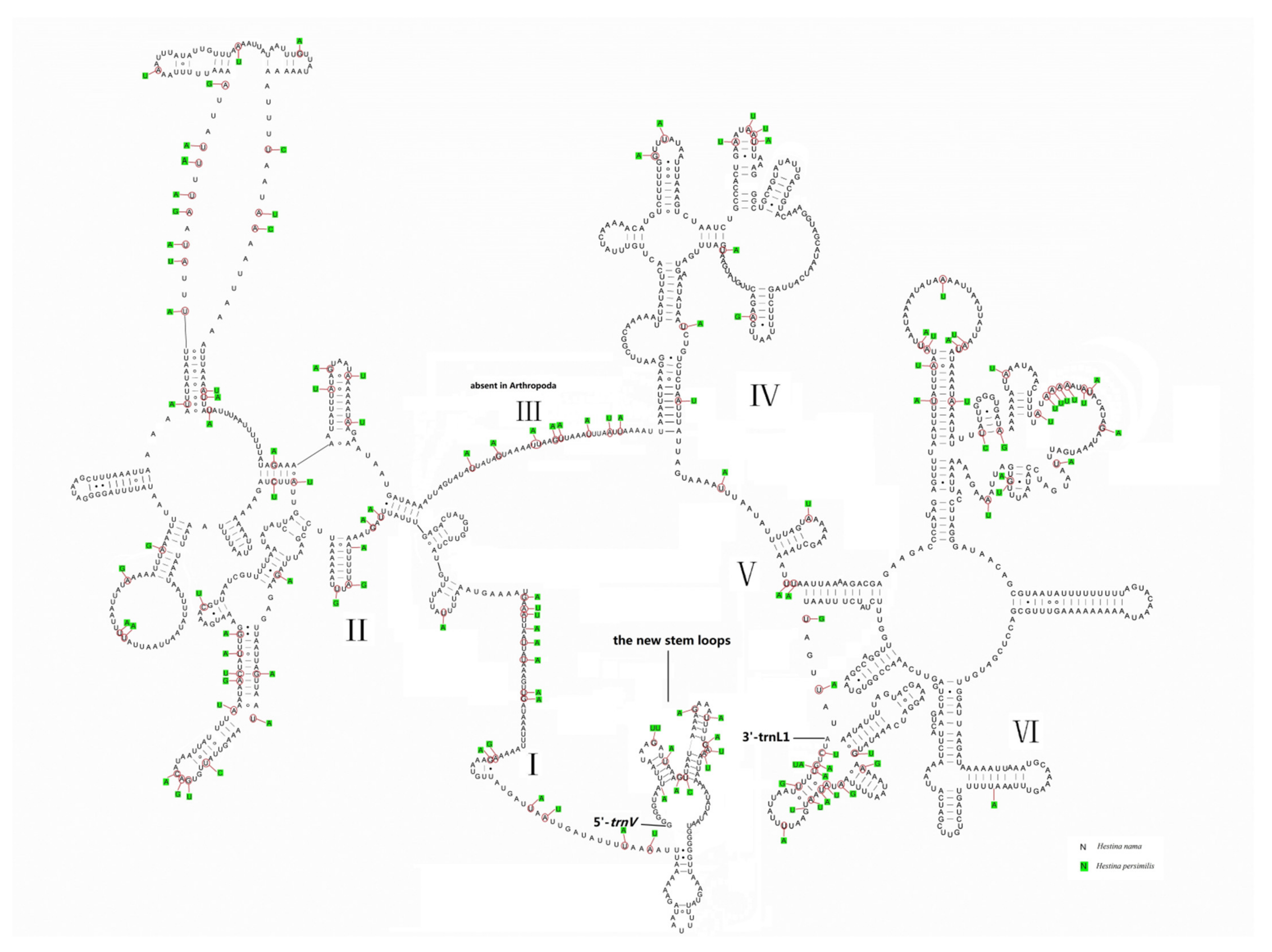
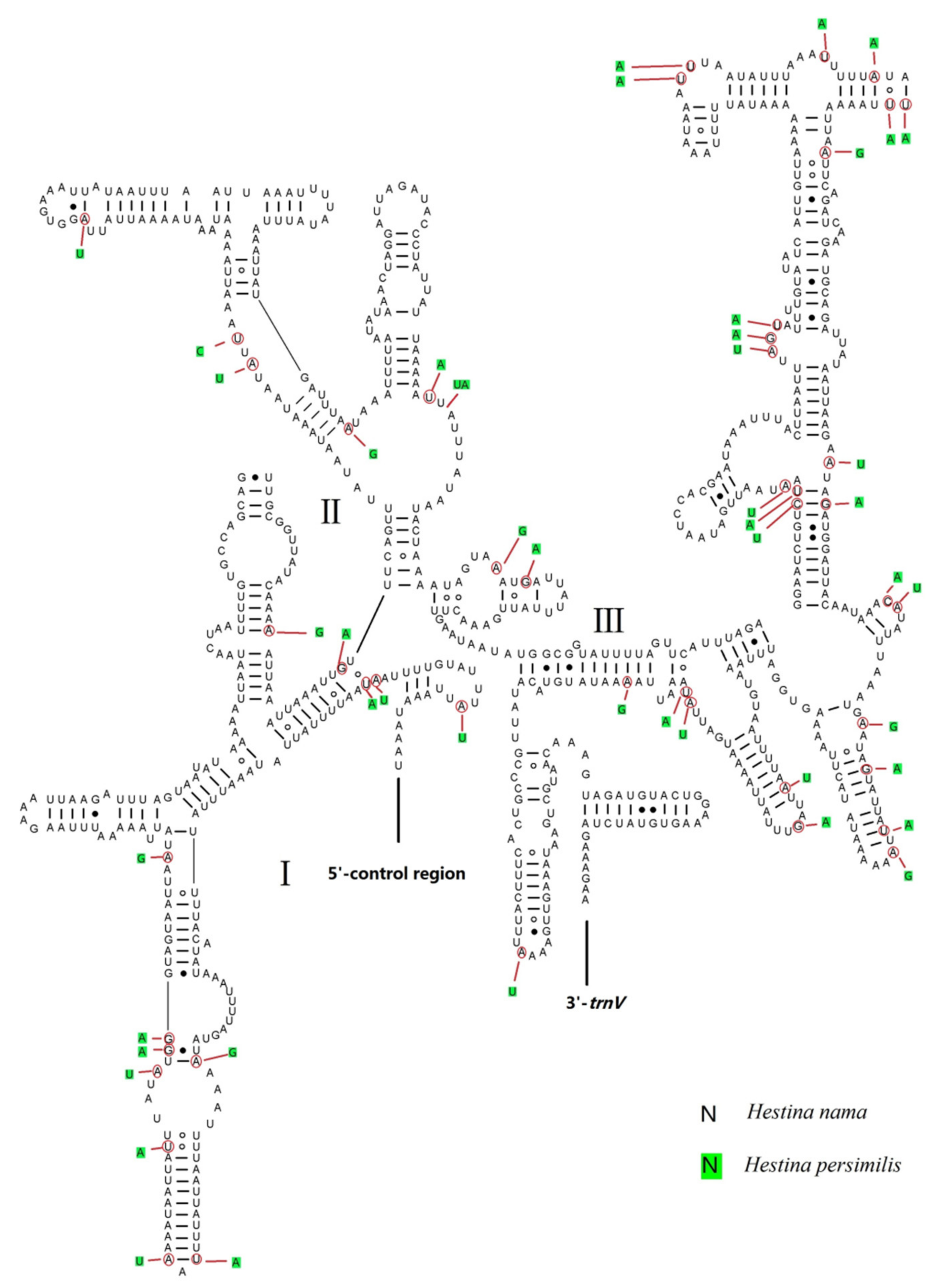
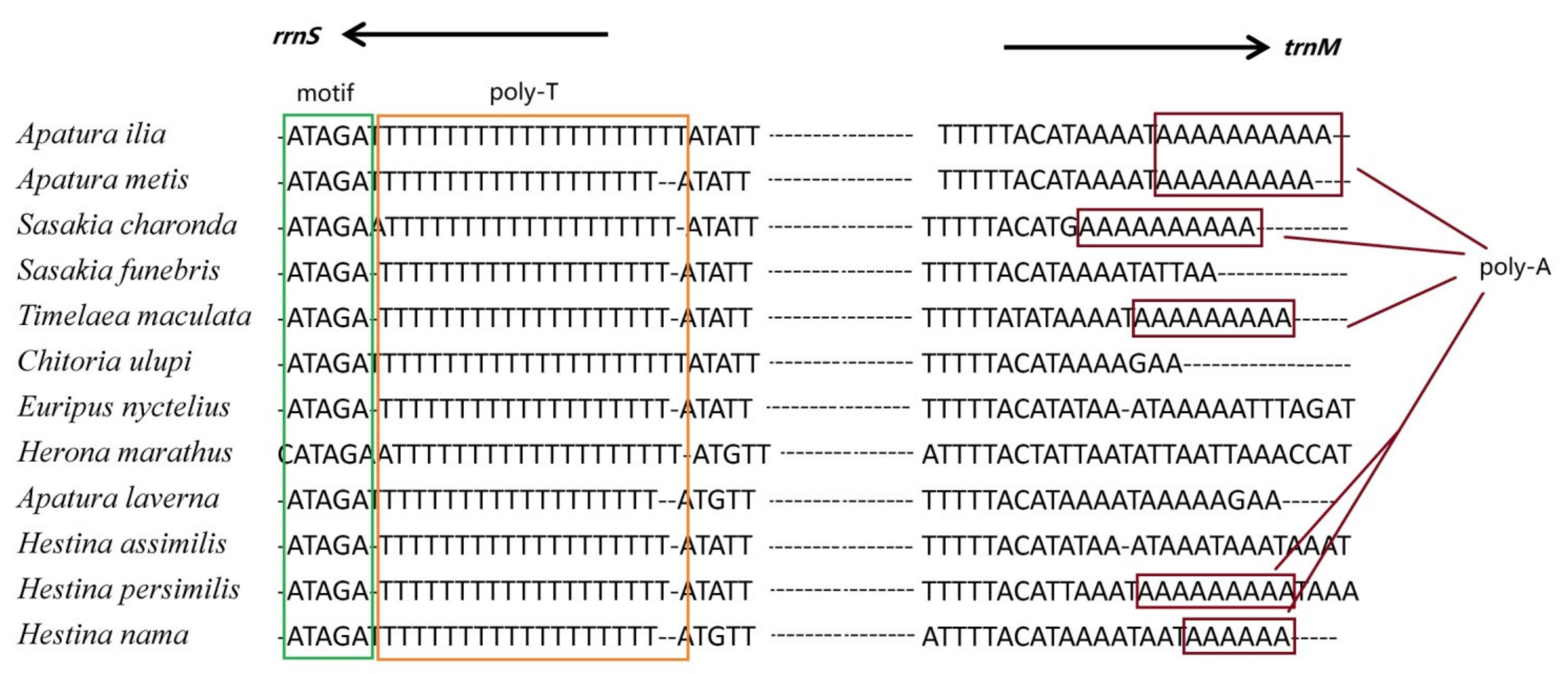
3.5. Putative Control Regions
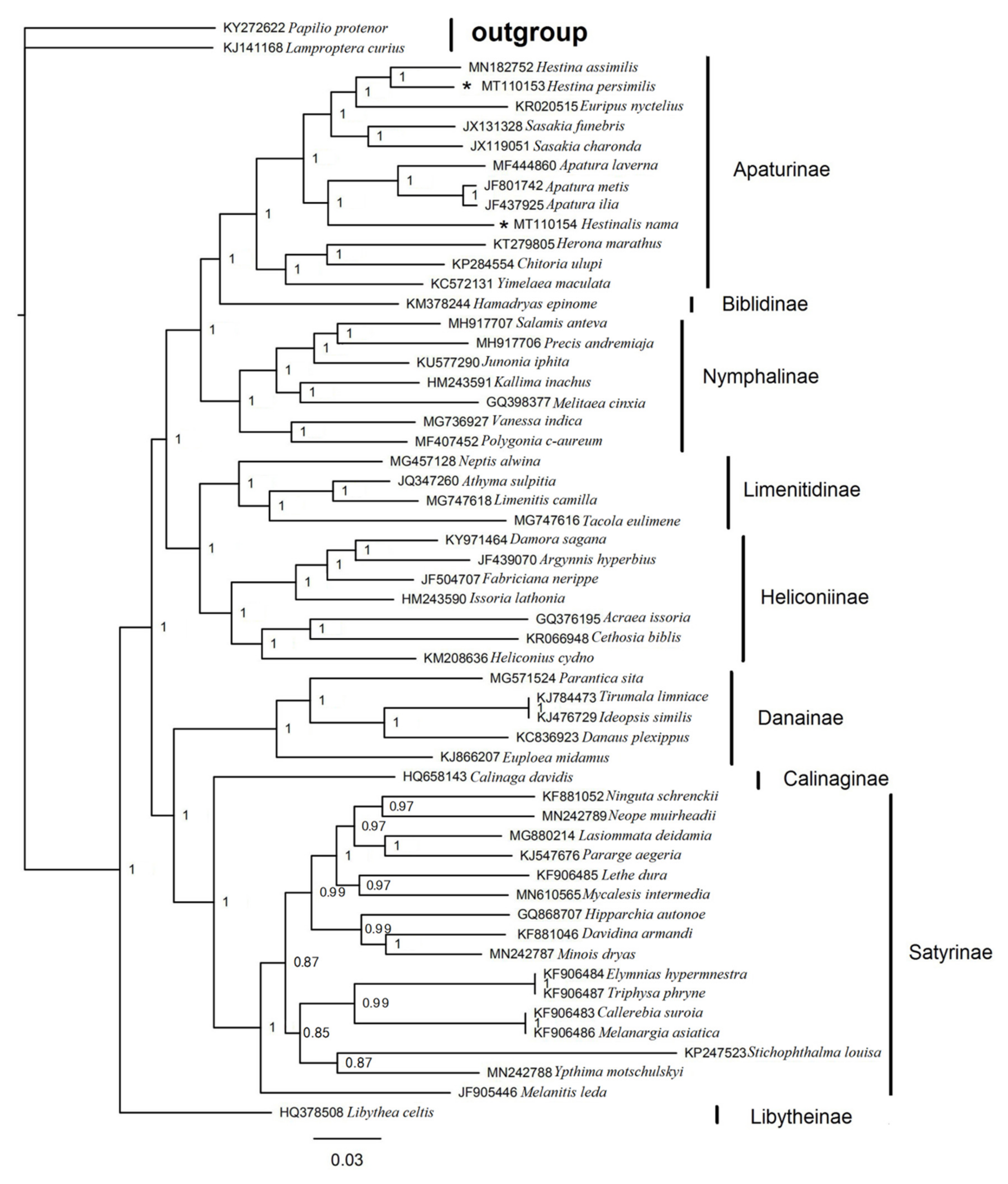
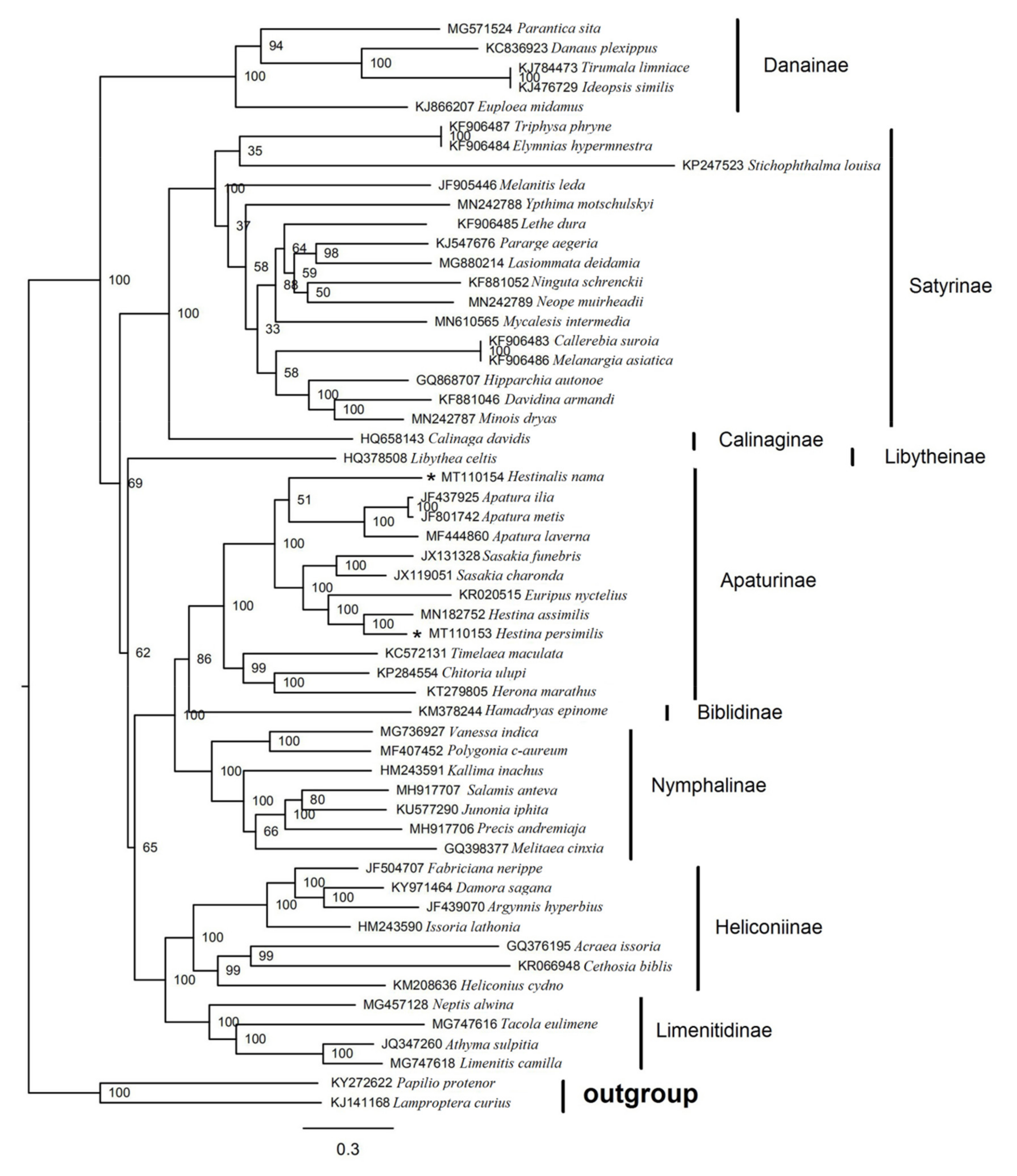
3.6. Phylogenetic Analysis
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Doubleday, E. List of the Specimens of Lepidopterous Insects in the Collection of the British Museum; Trustees of the British Museum: London, UK, 1844; Volume 1, p. 97. [Google Scholar]
- Chou, I. Monograph of Chinese Butterflies; Henan Scientific Technological Publishing House: Zhengzhou, China, 1994; pp. 451–456. [Google Scholar]
- Lee, C.L.; Zhu, B.Y. Atlas of Chinese Butterflies; Shanghai Far East Publishers: Shanghai, China, 1992; p. 136. [Google Scholar]
- Masui, A.; Bozano, G.C.; Floriani, A. Guide to the Butterflies of the Palearctic Region: Nymphalidae 4: Apaturinae; Omnes Artes: Milan, Italy, 2011; Volume 4, pp. 45–102. [Google Scholar]
- Lang, S.Y. The Nymphalidae of China (Lepidoptera, Rhopalocera); Tshikolovets Publications: Pardubice, Czech Republic, 2012; Volume 1, p. 21. [Google Scholar]
- Wu, C.S.; Hsu, Y.F. Butterflies of China; The straits Publishing &Distributing Group: Fuzhou, China, 2017; pp. 871–874. [Google Scholar]
- Simmons, R.B.; Weller, S.J. Utility evolution of cytochrome b in insects. Mol. Phylogenet. Evol. 2001, 20, 196–210. [Google Scholar] [CrossRef] [Green Version]
- Hebert, P.D.N.; Cywinska, A.; Ball, S.L.; de Waard, J.R. Biological identifications through DNA barcodes. Proc. R. Soc. Lond. B Bio. 2003, 270, 313–321. [Google Scholar] [CrossRef] [Green Version]
- Regier, J.G.; Mitter, C.; Zwick, A.; Bazinet, A.L.; Cummings, M.P.; Kawahara, A.Y.; Sohn, J.C.; Zwickl, D.J.; Cho, S.; Davis, D.R.; et al. A large–scale, higher–level, molecular phylogenetic study of the insect order Lepidoptera (moths butterflies). PLoS ONE 2013, 8, e58568. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.P.; Zhao, J.L.; Su, T.J.; Luo, A.R.; Zhu, C.D. The complete mitochondrial genome of Choristoneura longicellana (Lepidoptera: Tortricidae) phylogenetic analysis of Lepidoptera. Gene 2016, 591, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Dong, W.W.; Dong, S.Y.; Jiang, G.F.; Huang, G.H. Characterization of the complete mitochondrial genome of tea tussock moth, Euproctics pseudoconspersa (Lepidoptera: Lymantriidae) its phylogenetic implications. Gene 2016, 577, 37–46. [Google Scholar] [CrossRef] [PubMed]
