
Viral Prevalence and Genomic Xenology in the Coevolution of HzNV-2 (Nudiviridae) with Host Helicoverpa zea (Lepidoptera: Noctuidae)



Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Helicoverpa Collection and Identification
2.2. DNA Extraction
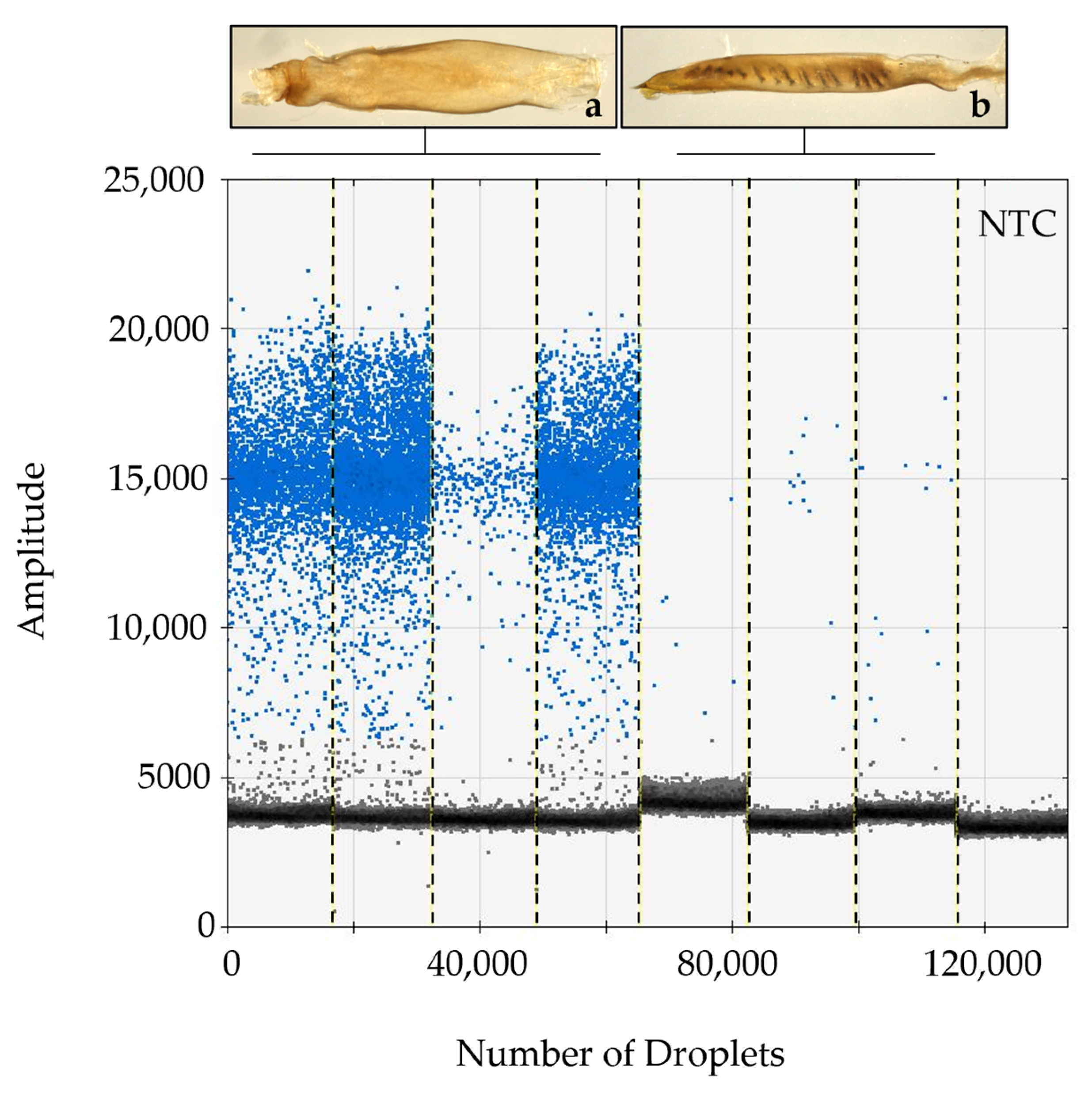
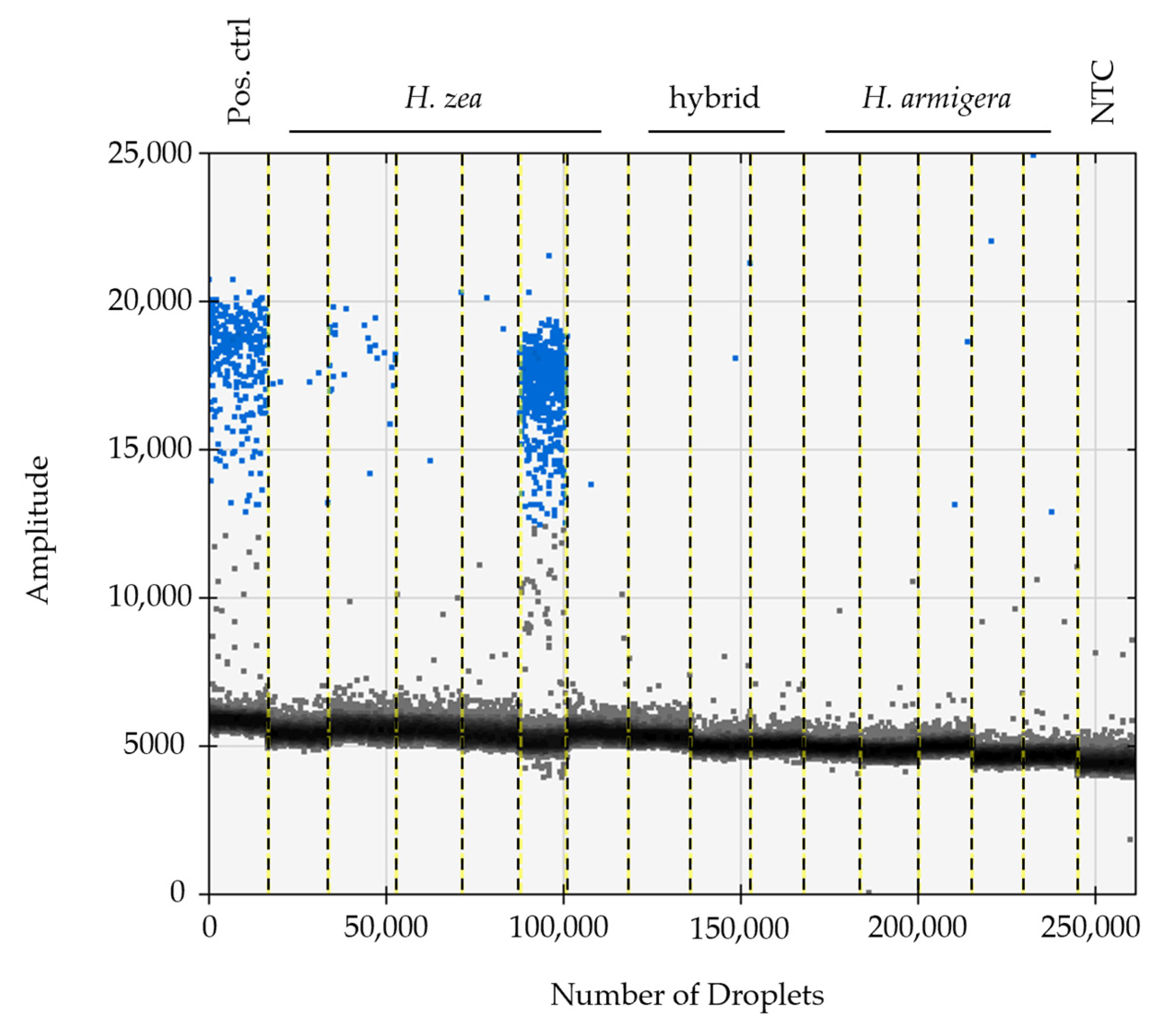
2.3. Droplet Digital PCR for Detection of HzNV-2 DNA
2.4. Genomic Sequence Comparisons
2.5. Phylogenetic Analyses
2.6. PCR Amplification and Sequencing of cSHMT Segments from H. armigera, H. zea, and Hybrids
3. Results
3.1. Specimen Identification, Confirmation of HzNV-2 in Agonadal Specimens, and Viral Prevalence Determined from Individual and Bulk Samples
3.2. Genomic Comparisons between HzNV-2, Host Species H. zea, and Closely Related Species H. armigera and C. virescens
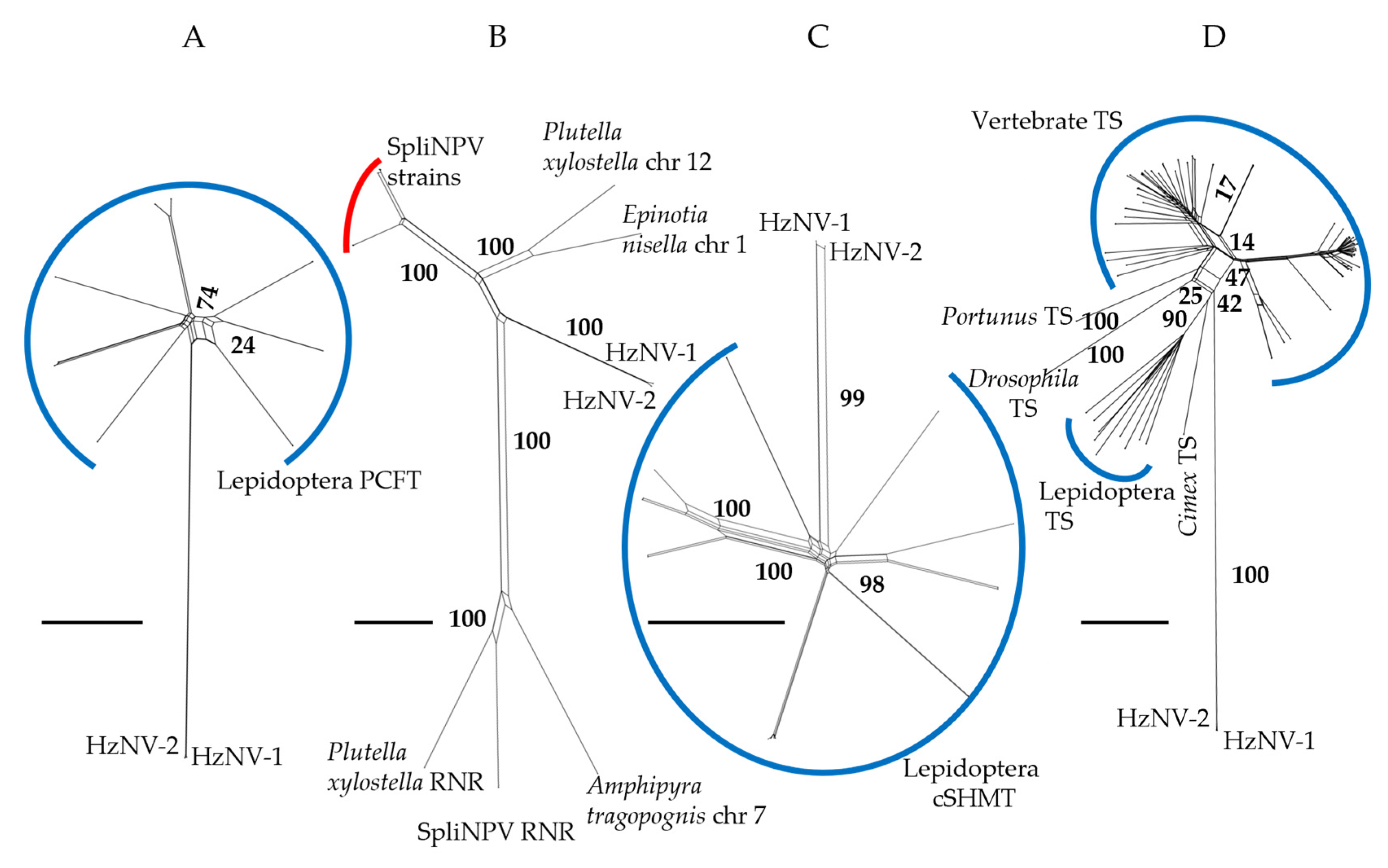
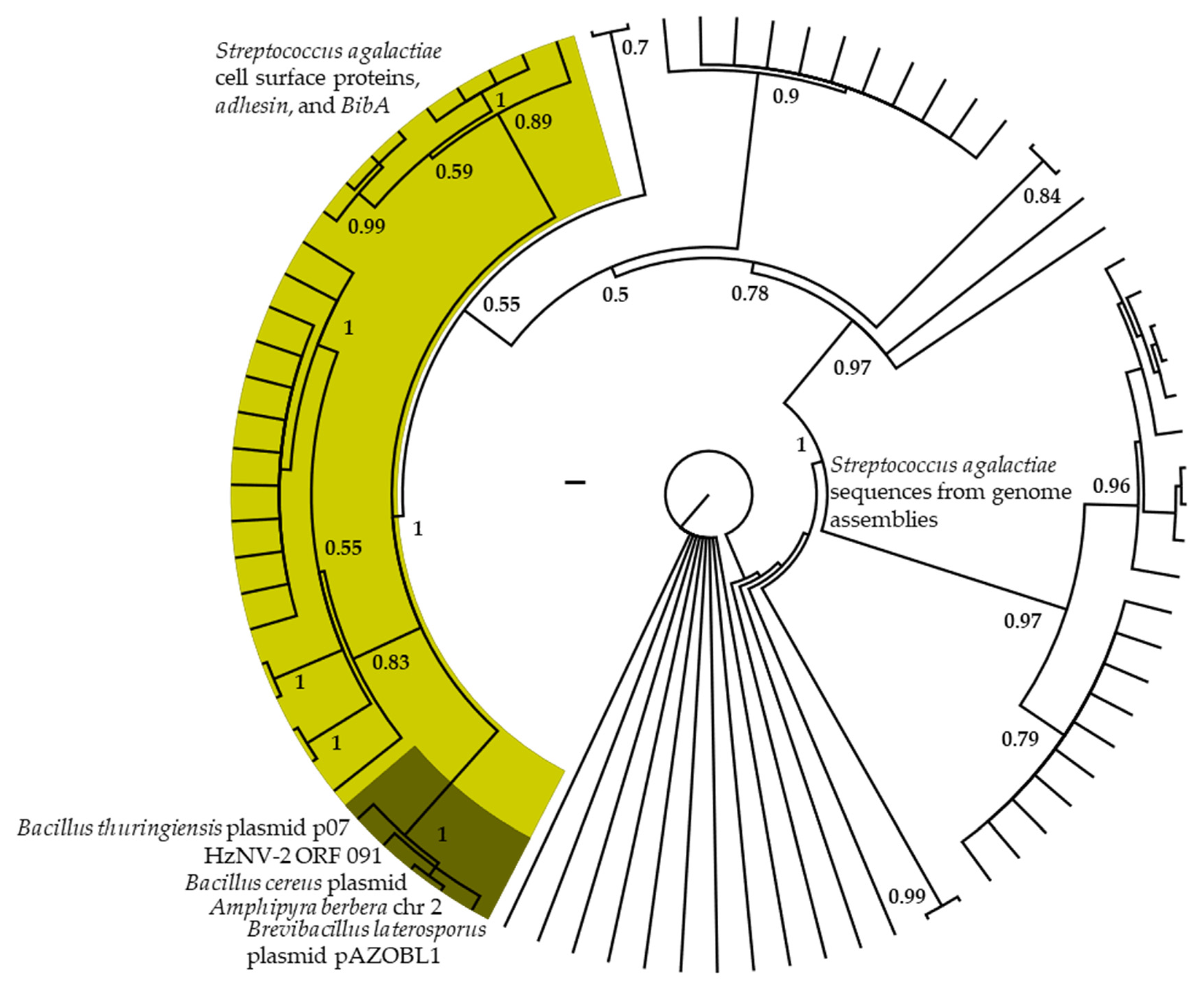
3.3. Phylogenetic Analyses of HzNV-2 Genes of Recent Exogenous Origin
3.4. Differences in Length and Nucleotide Content of cSHMT in HzNV-2, H. armigera, H. zea, and Hybrids
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Krupovic, M.; Bamford, D.H. Order to the viral universe. J. Virol. 2010, 84, 12476–12479. [Google Scholar] [CrossRef]
- Dougan, T.J.; Quake, S.R. Viral taxonomy derived from evolutionary genome relationships. PLoS ONE 2019, 14, e0220440. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Dolja, V.V. Virus world as an evolutionary network of viruses and capsidless selfish elements. Microbiol. Mol. Biol. R 2014, 78, 278–303. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.I.; Kazlauskas, D.; Iranzo, J.; Lucia-Sanz, A.; Kuhn, J.H.; Krupovic, M.; Dolja, V.V.; Koonin, E.V. Origins and Evolution of the Global RNA Virome. mBio 2018, 9, e02329-18. [Google Scholar] [CrossRef] [PubMed]
- Koonin, E.V.; Krupovic, M.; Yutin, N. Evolution of double-stranded DNA viruses of eukaryotes: From bacteriophages to transposons to giant viruses. Ann. N. Y. Acad. Sci. 2015, 1341, 10–24. [Google Scholar] [CrossRef]
- Smith, J.S.; Robinson, N.J. Age-specific prevalence of infection with herpes simplex virus types 2 and 1: A global review. J. Infect. Dis. 2002, 186 (Suppl. S1), S3–S28. [Google Scholar] [CrossRef]
- Doorbar, J. Latent papillomavirus infections and their regulation. Curr. Opin. Virol. 2013, 3, 416–421. [Google Scholar] [CrossRef]
- Bezier, A.; Annaheim, M.; Herbiniere, J.; Wetterwald, C.; Gyapay, G.; Bernard-Samain, S.; Wincker, P.; Roditi, I.; Heller, M.; Belghazi, M.; et al. Polydnaviruses of braconid wasps derive from an ancestral nudivirus. Science 2009, 323, 926–930. [Google Scholar] [CrossRef]
- Pichon, A. Recurrent DNA virus domestication leading to different parasite virulence strategies. Sci. Adv. 2015, 1, e1501150. [Google Scholar] [CrossRef]
- Petersen, J.M.; Bezier, A.; Drezen, J.M.; van Oers, M.M. The naked truth: An updated review on nudiviruses and their relationship to bracoviruses and baculoviruses. J. Invertebr. Pathol. 2022, 189, 107718. [Google Scholar] [CrossRef]
- James, C.; Harfouche, M.; Welton, N.J.; Turner, K.M.; Abu-Raddad, L.J.; Gottlieb, S.L.; Looker, K.J. Herpes simplex virus: Global infection prevalence and incidence estimates, 2016. Bull World Health Organ. 2020, 98, 315–329. [Google Scholar] [CrossRef] [PubMed]
- Lupiani, B.; Raina, A.K.; Huber, C. Development and use of a PCR assay for detection of the reproductive virus in wild populations of Helicoverpa zea (Lepidoptera: Noctuidae). J. Invertebr. Pathol. 1999, 73, 107–112. [Google Scholar] [CrossRef] [PubMed]
- Herzog, G.A.; Phillips, J.R. Manifestation of an abnormal reproductive system in a laboratory strain of the bollworm Heliothis zea Lepidoptera, Noctuidae. J. Georgia Entomol. Soc. 1982, 17, 506–513. [Google Scholar]
- Knell, R.J.; Webberley, K.M. Sexually transmitted diseases of insects: Distribution, evolution, ecology and host behaviour. Biol. Rev. 2004, 79, 557–581. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Burand, J.P.; Jehle, J.A. Nudivirus genomics: Diversity and classification. Virol. Sin. 2008, 22, 128–136. [Google Scholar] [CrossRef]
- Burand, J.P.; Kim, W.; Afonso, C.L.; Tulman, E.R.; Kutish, G.F.; Lu, Z.; Rock, D.L. Analysis of the genome of the sexually transmitted insect virus Helicoverpa zea nudivirus 2. Viruses 2012, 4, 28–61. [Google Scholar] [CrossRef] [PubMed]
- Raina, A.K.; Adams, J.R. Gonad-specific virus of corn earworm. Nature 1998, 374, 770. [Google Scholar] [CrossRef]
