


The Landscape of the DNA Transposons in the Genome of the Horezu_LaPeri Strain of Drosophila melanogaster

Abstract
:Simple Summary
Abstract
1. Introduction
2. Materials and Methods
2.1. Nanopore Sequencing and Genome Assembly
2.2. Computational Environment
2.3. Identification and Mapping of DNA NT Insertions
2.4. Multiple Alignment of KP Elements
2.5. GO Enrichment Analysis
2.6. Global Evaluation of Transposable Elements
2.7. PCR Validation
3. Results
4. Discussion
Supplementary Materials
Author Contributions
Funding
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fedoroff, N.V. Presidential address. Transposable elements, epigenetics, and genome evolution. Science 2012, 338, 758–767. [Google Scholar] [CrossRef] [PubMed]
- Kaminker, J.S.; Bergman, C.M.; Kronmiller, B.; Carlson, J.; Svirskas, R.; Patel, S.; Frise, E.; Wheeler, D.A.; Lewis, S.E.; Rubin, G.M.; et al. The transposable elements of the Drosophila melanogaster euchromatin: A genomics perspective. Genome Biol. 2002, 3, RESEARCH0084. [Google Scholar] [CrossRef] [PubMed]
- Merel, V.; Boulesteix, M.; Fablet, M.; Vieira, C. Transposable elements in Drosophila. Mob. DNA 2020, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Daniels, S.B.; Peterson, K.R.; Strausbaugh, L.D.; Kidwell, M.G.; Chovnick, A. Evidence for horizontal transmission of the P transposable element between Drosophila species. Genetics 1990, 124, 339–355. [Google Scholar] [CrossRef]
- Andrews, J.D.; Gloor, G.B. A role for the KP leucine zipper in regulating P element transposition in Drosophila melanogaster. Genetics 1995, 141, 587–594. [Google Scholar] [CrossRef]
- Ruiz, M.T.; Carareto, C.M. Copy number of P elements, KP/full-sized P element ratio and their relationships with environmental factors in Brazilian Drosophila melanogaster populations. Heredity 2003, 91, 570–576. [Google Scholar] [CrossRef]
- Corish, P.; Black, D.M.; Featherston, D.W.; Merriam, J.; Dover, G.A. Natural repressors of P-induced hybrid dysgenesis in Drosophila melanogaster: A model for repressor evolution. Genet. Res. 1996, 67, 109–121. [Google Scholar] [CrossRef]
- Bergman, C.M.; Han, S.; Nelson, M.G.; Bondarenko, V.; Kozeretska, I. Genomic analysis of P elements in natural populations of Drosophila melanogaster. PeerJ 2017, 5, e3824. [Google Scholar] [CrossRef]
- Simmons, M.J.; Grimes, C.D.; Czora, C.S. Cytotype Regulation Facilitates Repression of Hybrid Dysgenesis by Naturally Occurring KP Elements in Drosophila melanogaster. G3 Genes Genomes Genet. 2016, 6, 1891–1897. [Google Scholar] [CrossRef]
- Black, D.M.; Jackson, M.S.; Kidwell, M.G.; Dover, G.A. KP elements repress P-induced hybrid dysgenesis in Drosophila melanogaster. EMBO J. 1987, 6, 4125–4135. [Google Scholar] [CrossRef]
- O’Hare, K.; Rubin, G.M. Structures of P transposable elements and their sites of insertion and excision in the Drosophila melanogaster genome. Cell 1983, 34, 25–35. [Google Scholar] [CrossRef] [PubMed]
- Linheiro, R.S.; Bergman, C.M. Testing the palindromic target site model for DNA transposon insertion using the Drosophila melanogaster P-element. Nucleic Acids Res. 2008, 36, 6199–6208. [Google Scholar] [CrossRef] [PubMed]
- Lerat, E. Identifying repeats and transposable elements in sequenced genomes: How to find your way through the dense forest of programs. Heredity 2010, 104, 520–533. [Google Scholar] [CrossRef] [PubMed]
- O’Neill, K.; Brocks, D.; Hammell, M.G. Mobile genomics: Tools and techniques for tackling transposons. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2020, 375, 20190345. [Google Scholar] [CrossRef]
- Lee, H.; Schatz, M.C. Genomic dark matter: The reliability of short read mapping illustrated by the genome mappability score. Bioinformatics 2012, 28, 2097–2105. [Google Scholar] [CrossRef]
- Benoit, M. In the Transcripts: Long-Read Transcriptomics Enables a Novel Type of Transposable Element Annotation in Plants. Plant Cell 2020, 32, 2661–2662. [Google Scholar] [CrossRef]
- Shahid, S.; Slotkin, R.K. The current revolution in transposable element biology enabled by long reads. Curr. Opin. Plant Biol. 2020, 54, 49–56. [Google Scholar] [CrossRef]
- Pervez, M.T.; Hasnain, M.J.U.; Abbas, S.H.; Moustafa, M.F.; Aslam, N.; Shah, S.S.M. A Comprehensive Review of Performance of Next-Generation Sequencing Platforms. Biomed Res. Int. 2022, 2022, 3457806. [Google Scholar] [CrossRef]
- Slatko, B.E.; Gardner, A.F.; Ausubel, F.M. Overview of Next-Generation Sequencing Technologies. Curr. Protoc. Mol. Biol. 2018, 122, e59. [Google Scholar] [CrossRef]
- Disdero, E.; Filee, J. LoRTE: Detecting transposon-induced genomic variants using low coverage PacBio long read sequences. Mob. DNA 2017, 8, 5. [Google Scholar] [CrossRef]
- Ecovoiu, A.A.; Bologa, A.M.; Chifiriuc, D.I.M.; Ciuca, A.M.; Constantin, N.D.; Ghionoiu, I.C.; Ghita, I.C.; Ratiu, A.C. Genome ARTIST_v2-An Autonomous Bioinformatics Tool for Annotation of Natural Transposons in Sequenced Genomes. Int. J. Mol. Sci. 2022, 23, 12686. [Google Scholar] [CrossRef] [PubMed]
- Smit, A.F.A.; Hubley, R.; Green, P. RepeatMasker Open-3.0. 1996–2010. Available online: http://www.repeatmasker.org (accessed on 26 February 2022).