- Ratnasingham, S.; Hebert, P.D.N. Bold: The Barcode of Life Data System. Mol. Ecol. Notes 2007, 7, 355–364. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lafontaine, J.D.; Fibiger, M. Revised higher classification of the Noctuoidea (Lepidoptera). Can. Entomol. 2006, 138, 610–635. [Google Scholar] [CrossRef]
- Mitter, C.; Davis, D.R.; Cummings, M.P. Phylogeny Evolution of Lepidoptera. Annu. Rev. Entomol. 2017, 62, 265–283. [Google Scholar] [CrossRef]
- Yang, M.S.; Song, L.; Shi, Y.X.; Li, J.H.; Zhang, Y.L.; Song, N. The first mitochondrial genome of the family Epicopeiidae higher–level phylogeny of Macroheterocera (Lepidoptera: Ditrysia). Int. J. Biol. Macromol. 2019, 136, 123–132. [Google Scholar] [CrossRef]
- Mueller, R.L. Evolutionary rates, divergence dates, the performance of mitochondrial genes in bayesian phylogenetic analysis. Syst. Biol. 2006, 55, 289–300. [Google Scholar] [CrossRef] [Green Version]
- Timmermans, M.J.T.N.; Lees, D.C.; Simonsen, T.J. Towards a mitogenomic phylogeny of Lepidoptera. Mol. Phylogenet. Evol. 2014, 79, 169–178. [Google Scholar] [CrossRef]
- Liu, N.Y.; Li, N.; Yang, P.Y.; Sun, C.Q.; Fang, J.; Wang, S.Y. The complete mitochondrial genome of Damora sagana phylogenetic analyses of the family Nymphalidae. Genes Genom. 2018, 40, 109–122. [Google Scholar] [CrossRef]
- Habib, M.; Lakra, W.S.; Mohindra, V.; Khare, P.; Barman, A.S.; Singh, A.; Lal, K.K.; Punia, P.; Khan, A.A. Evaluation of cytochrome b mtDNA sequences in genetic diversity studies of Channa marulius (Channidae: Perciformes). Mol. Biol. Rep. 2011, 38, 841–846. [Google Scholar] [CrossRef]
- Chris, S.; Thomas, R.B.; Francesco, F.; James, B.S.; Andrew, T.B. Incorporating molecular evolution into phylogenetic analysis, a new compilation of conserved polymerase chain reaction primers for animal mitochondrial DNA. Annu. Rev. Ecol. Evol. Syst. 2006, 37, 545–579. [Google Scholar]
- Simon, C.; Frati, F.; Bekenbach, A.; Crespi, B.; Liu, H.; Flook, P. Evolution, weighting, phylogenetic utility of mitochondrial genesequences a compilation of conserved polymerase chain reaction primers. Ann. Entomol. Soc. Am. 1994, 87, 651–701. [Google Scholar] [CrossRef]
- Lowe, T.M.; Eddy, S.R. tRNAscan–SE: A program for improved detection of transfer RNA genes in genomic sequence. Nucleic Acids Res. 1997, 25, 955–964. [Google Scholar] [CrossRef] [PubMed]