- Hamm, J.J.; Carpenter, J.E.; Styer, E.L. Oviposition day effect on incidence of agonadal progeny of Helicoverpa zea (Lepidoptera: Noctuiade) infected with a virus. Ann. Entomol. Soc. Am. 1996, 89, 266–275. [Google Scholar] [CrossRef]
- Raina, A.K.; Adams, J.R.; Lupiani, B.; Lynn, D.E.; Kim, W.J.; Burand, J.P.; Dougherty, E.M. Further characterization of the gonad-specific virus of corn earworm, Helicoverpa zea. J. Invertebr. Pathol. 2000, 76, 6–12. [Google Scholar] [CrossRef]
- Burand, J.P. Pathology and replication of the sexually transmitted insect virus HzNV-2. In Virology I: HIV and Related Issues; Iconcept Press: Kowloon, Hong Kong, 2013. [Google Scholar]
- Valencia-Montoya, W.A.; Elfekih, S.; North, H.L.; Meier, J.I.; Warren, I.A.; Tay, W.T.; Gordon, K.H.J.; Specht, A.; Paula-Moraes, S.V.; Rane, R.; et al. Adaptive introgression across semipermeable species boundaries between local Helicoverpa zea and invasive Helicoverpa armigera moths. Mol. Biol. Evol. 2020, 37, 2568–2583. [Google Scholar] [CrossRef]
- Hardwick, D.F. The biological status of “Heliothis stombleri”. Can. Entomol. 1970, 102, 339–341. [Google Scholar] [CrossRef]
- Pogue, M.G. A new synonym of Helicoverpa zea (Bodie) and differentiation of adult males of H. zea and H. armigera (Hubner) (Lepidoptera: Noctuidae: Heliothinae). Ann. Entomol. Soc. Am. 2004, 97, 1222–1226. [Google Scholar] [CrossRef]
- Balbi, E.I.; Flores, F.M.; Tosto, D.S.; Arneodo, J.D. Further description of Helicoverpa zea (Lepidoptera: Noctuidae) male genitalia and new genetic evidence of synonymy with respect to the anomalous form, “Heliothis stombleri”. J. Insect Sci. 2017, 17, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Nagoshi, R.N.; Gilligan, T.M.; Brambila, J. Combining Tpi and CO1 genetic markers to discriminate invasive Helicoverpa armigera from local Helicoverpa zea (Lepidoptera: Noctuidae) populations in the Southeastern United States. J. Econ. Entomol. 2016, 109, 2115–2124. [Google Scholar] [CrossRef]
- Wallin, I.E. Symbionticism and the Origin of Species; Williams and Wilkins Company: Philadelphia, PA, USA, 1927. [Google Scholar]
- Margulis, L.; Sagan, D. Acquiring Genomes: A Theory of the Origins of Species; Basic Books: New York, NY, USA, 2002. [Google Scholar]
- Brucker, R.M.; Bordenstein, S.R. Speciation by symbiosis. Trends Ecol. Evol. 2012, 27, 443–451. [Google Scholar] [CrossRef]
- Roossinck, M.J. The good viruses: Viral mutualistic symbioses. Nat. Rev. Microbiol. 2011, 9, 99–108. [Google Scholar] [CrossRef]
- Michel, G.; Tonon, T.; Scornet, D.; Cock, J.M.; Kloareg, B. Central and storage carbon metabolism of the brown alga Ectocarpus siliculosus: Insights into the origin and evolution of storage carbohydrates in Eukaryotes. New Phytol. 2010, 188, 67–81. [Google Scholar] [CrossRef]
- Aiewsakun, P.; Katzourakis, A. Endogenous viruses: Connecting recent and ancient viral evolution. Virology 2015, 479–480, 26–37. [Google Scholar] [CrossRef]
- Gilbert, C.; Cordaux, R. Viruses as vectors of horizontal transfer of genetic material in eukaryotes. Curr. Opin. Virol. 2017, 25, 16–22. [Google Scholar] [CrossRef]
- Irwin, N.A.T.; Pittis, A.A.; Richards, T.A.; Keeling, P.J. Systematic evaluation of horizontal gene transfer between eukaryotes and viruses. Nat. Microbiol. 2022, 7, 327–336. [Google Scholar] [CrossRef]
- Rodriguez-Valera, F.; Martin-Cuadrado, A.B.; Rodriguez-Brito, B.; Pasic, L.; Thingstad, T.F.; Rohwer, F.; Mira, A. OPINION Explaining microbial population genomics through phage predation. Nat. Rev. Microbiol. 2009, 7, 828–836. [Google Scholar] [CrossRef]
- Manry, J.; Laval, G.; Patin, E.; Fornarino, S.; Itan, Y.; Fumagalli, M.; Sironi, M.; Tichit, M.; Bouchier, C.; Casanova, J.L.; et al. Evolutionary genetic dissection of human interferons. J. Exp. Med. 2011, 208, 2747–2759. [Google Scholar] [CrossRef] [PubMed]
- Obbard, D.J.; Dudas, G. The genetics of host-virus coevolution in invertebrates. Curr. Opin. Virol. 2014, 8, 73–78. [Google Scholar] [CrossRef] [PubMed]
- Maori, E.; Tanne, E.; Sela, I. Reciprocal sequence exchange between non-retro viruses and hosts leading to the appearance of new host phenotypes. Virology 2007, 362, 342–349. [Google Scholar] [CrossRef] [PubMed]
- Braga, L.P.P.; Soucy, S.M.; Amgarten, D.E.; da Silva, A.M.; Setubal, J.C. Bacterial diversification in the light of the interactions with phages: The genetic symbionts and their role in ecological speciation. Front. Ecol. Evol. 2018, 6, 1–12. [Google Scholar] [CrossRef]
- Fridman, S.; Flores-Uribe, J.; Larom, S.; Alalouf, O.; Liran, O.; Yacoby, I.; Salama, F.; Bailleul, B.; Rappaport, F.; Ziv, T.; et al. A myovirus encoding both photosystem I and II proteins enhances cyclic electron flow in infected Prochlorococcus cells. Nature Microbiol. 2017, 2, 1350–1357. [Google Scholar] [CrossRef]