- Kuleshov, M.V.; Jones, M.R.; Rouillard, A.D.; Fernandez, N.F.; Duan, Q.; Wang, Z.; Koplev, S.; Jenkins, S.L.; Jagodnik, K.M.; Lachmann, A.; et al. Enrichr: A comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res. 2016, 44, W90–W97. [Google Scholar] [CrossRef] [PubMed]
- Bologa, A.M.; Stoica, I.; Ratiu, A.C.; Constantin, N.D.; Ecovoiu, A.A. ONT-Based Alternative Assemblies Impact on the Annotations of Unique versus Repetitive Features in the Genome of a Romanian Strain of Drosophila melanogaster. Int. J. Mol. Sci. 2022, 23, 14892. [Google Scholar] [CrossRef] [PubMed]
- Koren, S.; Walenz, B.P.; Berlin, K.; Miller, J.R.; Bergman, N.H.; Phillippy, A.M. Canu: Scalable and accurate long-read assembly via adaptive k-mer weighting and repeat separation. Genome Res. 2017, 27, 722–736. [Google Scholar] [CrossRef]
- Kolmogorov, M.; Yuan, J.; Lin, Y.; Pevzner, P.A. Assembly of long, error-prone reads using repeat graphs. Nat. Biotechnol. 2019, 37, 540–546. [Google Scholar] [CrossRef]
- Larkin, A.; Marygold, S.J.; Antonazzo, G.; Attrill, H.; Dos Santos, G.; Garapati, P.V.; Goodman, J.L.; Gramates, L.S.; Millburn, G.; Strelets, V.B.; et al. FlyBase: Updates to the Drosophila melanogaster knowledge base. Nucleic Acids Res. 2021, 49, D899–D907. [Google Scholar] [CrossRef]
- GitHub-Bergmanlab/Drosophila-Transposons: Drosophila Transposable Element Canonical Sequences. Available online: https://github.com/bergmanlab/drosophila-transposons (accessed on 7 December 2022).
- Shen, W.; Le, S.; Li, Y.; Hu, F. SeqKit: A Cross-Platform and Ultrafast Toolkit for FASTA/Q File Manipulation. PLoS ONE 2016, 11, e0163962. [Google Scholar] [CrossRef]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinformatics 2007, 23, 2947–2948. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- RStudio Team. RStudio: Integrated Development for R. RStudio, PBC, Boston, MA. 2020. Available online: http://www.rstudio.com/ (accessed on 26 February 2022).
- Smit, A.F.A.; Hubley, R. RepeatModeler Open-1.0. 2008–2015. Available online: http://www.repeatmasker.org (accessed on 26 February 2022).