- Bernt, M.; Donath, A.; Jühling, F.; Externbrink, F.; Florentz, C.; Fritzsch, G.; Pütz, J.; Middendorf, M.; Stadler, P.F. MITOS: Improved de novo etazoan mitochondrial genome annotation. Mol. Phylogenet. Evol. 2013, 69, 313–319. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Calcott, B.; Ho, S.Y.W.; Guindon, S. Partitionfifinder: Combined selection of partitioning schemes substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New methods for selecting partitioned models of evolution for molecular morphological phylogenetic analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms methods to estimate maximum–likelihood phylogenies: Assessing the performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [Green Version]
- Miller, M.A.; Pfeiffer, W.T.; Schwartz, T. Creating the CIPRES Science Gateway for Inference of Large Phylogenetic Trees; Gateway Computing Environments Workshop (GCE): New Orleans, LA, USA, 2010; pp. 1–7. [Google Scholar]
- Stamatakis, A. RAxML version 8: A tool for phylogenetic analysis post–analysis large phylogenies. Bioinformatics 2014, 30, 1312–1313. [Google Scholar] [CrossRef]
- Rodríguez–Trelles, F.; Tarrío, R.; Ayala, F.J. Fluctuating mutation bias the evolution of base composition in Drosophila. J. Mol. Evol. 2000, 50, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clary, D.O.; Wolstenholme, D.R. The mitochondrial DNA molecular of Drosophila yakuba: Nucleotide sequence, gene organization, genetic code. J. Mol. Evol. 1985, 22, 252–271. [Google Scholar] [CrossRef] [PubMed]
- Lu, H.F.; Su, T.J.; Luo, A.R.; Zhu, C.D.; Wu, C.S. Characterization of the complete mitochondrion genome of Diurnal moth Amata emma (Butler) (Lepidoptera: Erebidae) its phylogenetic implications. PLoS ONE 2013, 8, e72410. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.L.; Zhang, Q.L.; Guo, Z.L.; Wang, J.; Shen, Y.Y. The complete mitochondrial genome of Corizus tetraspilus (Hemiptera: Rhopalidae) phylogenetic analysis of Pentatomomorpha. PLoS ONE 2015, 10, e0129003. [Google Scholar]
- Hershberg, R.; Petrov, D.A. Selection on codon bias. Annu. Rev. Genet. 2008, 42, 287–299. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Plotkin, J.B.; Kudla, G. Synonymous but not the same: The causes consequences of codon bias. Nat. Rev. Genet. 2011, 12, 32–42. [Google Scholar] [CrossRef] [Green Version]