- Schulz, F.; Yutin, N.; Ivanova, N.N.; Ortega, D.R.; Lee, T.K.; Vierheilig, J.; Daims, H.; Horn, M.; Wagner, M.; Jensen, G.J.; et al. Giant viruses with an expanded complement of translation system components. Science 2017, 356, 82–85. [Google Scholar] [CrossRef]
- Krupovic, M.; Dolja, V.V.; Koonin, E.V. Origin of viruses: Primordial replicators recruiting capsids from hosts. Nat. Rev. Microbiol. 2019, 17, 449–458. [Google Scholar] [CrossRef]
- Duffy, S.; Shackelton, L.A.; Holmes, E.C. Rates of evolutionary change in viruses: Patterns and determinants. Nat. Rev. Genet. 2008, 9, 267–276. [Google Scholar] [CrossRef]
- Belshaw, R.; Sanjuan, R.; Pybus, O.G. Viral mutation and substitution: Units and levels. Curr. Opin. Virol. 2011, 1, 430–435. [Google Scholar] [CrossRef]
- Moreira, D.; Lopez-Garcia, P. Evolution of viruses and cells: Do we need a fourth domain of life to explain the origin of eukaryotes? Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140327. [Google Scholar] [CrossRef] [PubMed]
- Niikura, M.; Dodgson, J.; Cheng, H. Direct evidence of host genome acquisition by the alphaherpesvirus Marek’s disease virus. Arch. Virol. 2006, 151, 537–549. [Google Scholar] [CrossRef] [PubMed]
- Tang, S.; Patel, A.; Krause, P.R. Herpes simplex virus ICP27 regulates alternative pre-mRNA polyadenylation and splicing in a sequence-dependent manner. Proc. Natl. Acad. Sci. USA 2016, 113, 12256–12261. [Google Scholar] [CrossRef] [PubMed]
- Brambila, J. Instructions for Dissecting Male Genitalia of Helicoverpa (Lepidoptera: Noctuidae) to Separate H. zea from H. armigera (Hubner); USDA-APHIS-PPQ: Gainesville, FL, USA, 2009. [Google Scholar]
- Gilligan, T.M.; Tembrock, L.R.; Farris, R.E.; Barr, N.B.; van der Straten, M.J.; van de Vossenberg, B.T.; Metz-Verschure, E. A multiplex real-time PCR assay to diagnose and separate Helicoverpa armigera and H. zea (Lepidoptera: Noctuidae) in the New World. PLoS ONE 2015, 10, e0142912. [Google Scholar] [CrossRef]
- Zink, F.A.; Tembrock, L.R.; Timm, A.E.; Farris, R.E.; Perera, O.P.; Gilligan, T.M. A droplet digital PCR (ddPCR) assay to detect Helicoverpa armigera (Lepidoptera: Noctuidae) in bulk trap samples. PLoS ONE 2017, 12, e0178704. [Google Scholar] [CrossRef]
- Tembrock, L.R.; Farris, R.E.; Ledezma, L.; Barr, N.B.; Gilligan, T.M. A real-Time PCR assay for the separation of Autographa gamma (Noctuidae: Plusiinae) from morphologically similar species in North America. J. Econ. Entomol. 2017, 110, 2609–2617. [Google Scholar] [CrossRef]
- Gloor, G.B.; Preston, C.R.; Johnson-Schlitz, D.M.; Nassif, N.A.; Phillis, R.W.; Benz, W.K.; Robertson, H.M.; Engels, W.R. Type I repressors of P element mobility. Genetics 1993, 135, 81–95. [Google Scholar] [CrossRef]
- Perera, O.P.; Allen, K.C.; Jain, D.; Purcell, M.; Little, N.S.; Luttrell, R.G. Rapid identification of Helicoverpa armigera and Helicoverpa zea (Lepidoptera: Noctuidae) using ribosomal RNA internal transcribed spacer 1. J. Insect Sci. 2015, 15, 155. [Google Scholar] [CrossRef]
- Zink, F.A.; Tembrock, L.R.; Timm, A.E.; Gilligan, T.M. A ddPCR assay for identification of Autographa gamma (Noctuidae: Plusiinae) in bulk trap samples. J. Econ. Entomol. 2018, 111, 1490–1495. [Google Scholar] [CrossRef]
- Jones, M.; Williams, J.; Gartner, K.; Phillips, R.; Hurst, J.; Frater, J. Low copy target detection by droplet digital PCR through application of a novel open access bioinformatic pipeline, ‘definetherain’. J. Virol. Methods 2014, 202, 46–53. [Google Scholar] [CrossRef]
- Altschul, S.F.; Gish, W.; Miller, W.; Myers, E.W.; Lipman, D.J. Basic local alignment search tool. J. Mol. Biol. 1990, 215, 403–410. [Google Scholar] [CrossRef] [PubMed]
- Pertsemlidis, A.; Fondon, J.W., 3rd. Having a BLAST with bioinformatics (and avoiding BLASTphemy). Genome Biol. 2001, 2, REVIEWS2002. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed]
- Stahlke, A.R.; Chang, J.; Tembrock, L.R.; Sim, S.B.; Chudalayandi, S.; Geib, S.M.; Scheffler, B.E.; Perera, O.P.; Gilligan, T.M.; Childers, A.K.; et al. A chromosome-scale genome assembly of a Helicoverpa zea strain resistant to Bacillus thuringiensis Cry1Ac insecticidal protein. Genome Biol. Evol. 2023, 15, evac131. [Google Scholar] [CrossRef] [PubMed]