- Price, A.L.; Jones, N.C.; Pevzner, P.A. De novo identification of repeat families in large genomes. Bioinformatics 2005, 21 (Suppl. S1), i351–i358. [Google Scholar] [CrossRef]
- Bao, Z.; Eddy, S.R. Automated de novo identification of repeat sequence families in sequenced genomes. Genome Res. 2002, 12, 1269–1276. [Google Scholar] [CrossRef] [PubMed]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef] [PubMed]
- Ullastres, A.; Merenciano, M.; Gonzalez, J. Regulatory regions in natural transposable element insertions drive interindividual differences in response to immune challenges in Drosophila. Genome Biol. 2021, 22, 265. [Google Scholar] [CrossRef]
- Gonzalez, J.; Petrov, D.A. The adaptive role of transposable elements in the Drosophila genome. Gene 2009, 448, 124–133. [Google Scholar] [CrossRef]
- Kwon, D.; Mucci, D.; Langlais, K.K.; Americo, J.L.; DeVido, S.K.; Cheng, Y.; Kassis, J.A. Enhancer-promoter communication at the Drosophila engrailed locus. Development 2009, 136, 3067–3075. [Google Scholar] [CrossRef]
- Caizzi, R.; Caggese, C.; Pimpinelli, S. Bari-1, a new transposon-like family in Drosophila melanogaster with a unique heterochromatic organization. Genetics 1993, 133, 335–345. [Google Scholar] [CrossRef]
- Marsano, R.M.; Milano, R.; Minervini, C.; Moschetti, R.; Caggese, C.; Barsanti, P.; Caizzi, R. Organization and possible origin of the Bari-1 cluster in the heterochromatic h39 region of Drosophila melanogaster. Genetica 2003, 117, 281–289. [Google Scholar] [CrossRef]
- Palazzo, A.; Caizzi, R.; Moschetti, R.; Marsano, R.M. What Have We Learned in 30 Years of Investigations on Bari Transposons? Cells 2022, 11, 583. [Google Scholar] [CrossRef]
- Palazzo, A.; Lovero, D.; D’Addabbo, P.; Caizzi, R.; Marsano, R.M. Identification of Bari Transposons in 23 Sequenced Drosophila Genomes Reveals Novel Structural Variants, MITEs and Horizontal Transfer. PLoS ONE 2016, 11, e0156014. [Google Scholar] [CrossRef] [PubMed]
- Marsano, R.M.; Marconi, S.; Moschetti, R.; Barsanti, P.; Caggese, C.; Caizzi, R. MAX, a novel retrotransposon of the BEL-Pao family, is nested within the Bari1 cluster at the heterochromatic h39 region of chromosome 2 in Drosophila melanogaster. Mol. Genet. Genom. 2004, 270, 477–484. [Google Scholar] [CrossRef] [PubMed]
- RepeatMasker. Available online: http://www.repeatmasker.org/species/dm.html (accessed on 28 February 2022).
- Smith, C.D.; Shu, S.; Mungall, C.J.; Karpen, G.H. The Release 5.1 annotation of Drosophila melanogaster heterochromatin. Science 2007, 316, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Dimitri, P.; Caizzi, R.; Giordano, E.; Carmela Accardo, M.; Lattanzi, G.; Biamonti, G. Constitutive heterochromatin: A surprising variety of expressed sequences. Chromosoma 2009, 118, 419–435. [Google Scholar] [CrossRef]
- Coulthard, A.B.; Alm, C.; Cealiac, I.; Sinclair, D.A.; Honda, B.M.; Rossi, F.; Dimitri, P.; Hilliker, A.J. Essential loci in centromeric heterochromatin of Drosophila melanogaster. I: The right arm of chromosome 2. Genetics 2010, 185, 479–495. [Google Scholar] [CrossRef]