- Boore, J.L. Animal mitochondrial genomics. Nucleic Acids Res. 1999, 27, 1767–1780. [Google Scholar] [CrossRef] [Green Version]
- Yong, H.S.; Song, S.L.; Lim, P.E.; Eamsobhana, P.; Suana, I.W. Complete Mitochondrial Genome of Three Bactrocera Fruit Flies of Subgenus Bactrocera (Diptera: Tephritidae) and Their Phylogenetic Implications. PLoS ONE 2016, 11, e0148201. [Google Scholar] [CrossRef] [PubMed]
- Zhang, K.J.; Zhu, W.C.; Rong, X.; Liu, J.; Ding, X.L.; Hong, X.Y. The complete mitochondrial genome sequence of Sogatella furcifera (Horváth) a comparative mitogenomic analysis of three predominant rice planthoppers. Gene 2014, 533, 100–109. [Google Scholar] [CrossRef]
- Nina, V.V.; Sofiya, L.; Derek, W.; Raman, S.; Yury, B.; Dmitrii, Z. Characteristic variability of five complete aphid mitochondrial genomes: Aphis fabae mordvilkoi, Aphis craccivora, Myzus persicae, Therioaphis tenera and Appendiseta robiniae (Hemiptera; Sternorrhyncha; Aphididae). Int. J. Biol. Macromol. 2020, 149, 187–206. [Google Scholar]
- Gillespie, J.J.; Johnston, J.S.; Cannone, J.J.; Gutell, R.R. Characteristics of the nuclear (18S, 5.8S, 28S and 5S) and mitochondrial (12S and 16S) rRNA genes of Apis mellifera (Insecta: Hymenoptera): Structure, organization, and retrotransposable elements. Insect Mol. Biol. 2006, 15, 657–686. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gong, Y.J.; Shi, B.C.; Kang, Z.J.; Zhang, F.; Wei, S.J. The complete mitochondrial genome of the oriental fruit moth Grapholita molesta (Busck) (Lepidoptera: Tortricidae). Mol. Biol. Rep. 2011, 39, 2893–2900. [Google Scholar] [CrossRef] [Green Version]
- Moritz, C.; Dowling, T.E.; Brown, W.M. Evolution of animal mitochondrial DNA: Relevance for population biology systematics. Annu. Rev. Ecol. Syst. 1987, 18, 269–292. [Google Scholar] [CrossRef]
- Sheffield, N.C.; Song, H.; Cameron, S.L.; Whiting, M.F. A comparative analysis of mitochondrial genomes in coleoptera genome descriptions of six new beetles. Mol. Biol. Evol. 2008, 25, 2499–2509. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.P.; Zhao, J.L.; Su, T.J.; Li, J.; Yu, F.; Chesters, D.; Fan, R.J.; Chen, M.C.; Wu, C.S.; Zhu, C.D. The Complete mitochondrial genome of Leucoptera malifoliella Costa (Lepidoptera: Lyonetiidae). DNA Cell Biol. 2012, 31, 1508–1522. [Google Scholar] [CrossRef] [Green Version]