- Wheeler, D.L.; Church, D.M.; Federhen, S.; Lash, A.E.; Madden, T.L.; Pontius, J.U.; Schuler, G.D.; Schriml, L.M.; Sequeira, E.; Tatusova, T.A.; et al. Database resources of the National Center for Biotechnology. Nucleic Acids Res. 2003, 31, 28–33. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Posada, D.; Crandall, K.A. MODELTEST: Testing the model of DNA substitution. Bioinformatics 1998, 14, 817–818. [Google Scholar] [CrossRef]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef]
- Swofford, D.L. PAUP*. Phylogenetic Analysis Using Parsimony (and Other Methods); Sinauer Associates: Sunderland, UK, 2003. [Google Scholar]
- Katoh, K.; Misawa, K.; Kuma, K.; Miyata, T. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—New capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef] [PubMed]
- SantaLucia, J., Jr. A unified view of polymer, dumbbell, and oligonucleotide DNA nearest-neighbor thermodynamics. Proc. Natl. Acad. Sci. USA 1998, 95, 1460–1465. [Google Scholar] [CrossRef]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef] [PubMed]
- Cairns, J.; Freire-Pritchett, P.; Wingett, S.W.; Varnai, C.; Dimond, A.; Plagnol, V.; Zerbino, D.; Schoenfelder, S.; Javierre, B.M.; Osborne, C.; et al. CHiCAGO: Robust detection of DNA looping interactions in Capture Hi-C data. Genome Biol. 2016, 17, 127. [Google Scholar] [CrossRef] [PubMed]
- Putnam, N.H.; O’Connell, B.L.; Stites, J.C.; Rice, B.J.; Blanchette, M.; Calef, R.; Troll, C.J.; Fields, A.; Hartley, P.D.; Sugnet, C.W.; et al. Chromosome-scale shotgun assembly using an in vitro method for long-range linkage. Genome Res. 2016, 26, 342–350. [Google Scholar] [CrossRef]
- Chen, S.; Li, X.C. Transposable elements are enriched within or in close proximity to xenobiotic-metabolizing cytochrome P450 genes. BMC Evol. Biol. 2007, 7, 46. [Google Scholar] [CrossRef]
- Klai, K.; ChEnais, B.; Zidi, M.; Djebbi, S.; Caruso, A.; Denis, F.; Confais, J.; Badawi, M.; Casse, N.; Mezghani Khemakhem, M. Screening of Helicoverpa armigera mobilome revealed transposable element insertions in insecticide resistance genes. Insects 2020, 11, 879. [Google Scholar] [CrossRef]
- Gray, Y.H. It takes two transposons to tango: Transposable-element-mediated chromosomal rearrangements. Trends Genet. 2000, 16, 461–468. [Google Scholar] [CrossRef]
- Tu, Z. Insect transposable elements. In Insect Molecular Biology and Biochemistry; Gilbert, L.I., Ed.; Elsevier Academic Press: Amsterdam, The Netherlands, 2012. [Google Scholar]
- Zink, F.A.; Tembrock, L.R.; Timm, A.E.; Gilligan, T.M. A duplex ddPCR assay for simultaneously detecting Ips sexdentatus and Ips typographus (Coleoptera: Curculionidae) in bulk trap samples. Can. J. For. Res. 2019, 49, 903–914. [Google Scholar] [CrossRef]
- Franke, G.N.; Maier, J.; Wildenberger, K.; Cross, M.; Giles, F.J.; Muller, M.C.; Hochhaus, A.; Niederwieser, D.; Lange, T. Comparison of real-time quantitative PCR and digital droplet PCR for BCR-ABL1 monitoring in patients with chronic myeloid leukemia. J. Mol. Diagn. 2020, 22, 81–89. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.L.; Wu, C.P.; Liu, C.Y.Y.; Hsu, P.W.C.; Wu, E.C.; Chao, Y.C. A non-coding RNA of insect HzNV-1 virus establishes latent viral infection through microRNA. Sci. Rep. 2011, 1, 60. [Google Scholar] [CrossRef]
- Tuxen, S.L. Taxonomists Glossary of Genitalia of Insects; Munksgaard: Copenhagen, Denmark, 1970. [Google Scholar]
- Masly, J.P. 170 years of “lock-and-key”: Genital morphology and reproductive isolation. Int. J. Evol. Biol. 2012, 2012, 247352. [Google Scholar] [CrossRef] [PubMed]
- Williams, T. Natural invertebrate hosts of iridoviruses (Iridoviridae). Neotrop. Entomol. 2008, 37, 615–632. [Google Scholar] [CrossRef] [PubMed]
- Herniou, E.A.; Huguet, E.; Theze, J.; Bezier, A.; Periquet, G.; Drezen, J.M. When parasitic wasps hijacked viruses: Genomic and functional evolution of polydnaviruses. Philos. Trans. R. Soc. B 2013, 368, 20130051. [Google Scholar] [CrossRef] [PubMed]
- DaPalma, T.; Doonan, B.P.; Trager, N.M.; Kasman, L.M. A systematic approach to virus-virus interactions. Virus Res. 2010, 149, 1–9. [Google Scholar] [CrossRef]
- Diesend, J.; Kruse, J.; Hagedorn, M.; Hammann, C. Amoebae, giant viruses, and virophages make up a complex, multilayered threesome. Front. Cell Inf. Microbiol. 2018, 7, 527. [Google Scholar] [CrossRef]
- Lundin, D.; Gribaldo, S.; Torrents, E.; Sjoberg, B.M.; Poole, A.M. Ribonucleotide reduction—Horizontal transfer of a required function spans all three domains. BMC Evol. Biol. 2010, 10, 383. [Google Scholar] [CrossRef]
- Shackelton, L.A.; Holmes, E.C. The evolution of large DNA viruses: Combining genomic information of viruses and their hosts. Trends Microbiol. 2004, 12, 458–465. [Google Scholar] [CrossRef]