- Marsano, R.M.; Dimitri, P. Constitutive Heterochromatin in Eukaryotic Genomes: A Mine of Transposable Elements. Cells 2022, 11, 761. [Google Scholar] [CrossRef]
- Dimitri, P.; Arcà, B.; Berghella, L.; Mei, E. High genetic instability of heterochromatin after transposition of the LINE-like I factor in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 1997, 94, 8052–8057. [Google Scholar] [CrossRef]
- Dimitri, P.; Junakovic, N. Revising the selfish DNA hypothesis: New evidence on accumulation of transposable elements in heterochromatin. TiG 1999, 15, 123–124. [Google Scholar] [CrossRef]
- Dimitri, P.; Junakovic, N.; Arcà, B. Colonization of heterochromatic genes by transposable elements in Drosophila. Mol. Biol. Evol. 2003, 20, 503–512. [Google Scholar] [CrossRef]
- Kelleher, E.S. Reexamining the P-Element Invasion of Drosophila melanogaster Through the Lens of piRNA Silencing. Genetics 2016, 203, 1513–1531. [Google Scholar] [CrossRef]
- Engels, W.R. The origin of P elements in Drosophila melanogaster. Bioessays 1992, 14, 681–686. [Google Scholar] [CrossRef] [PubMed]
- Itoh, M.; Woodruff, R.C.; Leone, M.A.; Boussy, I.A. Genomic P elements and P m characteristics of eastern Australian populations of Drosophila melanogaster. Genetica 1999, 106, 231–245. [Google Scholar] [CrossRef] [PubMed]
- Anxolabehere, D.; Nouaud, D.; Periquet, G.; Tchen, P. P-element distribution in Eurasian populations of Drosophila melanogaster: A genetic and molecular analysis. Proc. Natl. Acad. Sci. USA 1985, 82, 5418–5422. [Google Scholar] [CrossRef] [PubMed]
- Boussy, I.A.; Healy, M.J.; Oakeshott, J.G.; Kidwell, M.G. Molecular analysis of the P m gonadal dysgenesis cline in eastern Australian Drosophila melanogaster. Genetics 1988, 119, 889–902. [Google Scholar] [CrossRef] [PubMed]
- Sakoyama, Y.; Todo, T.; Ishiwa-Chigusa, S.; Honjo, T.; Kondo, S. Structures of defective P transposable elements prevalent in natural Q and Q-derived M strains of Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 1985, 82, 6236–6239. [Google Scholar] [CrossRef]
- Weilguny, L.; Vlachos, C.; Selvaraju, D.; Kofler, R. Reconstructing the Invasion Route of the P-Element in Drosophila melanogaster Using Extant Population Samples. Genome Biol. Evol. 2020, 12, 2139–2152. [Google Scholar] [CrossRef]
- Caggese, C.; Pimpinelli, S.; Barsanti, P.; Caizzi, R. The distribution of the transposable element Bari-1 in the Drosophila melanogaster and Drosophila simulans genomes. Genetica 1995, 96, 269–283. [Google Scholar] [CrossRef]
- Pimpinelli, S.; Berloco, M.; Fanti, L.; Dimitri, P.; Bonaccorsi, S.; Marchetti, E.; Caizzi, R.; Caggese, C.; Gatti, M. Transposable elements are stable structural components of Drosophila melanogaster heterochromatin. Proc. Natl. Acad. Sci. USA 1995, 92, 3804–3808. [Google Scholar] [CrossRef]
- Daborn, P.J.; Lumb, C.; Boey, A.; Wong, W.; Ffrench-Constant, R.H.; Batterham, P. Evaluating the insecticide resistance potential of eight Drosophila melanogaster cytochrome P450 genes by transgenic over-expression. Insect. Biochem. Mol. Biol. 2007, 37, 512–519. [Google Scholar] [CrossRef]