- Cao, Y.Q.; Ma, C.; Chen, J.Y.; Yang, D.R. The complete mitochondrial genomes of two ghost moths, Thitarodes renzhiensis and Thitarodes yunnanensis: The ancestral gene arrangement in Lepidoptera. BMC Genom. 2012, 13, 276. [Google Scholar] [CrossRef] [Green Version]
- Yang, M.; Shi, S.; Dai, P.; Song, L.; Liu, X. Complete mitochondrial genome of Palpita hypohomalia (Lepidoptera: Pyraloidea: Crambidae) and its phylogenetic implications. Eur. J. Entomol. 2018, 115, 708–717. [Google Scholar] [CrossRef]
- Tayler, M.F.; McKechnie, S.W.; Pierce, N.; Kreitman, M. The lepidopteran mitochondrial control region: Structure evolution. Mol. Biol. Evol. 1993, 10, 1259–1272. [Google Scholar]
- Clayton, D.A. Transcription replication of animal mitochondrial DNAs. Int. Rev. Cyt. 1992, 141, 217–232. [Google Scholar]
- Saito, S.; Tamura, K.; Aotsuka, T. Replication origin of mitochondrial DNA in insects. Genetics 2005, 171, 1695–1705. [Google Scholar] [CrossRef] [Green Version]
- Ye, W.; Dang, J.P.; Xie, L.D.; Huang, Y. Complete mitochondrial genome of Teleogryllus emma (Orthoptera: Gryllidae) with a new gene order in Orthoptera. Zool. Res. 2008, 29, 236–244. [Google Scholar] [CrossRef] [Green Version]
- Ehrlich, P.R. The comparative morphology, phylogeny higher classifification of the butterflflies (Lepidoptera: Papilionoidea). Univ. Kans. Sci. Bull. 1958, 39, 305–370. [Google Scholar]
- Scott, J.A. The phylogeny of butterflflies (Papilionoidea Hesperoidea). J. Res. Lepid. 1985, 23, 24–281. [Google Scholar]
- Kristensen, N.P.; Scoble, M.J.; Karsholt, O. Lepidoptera phylogeny systematic: The state of inventorying moth butterfly diversity. Zootaxa 2007, 1668, 699–747. [Google Scholar] [CrossRef] [Green Version]
- Wahlberg, N.; Weingartner, E.; Nylin, S. Towards a better understanding of the higher systematics of Nymphalidae (Lepidoptera: Papilionoidea). Mol. Phylogenet. Evol. 2003, 28, 473–484. [Google Scholar] [CrossRef]
- Wahlberg, N.; Wheat, C.W. Genomic outposts serve the phylogenomic pioneers: Designing novel nuclear markers for genomic DNA extractions of Lepidoptera. Syst. Biol. 2008, 57, 231–242. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wahlberg, N.; Leneveu, J.; Kodandaramaiah, U.; Peña, C.; Nylin, S.; Freitas, A.V.L.; Brower, A.V.Z. Nymphalid butterflies diversify following near demise at the Cretaceous/Tertiary boundary. Proceedings of the Royal Society of London Series B Biological Sciences. Proc. Biol. Sci. 2009, 276, 4295–4302. [Google Scholar] [PubMed] [Green Version]