- Ball, A.L. Virus replication strategies. In Fields Virology; Knipe, D.M., Howley, P.M., Eds.; Lippincott Williams & Wilkins: Philadelphia, PA, USA, 2007. [Google Scholar]
- Lembo, D.; Brune, W. Tinkering with a viral ribonucleotide reductase. Trends Biochem. Sci. 2009, 34, 25–32. [Google Scholar] [CrossRef]
- Carreras, C.W.; Santi, D.V. The catalytic mechanism and structure of thymidylate synthase. Annu. Rev. Biochem. 1995, 64, 721–762. [Google Scholar] [CrossRef] [PubMed]
- Arvizu-Flores, A.A.; Aispuro-Hernandez, E.; Garcia-Orozco, K.D.; Varela-Romero, A.; Valenzuela-Soto, E.; Velazquez-Contreras, E.F.; Rojo-Dominguez, A.; Yepiz-Plascencia, G.; Maley, F.; Sotelo-Mundo, R.R. Functional identity of the active sites of crustacean and viral thymidylate synthases. Comp. Biochem. Physiol. C Toxicol. Pharmacol. 2009, 150, 406–413. [Google Scholar] [CrossRef] [PubMed]
- Chabaud, S.; Lambert, H.; Sasseville, A.M.J.; Lavoie, H.; Guilbault, C.; Massie, B.; Landry, J.; Langelier, Y. The R1 subunit of herpes simplex virus ribonucleotide reductase has chaperone-like activity similar to Hsp27. Febs. Lett. 2003, 545, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Stover, P.; Schrich, V. Serine hydroxymethyltransferase catalyzes the hydrolysis of 5,10-methyltetrahydrofolate to 5-formyltetrahydrofolate. J. Biol. Chem. 1990, 265, 14227–14233. [Google Scholar] [CrossRef] [PubMed]
- Hou, Z.; Matherly, L.H. Biology of the major facilitative folate transporters SLC19A1 and SLC46A1. Curr. Top. Membr. 2014, 73, 175–204. [Google Scholar] [CrossRef]
- Hebbring, S.J.; Chai, Y.; Ji, Y.; Abo, R.P.; Jenkins, G.D.; Fridley, B.; Zhang, J.; Eckloff, B.W.; Wieben, E.D.; Weinshilboum, R.M. Serine hydroxymethyltransferase 1 and 2: Gene sequence variation and functional genomic characterization. J. Neurochem. 2012, 120, 881–890. [Google Scholar] [CrossRef]
- Newman, A.C.; Maddocks, O.D.K. One-carbon metabolism in cancer. Br. J. Cancer 2017, 116, 1499–1504. [Google Scholar] [CrossRef]
- Haque, M.R.; Hirowatari, A.; Nai, N.; Furuya, S.; Yamamoto, K. Serine hydroxymethyltransferase from the silkworm Bombyx mori: Identification, distribution, and biochemical characterization. Arch. Insect Biochem. Physiol. 2019, 102, e21594. [Google Scholar] [CrossRef]
- Zhang, G.; Hussain, M.; O’Neill, S.L.; Asgari, S. Wolbachia uses a host microRNA to regulate transcripts of a methyltransferase, contributing to dengue virus inhibition in Aedes aegypti. Proc. Natl. Acad. Sci. USA 2013, 110, 10276–10281. [Google Scholar] [CrossRef]
- Orr, M.W.; Mao, Y.; Storz, G.; Qian, S.B. Alternative ORFs and small ORFs: Shedding light on the dark proteome. Nucleic Acids Res. 2020, 48, 1029–1042. [Google Scholar] [CrossRef]
- Gillam, E.M.J.; Hunter, D.J.B. Chemical defense and exploitation biotransformation of xenobiotics by cytochrome P450 enzymes. In Metal Ions in Life Sciences; Sigel, A., Sigel, H., Sigel, R.K.O., Eds.; John Wiley and Sons: West Sussex, UK, 2007; Volume 3, pp. 477–560. [Google Scholar]
- Chen, S.; Yang, Y.H.; Wu, Y.D. Correlation between fenvalerate resistance and cytochrome P450-mediated O-demethylation activity in Helicoverpa armigera (Lepidoptera: Noctuidae). J. Econ. Entomol. 2005, 98, 943–946. [Google Scholar] [CrossRef]
- Epelboin, Y.; Wang, L.; Giai Gianetto, Q.; Choumet, V.; Gaborit, P.; Issaly, J.; Guidez, A.; Douche, T.; Chaze, T.; Matondo, M.; et al. CYP450 core involvement in multiple resistance strains of Aedes aegypti from French Guiana highlighted by proteomics, molecular and biochemical studies. PLoS ONE 2021, 16, e0243992. [Google Scholar] [CrossRef] [PubMed]
- Kim, Y.C.; Kim, S.Y.; Paek, K.H.; Choi, D.; Park, J.M. Suppression of CaCYP1, a novel cytochrome P450 gene, compromises the basal pathogen defense response of pepper plants. Biochem. Biophys. Res. Commun. 2006, 345, 638–645. [Google Scholar] [CrossRef] [PubMed]
- Behura, S.K.; Gomez-Machorro, C.; Harker, B.W.; deBruyn, B.; Lovin, D.D.; Hemme, R.R.; Mori, A.; Romero-Severson, J.; Severson, D.W. Global cross-talk of genes of the mosquito Aedes aegypti in response to dengue virus infection. PLoS Negl. Trop. Dis. 2011, 5, e1385. [Google Scholar] [CrossRef] [PubMed]
- Cowley, M.; Oakey, R.J. Transposable elements re-wire and fine-tune the transcriptome. PLoS Genet. 2013, 9, e1003234. [Google Scholar] [CrossRef]
- Noland, J.E.; Breitenbach, J.E.; Popham, H.J.; Hum-Musser, S.M.; Vogel, H.; Musser, R.O. Gut transcription in Helicoverpa zea is dynamically altered in response to baculovirus infection. Insects 2013, 4, 506–520. [Google Scholar] [CrossRef]