- Ly, S.; Pack, A.I.; Naidoo, N. The neurobiological basis of sleep: Insights from Drosophila. Neurosci. Biobehav. Rev. 2018, 87, 67–86. [Google Scholar] [CrossRef]
- Eriksson, A.; Raczkowska, M.; Navawongse, R.; Choudhury, D.; Stewart, J.C.; Tang, Y.L.; Wang, Z.; Claridge-Chang, A. Neuromodulatory circuit effects on Drosophila feeding behaviour and metabolism. Sci. Rep. 2017, 7, 8839. [Google Scholar] [CrossRef] [PubMed]
- MacMillan, H.A.; Knee, J.M.; Dennis, A.B.; Udaka, H.; Marshall, K.E.; Merritt, T.J.; Sinclair, B.J. Cold acclimation wholly reorganizes the Drosophila melanogaster transcriptome and metabolome. Sci. Rep. 2016, 6, 28999. [Google Scholar] [CrossRef] [PubMed]
- Shearer, P.W.; West, J.D.; Walton, V.M.; Brown, P.H.; Svetec, N.; Chiu, J.C. Seasonal cues induce phenotypic plasticity of Drosophila suzukii to enhance winter survival. BMC Ecol. 2016, 16, 11. [Google Scholar] [CrossRef]
- Senthilan, P.R.; Grebler, R.; Reinhard, N.; Rieger, D.; Helfrich-Forster, C. Role of Rhodopsins as Circadian Photoreceptors in the Drosophila melanogaster. Biology 2019, 8, 6. [Google Scholar] [CrossRef] [PubMed]
- Schlichting, M.; Grebler, R.; Peschel, N.; Yoshii, T.; Helfrich-Forster, C. Moonlight detection by Drosophila’s endogenous clock depends on multiple photopigments in the compound eyes. J. Biol. Rhythm. 2014, 29, 75–86. [Google Scholar] [CrossRef] [PubMed]
- Slobodian, M.R.; Petahtegoose, J.D.; Wallis, A.L.; Levesque, D.C.; Merritt, T.J.S. The Effects of Essential and Non-Essential Metal Toxicity in the Drosophila melanogaster Insect Model: A Review. Toxics 2021, 9, 269. [Google Scholar] [CrossRef] [PubMed]
- Harbison, S.T.; Kumar, S.; Huang, W.; McCoy, L.J.; Smith, K.R.; Mackay, T.F.C. Genome-Wide Association Study of Circadian Behavior in Drosophila melanogaster. Behav. Genet 2019, 49, 60–82. [Google Scholar] [CrossRef]
- Mandilaras, K.; Missirlis, F. Genes for iron metabolism influence circadian rhythms in Drosophila melanogaster. Metallomics 2012, 4, 928–936. [Google Scholar] [CrossRef]
- Xu, K.; DiAngelo, J.R.; Hughes, M.E.; Hogenesch, J.B.; Sehgal, A. The circadian clock interacts with metabolic physiology to influence reproductive fitness. Cell Metab. 2011, 13, 639–654. [Google Scholar] [CrossRef]
- Gonzalez, J.; Lenkov, K.; Lipatov, M.; Macpherson, J.M.; Petrov, D.A. High rate of recent transposable element-induced adaptation in Drosophila melanogaster. PLoS Biol. 2008, 6, e251. [Google Scholar] [CrossRef]
- Kumar, S.; Tunc, I.; Tansey, T.R.; Pirooznia, M.; Harbison, S.T. Identification of Genes Contributing to a Long Circadian Period in Drosophila Melanogaster. J. Biol. Rhythm. 2021, 36, 239–253. [Google Scholar] [CrossRef] [PubMed]
- Fonseca, P.M.; Moura, R.D.; Wallau, G.L.; Loreto, E.L.S. The mobilome of Drosophila incompta, a flower-breeding species: Comparison of transposable element landscapes among generalist and specialist flies. Chromosome Res. 2019, 27, 203–219. [Google Scholar] [CrossRef] [PubMed]
- Shao, F.; Han, M.; Peng, Z. Evolution and diversity of transposable elements in fish genomes. Sci. Rep. 2019, 9, 15399. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