- Freitas, A.V.L.; Brown, K.S. Phylogeny of the Nymphalidae (Lepidoptera). Syst. Biol. 2004, 53, 363–383. [Google Scholar] [CrossRef] [Green Version]
- Zhang, M.; Cao, T.W.; Zhang, R.; Guo, Y.P.; Duan, Y.H.; Ma, E.B. Phylogeny of Apaturinae Butterflies (Lepidoptera: Nymphalidae) based on mitochondrial cytochrome oxidase I gene. J. Genet Genom. 2007, 34, 812–823. [Google Scholar] [CrossRef]
- Ohshima, I.; Tanikawadodo, Y.; Saigusa, T.; Nishiyama, T.; Kitani, M.; Hasebe, M.; Mohri, H. Phylogeny, biogeography, host–plant association in the subfamily Apaturinae (Insecta: Lepidoptera: Nymphalidae) inferred from eight nuclear seven mitochondrial genes. Mol. Phylogenet. Evol. 2010, 57, 1026–1036. [Google Scholar] [CrossRef] [PubMed]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Direction | Location | Size | Anticodon | Start Codon | Stop Codon | Intergenic Nucleotides |
|---|---|---|---|---|---|---|---|
| trnM | F | 1–68 | 68 | CAT 32–34 | |||
| trnI | F | 69–134 | 66 | GAT 98–100 | 0 | ||
| trnQ | R | 132–200 | 69 | TTG 159–161 | −3 | ||
| nad2 | F | 292–1305 | 1014 | ATT | TAA | 91 | |
| trnW | F | 1304–1371 | 68 | TCA1335–1337 | −2 | ||
| trnC | R | 1364–1427 | 64 | GCA 1397–1399 | −8 | ||
| trnY | R | 1428–1492 | 65 | GTA 1359–1461 | 0 | ||
| cox1 | F | 1498–3033 | 1536 | CGA | TAA | 5 | |
| trnL2 (UUR) | F | 3029–3095 | 67 | TAA 3059–3061 | −5 | ||
| cox2 | F | 3096–3774 | 679 | ATG | T | 0 | |
| trnK | F | 3772–3842 | 71 | CTT 3802–3804 | −3 | ||
| trnD | F | 3842–3907 | 66 | GTC 3872–3874 | −1 | ||
| atp8 | F | 3908–4069 | 162 | ATC | TAA | 0 | |
| atp6 | F | 4063–4737 | 675 | ATG | TAA | −7 | |
| cox3 | F | 4737–5525 | 789 | ATG | TAA | −1 | |
| trnG | F | 5528–5594 | 67 | TCC 5558–5560 | 2 | ||
| nad3 | F | 5595–5948 | 354 | ATT | TAG | 0 | |
| trnA | F | 5947–6014 | 68 | TGC 5976–5978 | −2 | ||
| trnR | F | 6014–6077 | 64 | TCG 6040–6042 | −1 | ||
| trnN | F | 6090–6155 | 66 | GTT 6121–6123 | 12 | ||
| trnS1 (AGN) | F | 6154–6213 | 60 | GCT 6171–6173 | −2 | ||
| trnE | F | 6216–6280 | 65 | TTC 6245–6247 | 2 | ||
| trnF | R | 6279–6342 | 64 | GAA 6310–6312 | −2 | ||
| nad5 | R | 6317–8077 | 1761 | ATT | TAA | −26 | |
| trnH | R | 8075–8141 | 67 | GTG 8109–8111 | −3 | ||
| nad4 | R | 8142–9480 | 1339 | ATG | T | 0 | |