- Shrestha, A.; Bao, K.; Chen, W.; Wang, P.; Fei, Z.; Blissard, G.W. Transcriptional responses of the Trichoplusia ni midgut to oral infection by the baculovirus Autographa californica multiple nucleopolyhedrovirus. J. Virol. 2019, 93, e00353-19. [Google Scholar] [CrossRef]
- Roy, M.; Viginier, B.; Saint-Michel, E.; Arnaud, F.; Ratinier, M.; Fablet, M. Viral infection impacts transposable element transcript amounts in Drosophila. Proc. Natl. Acad. Sci. USA 2020, 117, 12249–12257. [Google Scholar] [CrossRef]
- MacFarlane, A.J.; Liu, X.; Perry, C.A.; Flodby, P.; Allen, R.H.; Stabler, S.P.; Stover, P.J. Cytoplasmic serine hydroxymethyltransferase regulates the metabolic partitioning of methylenetetrahydrofolate but is not essential in mice. J. Biol. Chem. 2008, 283, 25846–25853. [Google Scholar] [CrossRef]
- Anderson, D.D.; Stover, P.J. SHMT1 and SHMT2 are functionally redundant in nuclear de novo thymidylate biosynthesis. PLoS ONE 2009, 4, e5839. [Google Scholar] [CrossRef]
- Florio, R.; di Salvo, M.L.; Vivoli, M.; Contestabile, R. Serine hydroxymethyltransferase: A model enzyme for mechanistic, structural, and evolutionary studies. Biochim. Biophys. Acta 2011, 1814, 1489–1496. [Google Scholar] [CrossRef]
- Anderson, D.D.; Woeller, C.F.; Chiang, E.P.; Shane, B.; Stover, P.J. Serine hydroxymethyltransferase anchors de novo thymidylate synthesis pathway to nuclear lamina for DNA synthesis. J. Biol. Chem. 2012, 287, 7051–7062. [Google Scholar] [CrossRef] [PubMed]
- Visentin, M.; Zhao, R.; Goldman, I.D. The antifolates. Hematol. Oncol. Clin. North. Am. 2012, 26, 629–648. [Google Scholar] [CrossRef] [PubMed]
- Sugai, E.; Ashoush, I. Male sterility in the silkworm, Bombyx mori L., induced by aminopterin (Lepidoptera: Bombycidae). App Entomol. Zool. 1970, 5, 202–207. [Google Scholar] [CrossRef]
- Dramsi, S.; Bierne, H. Spatial organization of cell wall-anchored proteins at the surface of gram-positive bacteria. Curr. Top. Microbiol. 2017, 404, 177–201. [Google Scholar] [CrossRef]
- Fischetti, V.A. Surface proteins on gram-positive bacteria. Microbiol. Spectr. 2019, 7, 1–27. [Google Scholar] [CrossRef]
- Diaz, E.; Lopez, R.; Garcia, J.L. Chimeric phage-bacterial enzymes: A clue to the modular evolution of genes. Proc. Natl. Acad. Sci. USA 1990, 87, 8125–8129. [Google Scholar] [CrossRef]
- Garcia, J.L.; Sanchez-Beato, A.R.; Medrano, F.J.; Lopez, R. Versatility of choline-binding domain. Microb. Drug Resist. 1998, 4, 25–36. [Google Scholar] [CrossRef]
- Calatayud, S.; Garcia-Risco, M.; Capdevila, M.; Canestro, C.; Palacios, O.; Albalat, R. Modular evolution and population variability of Oikopleura dioica metallothioneins. Front. Cell Dev. Biol. 2021, 9, 702688. [Google Scholar] [CrossRef]
- Wang, X.; Zhang, Y.; Zhang, R.; Zhang, J. The diversity of pattern recognition receptors (PRRs) involved with insect defense against pathogens. Curr. Opin. Insect Sci. 2019, 33, 105–110. [Google Scholar] [CrossRef]
- Filee, J.; Siguier, P.; Chandler, M. I am what I eat and I eat what I am: Acquisition of bacterial genes by giant viruses. Trends Genet. 2007, 23, 10–15. [Google Scholar] [CrossRef] [PubMed]
- Bertelli, C.; Greub, G. Lateral gene exchanges shape the genomes of amoeba-resisting microorganisms. Front. Cell Infect. Microbiol. 2012, 2, 110. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, G.; Silva, L.; Leao, T.; Mougari, S.; da Fonseca, F.G.; Kroon, E.G.; La Scola, B.; Abrahao, J.S. Tupanvirus-infected amoebas are induced to aggregate with uninfected cells promoting viral dissemination. Sci. Rep. 2019, 9, 183. [Google Scholar] [CrossRef] [PubMed]
- Ruiu, L. Brevibacillus laterosporus, a pathogen of invertebrates and a broad-spectrum antimicrobial species. Insects 2013, 4, 476–492. [Google Scholar] [CrossRef]
- McCullers, J.A. The co-pathogenesis of influenza viruses with bacteria in the lung. Nat. Rev. Microbiol. 2014, 12, 252–262. [Google Scholar] [CrossRef]
- Klages-Mundt, N.L.; Li, L. Formation and repair of DNA-protein crosslink damage. Sci. China Life Sci. 2017, 60, 1065–1076. [Google Scholar] [CrossRef]
- Novarina, D.; Desai, R.; Vaisica, J.A.; Ou, J.; Bellaoui, M.; Brown, G.W.; Chang, M. A genome-wide screen for genes affecting spontaneous direct-repeat recombination in Saccharomyces cerevisiae. G3 (Bethesda) 2020, 10, 1853–1867. [Google Scholar] [CrossRef]
- Makarenkov, V.; Mazoure, B.; Rabusseau, G.; Legendre, P. Horizontal gene transfer and recombination analysis of SARS-CoV-2 genes helps discover its close relatives and shed light on its origin. BMC Ecol. Evol. 2021, 21, 5. [Google Scholar] [CrossRef]