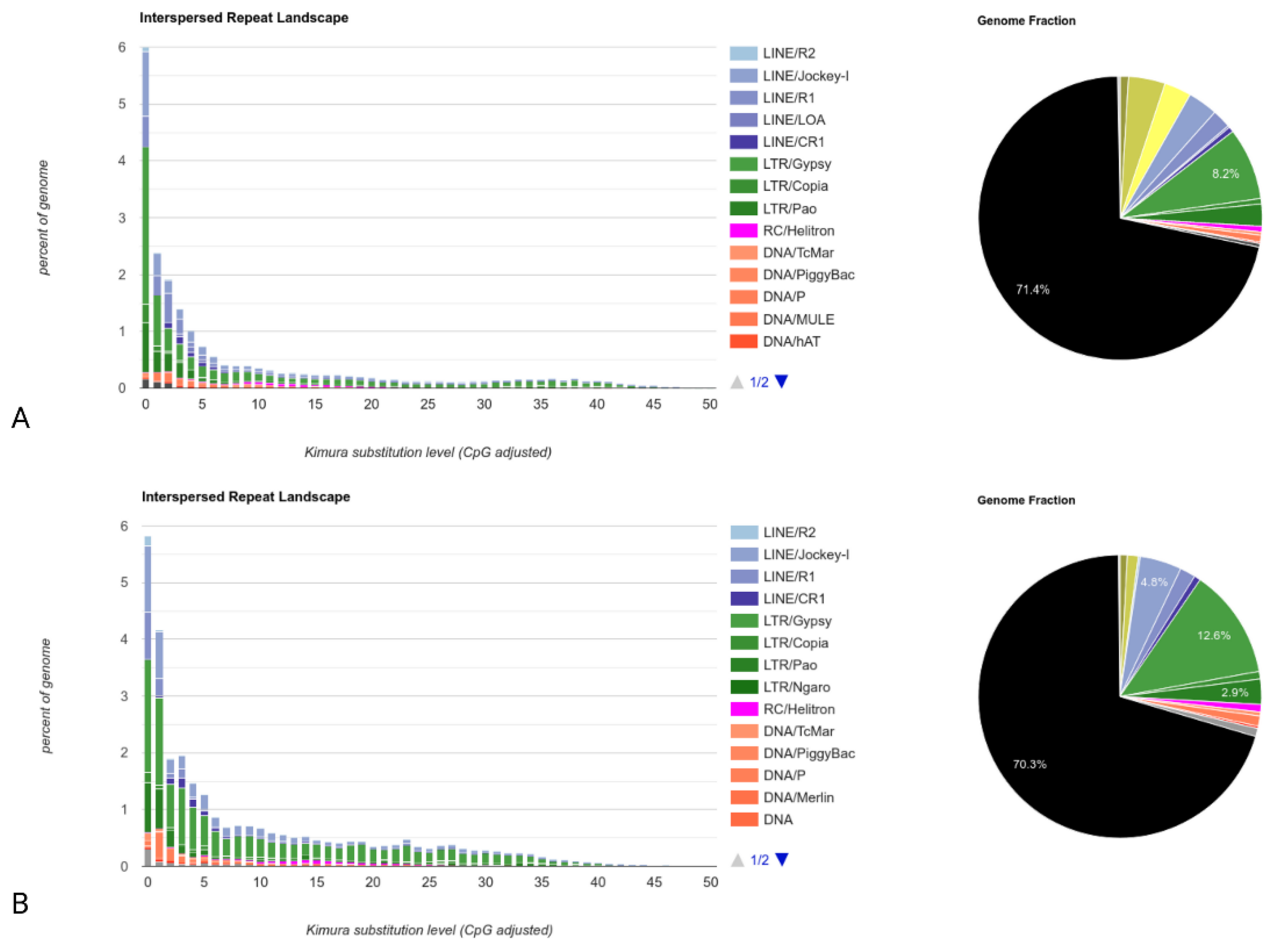{kind=link}
{kind=link}
| DNA NT Name | NT Length (bp) | TIR Length (bp) | Copy Number in D. melanogaster r6.48 |
|---|---|---|---|
| Bari1 | 1728 | 26 | 7 |
| Bari2 | 1064 | 253 | 5 |
| HB | 1653 | 29–31 | 60 |
| hobo | 2959 | 12 | 60 |
| hopper | 1435 | 33 | 26 |
| mariner2 | 912 | 29 | 23 |
| NOF | 4347 | 308 | 8 |
| P-element | 2907 | 31 | 0 |
| pogo | 2121 | 26 | 50 |
| S-element | 1736 | 234 | 187 |
| S2 | 1735 | 233 | 16 |
| Tc1 | 1666 | 26 | 31 |
| Tc3 | 1743 | 43 | 19 |
| transib1 | 2167 | 43 | 3 |
| transib2 | 2844 | 42 | 27 |
| transib3 | 2883 | 45 | 13 |
| transib4 | 2656 | 40 | 8 |
| DNA NT | Horezu_LaPeri-Specific Insertions | Conserved Insertions | Total Mapped Insertions | Unannotated Conserved Insertions | Ambiguous Insertions | Unresolvable Insertions |
|---|---|---|---|---|---|---|
| Bari1 | 3 | 3 | 6 | 1 | 1 | 1 |
| Bari2 | 0 | 4 | 4 | 2 | 0 | 0 |
| HB | 1 | 46 | 47 | 10 | 0 | 1 |
| hobo | 26 | 12 | 38 | 9 | 0 | 0 |
| hopper | 13 | 26 | 39 | 14 | 0 | 1 |
| mariner2 | 1 | 34 | 35 | 16 | 0 | 0 |
| NOF | 1 | 2 | 3 | 1 | 0 | 0 |
| P-element | 16 | 0 | 16 | 0 | 0 | 0 |
| pogo | 18 | 4 | 22 | 0 | 1 | 2 |
| S-element | 12 | 74 | 86 | 39 | 2 | 10 |
| S2 | 0 | 16 | 16 | 7 | 0 | 2 |
| Tc1 | 1 | 43 | 44 | 15 | 0 | 3 |
| Tc3 | 3 | 9 | 12 | 4 | 0 | 0 |
| transib1 | 19 | 12 | 31 | 10 | 0 | 2 |
| transib2 | 6 | 34 | 40 | 20 | 2 | 2 |
| transib3 | 0 | 22 | 22 | 13 | 0 | 0 |
| transib4 | 0 | 8 | 8 | 2 | 1 | 2 |
| NT Name | Contig Number | Insertion Coordinate; Host Chromosome | Hit/Close Genes/NTs |
|---|---|---|---|
| Bari1 | 892 | 3,657,194; X | AstA-R1 |
| 2963 | 20,848,451; 3R | close to CG15696 and RpS30 | |
| 3495 | 23,430,916; 2L | -* | |
| HB | 2615 | 22,744,158; 3L | CG7369* NTs hotspot |
| hobo | 37 | 11,731,193; 3R | CG12594 |
| 142 | 8,265,913; 3L | Dscam4 | |
| 153 | 13,627,239; 3L | bru3 | |
| 166 | 31,134,735; 3R | Gycβ100B | |
| 364 | 11,729,899; X | inaF-A inaF-B inaF-C CG15221 | |
| 429 | 16,058,145; 2L | beat-Ia | |
| 1066 | 18,846,823; 3R | Octα2R | |
| 1121 | 11,868,933; 2L | Pde1c | |
| 2025, 2026 | 8,555,248; X | IntS4 | |
| 2056 | 12,963,046; 2L | - | |
| 2063 | 13,826,443; 3L | - | |
| 2110 | 13,678,976; 3L | bru3 | |
| 2111, 2421, 2422 | 14,043,231; 2R | mam | |
| 2124 | 8,322,604; 3L | - | |
| 2136 | 16,515,554; 3L | CG43373 | |
| 2166 | 17,551,994; 2L | - | |
| 2167 | 19,461,109; 2L | - | |
| 2316 | 17,384,934; 2R | lncRNA:CR44344 | |
| 2343 | 17,184,472; 2R | lncRNA:CR44387 | |
| 2484 | 20,235,449; 3R | Nlg4 | |
| 2673 | 5,866,909; 2L | rau | |
| 5,649,715; 2L | DIP-θ | ||
| 2707 | 15,577,454; 2R | - | |
| 2762 | 9,291,978; 3R | - | |
| 2788 | 22,850,595; 3R | lncRNA:CR43846 | |
| 3145 | 16,137,749; 2L | lncRNA:CR44871 | |
| hopper | 165 | 18,193,772; X | Frq2 |
| 526 | 16,891,891; 2R | CG8910 | |
| 1264 | 22,559,246; X | 1731{}3268* NTs hotspot | |
| 1938 | 5,438,793; 3L | DIP-δ | |
| 1989 | 20,939,569; 2L | - | |
| 2020 | 27,859,138; 3R | CG34353 | |
| 27,974,479; 3R | βTub97EF | ||
| 2615 | 22,635,736; 3L | CG14459 | |
| 2675 | 21,846,566; 3L | mub | |