| nad4L | R | 9482–9772 | 291 | ATA | TAA | 1 | |
| trnT | F | 9780–9844 | 65 | TGT 9811–9813 | 7 | ||
| trnP | R | 9845–9908 | 64 | TGG 9877–9879 | 0 | ||
| nad6 | F | 9911–10438 | 528 | ATA | TAA | 2 | |
| cob | F | 10,442–11,593 | 1152 | ATG | TAA | 3 | |
| trnS2 (UCN) | F | 11,596–11,662 | 67 | TGA 11,625–11,627 | 2 | ||
| nad1 | R | 11,685–12,626 | 942 | ATG | TAA | 22 | |
| trnL1 (CUN) | R | 12,628–12,702 | 75 | TAG 12,671–12,673 | 1 | ||
| rrnL | R | 12,703–14,036 | 1334 | 0 | |||
| trnV | R | 14,037–14,100 | 64 | TAC 14,069–14,071 | 0 | ||
| rrnS | R | 14,101–14,876 | 776 | 0 | |||
| Control region | 14,877–15,252 | 376 | 0 |
| Gene | Direction | Location | Size | Anticodon | Start Codon | Stop Codon | Intergenic Nucleotides |
|---|---|---|---|---|---|---|---|
| trnM | F | 1–68 | 68 | CAT 32–34 | |||
| trnI | F | 69–133 | 65 | GAT 99–101 | 0 | ||
| trnQ | R | 131–199 | 69 | TTG 158–160 | −3 | ||
| nad2 | F | 269–1282 | 1013 | ATT | TAA | 69 | |
| trnW | F | 1281–1348 | 68 | TCA 1312–1314 | −2 | ||
| trnC | R | 1341–1403 | 63 | GCA 1372–1374 | −8 | ||
| trnY | R | 1404–1468 | 65 | GTA 1435–1437 | 0 | ||
| cox1 | F | 1474–3009 | 1536 | CGA | TAA | 5 | |
| trnL2 (UUR) | F | 3005–3071 | 67 | TAA 3035–3037 | −5 | ||
| cox2 | F | 3072–3750 | 679 | ATG | T | 0 | |
| trnK | F | 3748–3818 | 71 | CTT 3778–3780 | −3 | ||
| trnD | F | 3818–3883 | 66 | GTC 3848–3850 | −1 | ||
| atp8 | F | 3884–4042 | 159 | ATC | TAA | 0 | |
| atp6 | F | 4036–4713 | 678 | ATG | TAA | −7 | |
| cox3 | F | 4713–5501 | 789 | ATG | TAA | −1 | |
| trnG | F | 5504–5568 | 65 | TCC 5534–5536 | 2 | ||
| nad3 | F | 5566–5922 | 357 | ATA | TAG | −3 | |
| trnA | F | 5921–5987 | 67 | TGC 5953–5955 | −2 | ||
| trnR | F | 5987–6052 | 66 | TCG 6014–6016 | −1 | ||
| trnN | F | 6053–6118 | 66 | GTT 6084–6086 | 0 | ||
| trnS1 (AGN) | F | 6117–6176 | 60 | GCT 6134–6136 | −2 | ||
| trnE | F | 6180–6243 | 64 | TTC 6108–6210 | 3 | ||
| trnF | R | 6244–6308 | 65 | GAA 6276–6278 | 0 | ||
| nad5 | R | 6308–8044 | 1737 | ATT | TAA | −1 | |
| trnH | R | 8042–8106 | 65 | GTG 8071–8073 | −3 | ||
| nad4 | R | 8107–9445 | 1339 | ATG | T | 0 | |
| nad4L | R | 9447–9731 | 285 | ATG | TAA | 1 | |
| trnT | F | 9744–9807 | 64 | TGT 9774–9776 | 12 | ||
| trnP | R | 9808–9871 | 64 | TGG 9840–9842 | 0 | ||
| nad6 | F | 9874–10401 | 528 | ATA | TAA | 2 | |
| cob | F | 10,406–11,554 | 1149 | ATG | TAA | 4 | |