- Pal, C.; Macia, M.D.; Oliver, A.; Schachar, I.; Buckling, A. Coevolution with viruses drives the evolution of bacterial mutation rates. Nature 2007, 450, 1079–1081. [Google Scholar] [CrossRef]
- M’Gonigle, L.K.; Shen, J.J.; Otto, S.P. Mutating away from your enemies: The evolution of mutation rate in a host-parasite system. Theor. Popul. Biol. 2009, 75, 301–311. [Google Scholar] [CrossRef]
- Streicker, D.G.; Winternitz, J.C.; Satterfield, D.A.; Condori-Condori, R.E.; Broos, A.; Tello, C.; Recuenco, S.; Velasco-Villa, A.; Altizer, S.; Valderrama, W. Host-pathogen evolutionary signatures reveal dynamics and future invasions of vampire bat rabies. Proc. Natl. Acad. Sci. USA 2016, 113, 10926–10931. [Google Scholar] [CrossRef] [PubMed]
- Dominguez-Andres, J.; Netea, M.G. Impact of historic migrations and evolutionary processes on human immunity. Trends Immunol. 2019, 40, 1105–1119. [Google Scholar] [CrossRef] [PubMed]
- Goic, B.; Stapleford, K.A.; Frangeul, L.; Doucet, A.J.; Gausson, V.; Blanc, H.; Schemmel-Jofre, N.; Cristofari, G.; Lambrechts, L.; Vignuzzi, M.; et al. Virus-derived DNA drives mosquito vector tolerance to arboviral infection. Nat. Commun. 2016, 7, 12410. [Google Scholar] [CrossRef] [PubMed]
- Pearce, S.L.; Clarke, D.F.; East, P.D.; Elfekih, S.; Gordon, K.H.J.; Jermiin, L.S.; McGaughran, A.; Oakeshott, J.G.; Papanicolaou, A.; Perera, O.P.; et al. Genomic innovations, transcriptional plasticity and gene loss underlying the evolution and divergence of two highly polyphagous and invasive Helicoverpa pest species. BMC Biol. 2017, 15, 63. [Google Scholar] [CrossRef]
- Ardisson-Araujo, D.M.P.; Sosa-Gómez, D.R.; Melo, F.L.; Bao, S.N. Characterization of the Helicoverpa zea single nucleopolyhedrovirus isolated in Brazil during the first old world bollworm (Noctuidae: Helicoverpa armigera) nationwide outbreak. Virus Rev. Res. 2015, 20, 1–4. [Google Scholar] [CrossRef]
- Zhou, M.; Sun, X.; Sun, X.; Vlak, J.M.; Hu, Z.; van der Werf, W. Horizontal and vertical transmission of wild-type and recombinant Helicoverpa armigera single-nucleocapsid nucleopolyhedrovirus. J. Invertebr. Pathol. 2005, 89, 165–175. [Google Scholar] [CrossRef]
- Stork, N.E. How many species of insects and other terrestrial arthropods are there on earth? Annu. Rev. Entomol. 2018, 63, 31–45. [Google Scholar] [CrossRef]
- Anthony, S.J.; Epstein, J.H.; Murray, K.A.; Navarrete-Macias, I.; Zambrana-Torrelio, C.M.; Solovyov, A.; Ojeda-Flores, R.; Arrigo, N.C.; Islam, A.; Ali Khan, S.; et al. A strategy to estimate unknown viral diversity in mammals. mBio 2013, 4, e00598-13. [Google Scholar] [CrossRef]
- Junglen, S.; Drosten, C. Virus discovery and recent insights into virus diversity in arthropods. Curr. Opin. Microbiol. 2013, 16, 507–513. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name | Description | Sequence | Tm (°C) | Source |
|---|---|---|---|---|
| P4-I | HzNV-2 detection | 5′-GCACGATTCGTAATGTTC-3′ | 51.8 * | [12] |
| P4-II | HzNV-2 detection | 5′-GCACACCTATCAATCACC-3′ | 52.8 * | [12] |
| P13-I | HzNV-2 detection | 5′-TCGATGCCGTAATACC-3′ | 49.7 * | [12] |
| P13-II | HzNV-2 detection | 5′-GTCGCTGAATCAAGTCTG-3′ | 53.2 * | [12] |
| Hz_SHMT_1738F | cSHMT TE insert intron 7 | 5′-CCAGCGCCTCTGCAAAGG-3′ | 61.4 | This Study |
| Hza_SHMT_24R | cSHMT TE insert intron 7 | 5′-TAAATGTTAAGCTGTTRTCCTCT-3′ | 53.2–55.9 | This Study |
| Location in HzNV2 Genome | Annotation from Similar Sequences | H. zea | H. armigera | C. virescens | Best DNA Hit | Best Protein Hit | Putative Viral Function | Burand et al. Annotation |
|---|---|---|---|---|---|---|---|---|
| ORF Hz2V047 (96009-98822) | Ribonucleotide reductase | 1 × 10−30 | 6 × 10−34 | 8 × 10−21 | Spodoptera littoralis nucleopolyhedrovirus isolate SpliNPV-Tun2 (2 × 10−130/66/67.1) | Ribonucleoside-diphosphate reductase large subunit, Hyposmocoma kahamanoa (0.0) | Nucelotide anabolism, inhibit host cell signalling | Ribonuclease reductase |
| ORF Hz2V066 (127832-126510) | Serine hydroxymethyltransferase | 7 × 10−27 | 5 × 10−22 | 6 × 10−23 | Serine hydroxymethyltransferase, cytosolic transcript variant × 2, Helicoverpa armigera (5 × 10−113/88/67.2) | Serine hydroxymethyltransferase, cytosolic isoform × 1, Helicoverpa armigera (0.0) | Nucelotide anabolism, one-carbon metabolism | Serine hydroxymethyltransferase |
| ORF Hz2V023 (49772-51307) | Proton-coupled folate transporter | 2 × 10−21 | 2 × 10−20 | 3 × 10−20 | Proton-coupled folate transporter, Helicoverpa armigera (3.0E-166/81/70.8) | Proton-coupled folate transporter, Helicoverpa armigera (0.0) | Folate transport | Membrane transporter |