| 2725 | 2,586,216; 2R | 1360{}6340* | |
| 2760 | 12,301,235; 3L | lncRNA:CR44550 | |
| 3122 | 24,568,086; 3L | -* | |
| 3153 | 11,661,763; 3L | CG32085 | |
| mariner2 | 744 | 2,671,627; 3R | Pzl* |
| NOF | 2967 | 19,470,432; 3R | Dys |
| P-element | 477 | 29,765,960; 3R | close to kay |
| 491 | 21,347,168; 3L | ebd2 CG32436 | |
| 869 | 10,547,695; X | spri | |
| 1601 | 3,250,477; 3L | lncRNA:CR43626 | |
| 1937 | 959,438; 3L | Glut1 | |
| 1938 | 1,688,826; 3L | CG7991 | |
| 2081 | 7,489,940; 2R | close to Coop | |
| 2082 | 21,697,891; 2L | nolo NTs hotspot | |
| 2111 | 14,928,221; 2R | Kank | |
| 2122 | 13,477,242; 3L | close to CG10089 | |
| 2175 | 289,069; 2R | -* | |
| 2267 | 9,698,895; 2L | Pka-C1 | |
| 2837 | 3,059,410; Y | -* | |
| 2907 | 3,063,077; 2R | 1360{}6347* | |
| 2967 | 19,180,631; 3R | close to CG5555, CG14282, myd | |
| 3114 | 11,201,707; X | gypsy5{}103 | |
| pogo | 166 | 30,374,902; 3R | close to hdc |
| 222 | 15,035,178; 3L | Sytβ | |
| 235 | 21,531,991; 2R | Egfr lncRNA:CR44725 | |
| 941 | 15,069,400; X | Lsd-2 | |
| 1021 | 15,190,453; 2R | close to igl | |
| 1202 | 21,254,529; X | Mnr | |
| 1938 | 1,684,964; 3L | CG7991 | |
| 1972 | 14,688,291; 3R | Meltrin | |
| 1993 | 22,113,023; 2L | NTs hotspot* | |
| 2040 | 2,801,161; 3R | Pzl* | |
| 2081 | 7,839,017; 2R | Dgk | |
| 2139 | 10,889,337; 3L | close to OXA1L | |
| 2166 | 17,730,485; 2L | CadN | |
| 2280 | 18,913,439; 3R | close to Sgsh | |
| 2410 | 20,456,532; 2L | CG31687 | |
| 2615 | 22,676,271; 3L | Jhbp6 | |
| 2768 | 17,503,189; 3L | Oatp74D | |
| 3136 | 24,834,544; 3L | -* | |
| S-element | 67 | 9,950,745; 2R | Mef2 |
| 415 | 23,313,720; 2L | -* | |
| 940 | 1,664,262; 3R | Myo81F* | |
| 1611 | 23,143,313; 3L | -* | |
| 2290 | 6,076,695; 2L | - | |
| 2353 | 3,827,600; 3R | - | |
| 2555 | 5,674,700; 2R | Kune* | |
| 2660 | 4,343,666; 2R | Gprk1* | |
| 2730 | 25,200,700; 3L | CR40354* | |
| 3019 | 20,099,984; X | 3S18{}177 | |
| 3138 | 5,701,549; 2R | vlc* | |
| 3528 | 23,400,788; 2L | -* | |
| Tc1 | 2168 | 19,339,397; 3L | close to CG32206 |
| Tc3 | 474 | 27,356,129; 3L | Dbp80* |
| 578 | 3,058,866; 3R | Pzl* | |
| 3086 | 2,884,603; 3R | Pzl* | |
| transib1 | 67 | 9,706,842; 2R | CG1773 |
| 189 | 21,115,833; 3L | ko | |
| 410 | 15,043,410; 2L | close to Su(H) | |
| 718 | 26,170,465; 3L | CR41320* | |
| 1760 | 2,221,072; 2L | Ir40a* | |
| 1954, 1955 | 282,647; 3L | RhoGEF3 | |
| 1973 | 14,535,266; 3R | Pde6 lncRNA:CR46023 | |
| 1974, 2041, 2042 | 5,533,505; 3L | close to CG13285 | |
| 2026 | 8,675,495; X | - | |
| 2069 | 16,135,549; 2R | spin | |
| 2137 | 3,473,173; 3L | CG42324 | |
| 2183 | 4,285,753; 3R | cpx | |
| 2302 | 11,134,998; 3R | close to mAcon2 | |
| 2660 | 4,322,642; 2R | Gprk1* | |
| 2877, 2878 | 15,028,387; 2L | CG33310 GABA-B-R1 | |
| 2932 | 23,188,307; 2L | -* | |
| 3138 | 5,787,158; 2R | -* | |
| 3782 | 22,314,057; 3L | Ten-m | |
| 3783 | 19,656,233; X | Elys | |
| transib2 | 2375 | 958,213; 2R | -* |
| 2528, 2530, 2531 | 2,831,535; Y | -* | |
| 2789 | 14,049,394; 3R | - | |
| 3233 | 3,146,809; Y | -* | |
| 3311 | 3,593,807; Y | ORY* | |
| 3389 | 22,662,667; 2L | CG40006* |