| trnS2 (UCN) | F | 11,561–11,624 | 64 | TGA 11,589–11,591 | 6 | ||
| nad1 | R | 11,638–12,579 | 942 | ATG | TAA | 13 | |
| trnL1 (CUN) | R | 12,581–12,654 | 74 | TAG 12,623–12,625 | 1 | ||
| rrnL | R | 12,655–13,981 | 1327 | 0 | |||
| trnV | R | 13,982–14,044 | 63 | TAC 14,014–14,016 | 0 | ||
| rrnS | R | 14,045–14,818 | 774 | 0 | |||
| Control region | 14,819–15,208 | 390 | 0 |
| Size (bp) | A% | T% | G% | C% | A + T% | G + C% | |
|---|---|---|---|---|---|---|---|
| mtDNA | 15,252 | 39.7 | 41.2 | 7.6 | 11.5 | 80.9 | 19.1 |
| PCGs | 11,222 | 33.8 | 45.9 | 10.4 | 9.9 | 79.7 | 20.3 |
| tRNA | 1459 | 41.7 | 39.7 | 11.0 | 7.7 | 81.4 | 18.7 |
| rrnL | 1334 | 44.5 | 39.8 | 10.5 | 5.2 | 84.3 | 15.7 |
| rrnS | 776 | 43.6 | 41.5 | 10.1 | 4.9 | 85.1 | 15 |
| Control region | 376 | 43.6 | 47.6 | 2.9 | 5.9 | 91.2 | 8.8 |
| Size (bp) | A% | T% | G% | C% | A + T% | G + C% | |
|---|---|---|---|---|---|---|---|
| mtDNA | 15,208 | 39.9 | 39.3 | 7.9 | 12.9 | 79.2 | 20.8 |
| PCGs | 11,192 | 32.8 | 44.8 | 11.4 | 11.0 | 77.6 | 22.4 |
| tRNA | 1449 | 42.0 | 39.3 | 10.8 | 7.9 | 81.3 | 18.7 |
| rrnL | 1327 | 40.2 | 43.4 | 5.3 | 11.1 | 83.6 | 16.4 |
| rrnS | 774 | 41.2 | 43.9 | 5.2 | 9.7 | 85.1 | 14.9 |
| Control region | 390 | 44.9 | 43.8 | 2.6 | 8.7 | 88.7 | 11.3 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wu, Y.; Fang, H.; Wen, J.; Wang, J.; Cao, T.; He, B. Mitochondrial Genomes of Hestina persimilis and Hestinalis nama (Lepidoptera, Nymphalidae): Genome Description and Phylogenetic Implications. Insects 2021, 12, 754. https://doi.org/10.3390/insects12080754
Wu Y, Fang H, Wen J, Wang J, Cao T, He B. Mitochondrial Genomes of Hestina persimilis and Hestinalis nama (Lepidoptera, Nymphalidae): Genome Description and Phylogenetic Implications. Insects. 2021; 12(8):754. https://doi.org/10.3390/insects12080754
Chicago/Turabian StyleWu, Yupeng, Hui Fang, Jiping Wen, Juping Wang, Tianwen Cao, and Bo He. 2021. "Mitochondrial Genomes of Hestina persimilis and Hestinalis nama (Lepidoptera, Nymphalidae): Genome Description and Phylogenetic Implications" Insects 12, no. 8: 754. https://doi.org/10.3390/insects12080754
APA StyleWu, Y., Fang, H., Wen, J., Wang, J., Cao, T., & He, B. (2021). Mitochondrial Genomes of Hestina persimilis and Hestinalis nama (Lepidoptera, Nymphalidae): Genome Description and Phylogenetic Implications. Insects, 12(8), 754. https://doi.org/10.3390/insects12080754