| ORF Hz2V035 (69749-70621) | Thymidylate synthase | 1 × 10−18 | 3 × 10−18 | 5 × 10−17 | Thymidylate synthase, Pectinophora gossypiella (3 × 10−90/90/70.3) | Thymidylate synthase, Manduca sexta (7 × 10−170) | Thymidine biosynthesis | Thymidylate synthesis |
| ORF Hz2V002 (7183-13047) hit associated with nested ORF 12222-11431 | unknown | 2 × 10−14 | 1 × 10−14 | no match | Chromosome 19, Perizoma flavofasciatum (2 × 10−34/60/74.6) | transcriptional regulatory protein AlgP, partial Biomphalaria glabrata (0.036) | unknown | Unidentified |
| ORF Hz2V098 (195699-199133) | unknown | 7 × 10−8 | 1 × 10−9 | no match | Chromsome7, Amphipyra tragopoginis (5 × 10−81/21/70) | Hypothetical protein Cantr_06524, Candida viswanathii (3 × 10−4) | unknown | Unidentified |
| ORF Hz2V091 (177136-178524) | Cell surface protein (bacterial origin) | 1 × 10−5 | 3 × 10−12 | no match | HD-771 plasmid p07, Bacillus thuringiensis (4 × 10−34/21/75.9) | Hypothetical protein BTXL6_27630, Bacillus thuringiensis (5 × 10−23) | Cell surface interaction | Unidentified |
| Between ORF Hz2V083 (158080-158743) and ORF Hz2V084 (163557-159694) | unknown | 1 × 10−6 | 6 × 10−9 | no match | Chromosome 6, Harpalus rufipes (1 × 10−21/12/68) | no match | unknown | Unidentified |
| ORF Hz2V062 (120105-118786) | Occlusion-dervived virus envelop protein e56 | 6 × 10−4 | 1 × 10−5 | no match | Chromosome 14, Lumbricus terrestris (1 × 10−7/9/84.6) | PIF-5a, Tipula oleracea nudivirus (9 × 10−38) | Viral envelope synthesis | Unidentified |
| Chr | Length (bp) | % Identity | e-Value | 5’ Position | Annotation from Reference Genome | Genic Location | Gene # |
|---|---|---|---|---|---|---|---|
| 1 | 63 | 89.71 | 2 × 10−17 | 10,131,593 | cytochrome p450 4C1-like | intron 7 of 7 | 1 |
| 5 | 183 | 73.30 | 4 × 10−32 | 13,156,359 | 138 bp from cytochrome p450 4C1-like | UTR | 2 |
| 5 | 62 | 91.05 | 5 × 10−18 | 13,139,843 | cytochrome p450 4C1-like isoform x1 | intron 3 of 9 | 2 |
| 5 | 58 | 84.75 | 9 × 10−09 | 13,139,943 | cytochrome p450 4C1-like isoform x1 | intron 3 of 9 into exon 4 of 10 | 2 |
| 9 | 33 | 84.21 | 0.029 | 1,586,828 | cytochrome p450 9E2-like | intron 2 of 9 | 3 |
| 9 | 83 | 94.05 | 7 × 10−29 | 1,590,440 | cytochrome p450 9E2-like | intron 6 of 9 | 3 |
| 9 | 31 | 87.50 | 0.029 | 1,590,561 | cytochrome p450 9E2-like | intron 6 of 9 | 3 |
| 15 | 170 | 73.14 | 2 × 10−29 | 11,280,289 | cytochrome p450 6K1-like | intron 7 of 8 | 4 |
| 19 | 73 | 83.33 | 5 × 10−18 | 4,236,290 | cytochrome p450 4G15-like | intron 5 of 10 | 5 |
| 19 | 168 | 75.15 | 9 × 10−34 | 4,240,146 | 119 bp from cytochrome p450 4G15-like | UTR | 5 |
| 19 | 50 | 86.54 | 1 × 10−6 | 4,250,534 | cytochrome p450 4C1-like | intron 4 of 10 | 6 |
| 23 | 44 | 85.71 | 4 × 10−7 | 7,407,358 | cytochrome p450 4V2-like | intron 4 of 9 | 7 |
| 23 | 171 | 71.75 | 6 × 10-24 | 7,426,917 | cytochrome p450 4C1-like | intron 3 of 9 into exon 4 of 10 | 8 |
| 23 | 41 | 85.71 | 7 × 10−4 | 7,447,063 | cytochrome p450 4C3-like isoform x1 | intron 2 of 10 | 9 |
| 23 | 63 | 86.77 | 9 × 10−15 | 7,447,155 | cytochrome p450 4C3-like isoform x1 | intron 2 of 10 | 9 |
| 23 | 45 | 82.00 | 7 × 10−4 | 9,722,754 | cytochrome p450 4C1-like | intron 7 of 9 | 10 |
| 23 | 44 | 85.71 | 4 × 10−7 | 9,722,850 | cytochrome p450 4C1-like | intron 7 of 9 | 10 |
| 23 | 82 | 87.06 | 1 × 10−19 | 9,783,243 | cytochrome p450 4V2-like | intron 7 of 10 | 11 |
| 23 | 78 | 87.34 | 2 × 10−18 | 9,820,391 | cytochrome p450 4C1-like | intron 2 of 9 | 12 |
| 30 | 70 | 97.00 | 5 × 10−18 | 2,071,964 | cytochrome p450 307A1 | intron 1 of 1 | 13 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tembrock, L.R.; Zink, F.A.; Gilligan, T.M. Viral Prevalence and Genomic Xenology in the Coevolution of HzNV-2 (Nudiviridae) with Host Helicoverpa zea (Lepidoptera: Noctuidae). Insects 2023, 14, 797. https://doi.org/10.3390/insects14100797
Tembrock LR, Zink FA, Gilligan TM. Viral Prevalence and Genomic Xenology in the Coevolution of HzNV-2 (Nudiviridae) with Host Helicoverpa zea (Lepidoptera: Noctuidae). Insects. 2023; 14(10):797. https://doi.org/10.3390/insects14100797
Chicago/Turabian StyleTembrock, Luke R., Frida A. Zink, and Todd M. Gilligan. 2023. "Viral Prevalence and Genomic Xenology in the Coevolution of HzNV-2 (Nudiviridae) with Host Helicoverpa zea (Lepidoptera: Noctuidae)" Insects 14, no. 10: 797. https://doi.org/10.3390/insects14100797
APA StyleTembrock, L. R., Zink, F. A., & Gilligan, T. M. (2023). Viral Prevalence and Genomic Xenology in the Coevolution of HzNV-2 (Nudiviridae) with Host Helicoverpa zea (Lepidoptera: Noctuidae). Insects, 14(10), 797. https://doi.org/10.3390/insects14100797