| GO Term | Genes | p Value |
|---|---|---|
| A. Horezu_LaPeri-specific NT insertions | ||
| cellular metal ion homeostasis (GO:0006875) | RhoGEF3, Pde6, Pde1c, CG8910, Glut1, GABA-B-R1, Dgk, cpx, CG34353 | 1.34 × 10−9 |
| temperature compensation of the circadian clock (GO:0010378) | bru3, Pde6, Pka-C1, CG8910, CG32085, Pde1c, GABA-B-R1, Dgk, CG34353 | 7.49 × 10−8 |
| female mating behavior (GO:0060180) | bru3, Pde6, Pka-C1, CG8910, Pde1c, Glut1, GABA-B-R1, mub, CG34353 | 7.49 × 10−8 |
| RNA–gene interaction with Putative Regulatory mir-4 | Mef2, bru3, Pka-C1, CG14459, Dbp80, CG7991, Dgk, cpx, CG42324, Nlg4, CG43373, CG8910, Pde1c, ORY, spin, GABA-B-R1, mam, mub, CG15221 | 4.87 × 10−7 |
| regulation of terminal button organization (GO:2000331) | CG8910, CG14459, Pde1c, Glut1, CadN, GABA-B-R1, mub, CG34353 | 1.00 × 10−6 |
| regulation of muscle tissue development (GO:1001861) | Mef2, Kank, Ten-m, CadN, beat-Ia, Egfr, Dys | 4.00 × 10−6 |
| modulation of chemical synaptic transmission (GO:0050804) | Pka-C1, cpx, Frq2, Nlg4, Dys | 7.00 × 106 |
| R7 cell differentiation (GO:0045466) | Ten-m, CadN, rau, Egfr | 2.20 × 10−5 |
| positive regulation of intracellular signal transduction (GO:1902533) | Mef2, rau, mam, cpx, Egfr | 0.00014 |
| 3′,5′-cyclic-GMP phosphodiesterase activity (GO:0047555) | Pde6, Pde1c | 0.000217 |
| rhodopsin biosynthetic process (GO:0016063) | Pka-C1, Gprk1, Dgk, Frq2, Egfr, AstA-R1 | 0.000305 |
| temperature response-defective phenotype | CG8910, GABA-B-R1, mub, CG34353 | 0.007 |
| B. Horezu_LaPeri specific and conserved NT insertions | ||
| response to alcohol (GO:0097305) | bru3, Pka-C1, nAChRalpha4, Syt7, CG8910, Snap25, GABA-B-R1, CG18208, mub, CG34353, CG17684, MFS17, Ten-a | 3.15 × 10−8 |
| sensory perception of touch (GO:0050975) | AGO2, hiw, Usp2, Ank | 0.0001834 |
| increased fecundity phenotype | Pde1c, AGO2, Parp, kl-5, Egfr, Ank | 0.006329 |
| radiation resistant phenotype | rl, zfh2, sxc | 0.009606 |
| circadian rhythm defective phenotype | RhoGEF3, CadN, Egfr, Dys, Ank | 0.0342 |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bologa, A.M.; Stoica, I.; Constantin, N.D.; Ecovoiu, A.A. The Landscape of the DNA Transposons in the Genome of the Horezu_LaPeri Strain of Drosophila melanogaster. Insects 2023, 14, 494. https://doi.org/10.3390/insects14060494
Bologa AM, Stoica I, Constantin ND, Ecovoiu AA. The Landscape of the DNA Transposons in the Genome of the Horezu_LaPeri Strain of Drosophila melanogaster. Insects. 2023; 14(6):494. https://doi.org/10.3390/insects14060494
Chicago/Turabian StyleBologa, Alexandru Marian, Ileana Stoica, Nicoleta Denisa Constantin, and Alexandru Al. Ecovoiu. 2023. "The Landscape of the DNA Transposons in the Genome of the Horezu_LaPeri Strain of Drosophila melanogaster" Insects 14, no. 6: 494. https://doi.org/10.3390/insects14060494
APA StyleBologa, A. M., Stoica, I., Constantin, N. D., & Ecovoiu, A. A. (2023). The Landscape of the DNA Transposons in the Genome of the Horezu_LaPeri Strain of Drosophila melanogaster. Insects, 14(6), 494. https://doi.org/10.3390/insects14060494