
G9a: An Emerging Epigenetic Target for Melanoma Therapy


Abstract
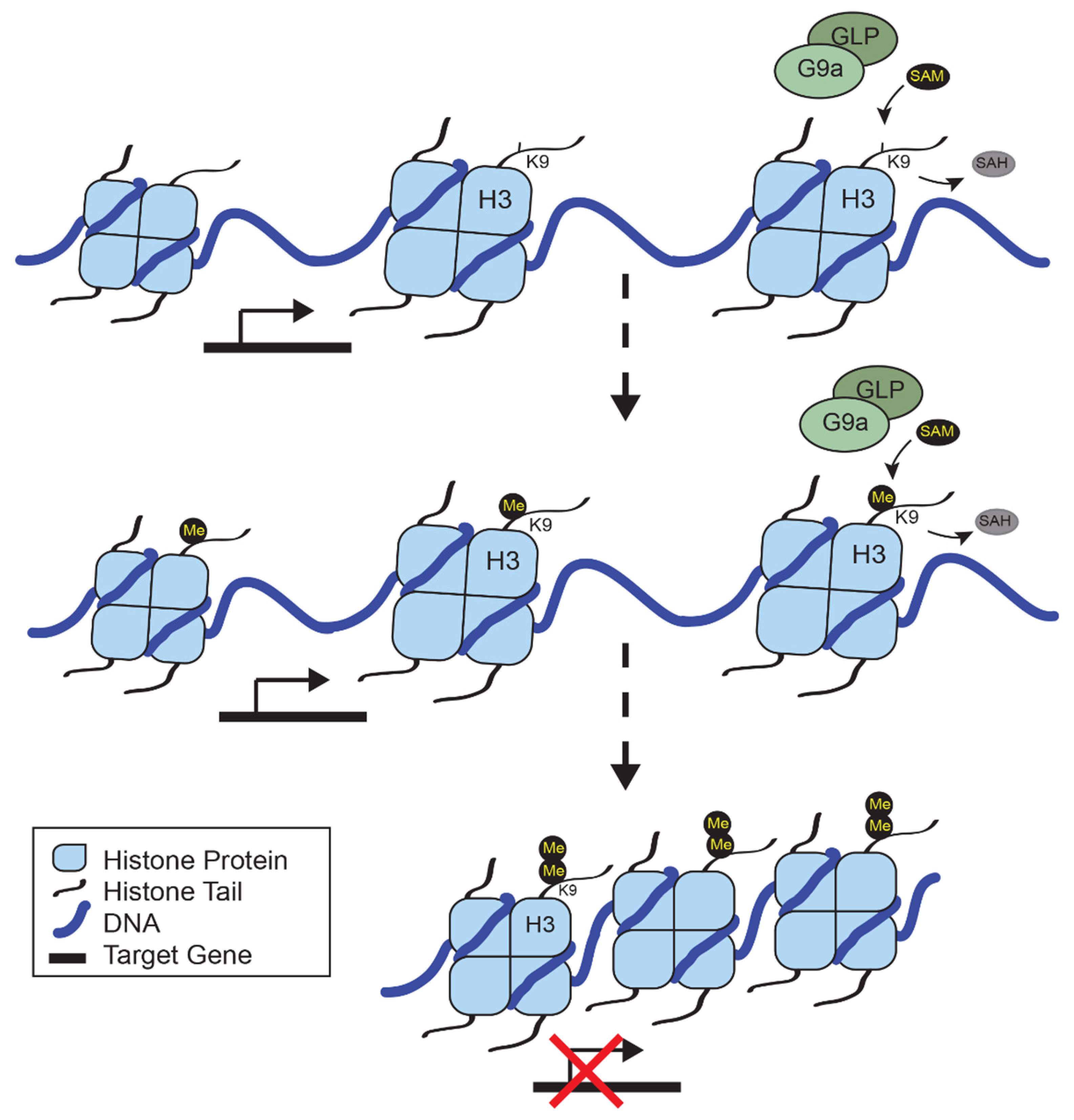
:1. Epigenetic Regulation by Histone Methyltransferases
2. The Role of G9a in Development and Differentiation
3. The Oncogenic Roles of G9a in Melanoma

{kind=link}
| Signaling Pathway/ Cellular Mechanism | Key Findings | Tumor Type | Reference |
|---|---|---|---|
| Wnt Signaling | The Wnt repressor, DKK1, is targeted by G9a repression | Melanoma | [21] |
| Wnt signaling promotes melanoma development and inhibits immunotherapy response | Melanoma | [23,24] | |
| G9a loss or inhibition of H3K9me2 causes upregulation of DKK1 | Neuroendocrine | [25] | |
| G9a is enriched at genes that negatively regulate Wnt signaling in patient-derived organoids | Colorectal Cancer | [26] | |
| Notch Signaling | Overexpression of G9a promotes upregulation of Notch1 signaling pathway | Melanoma | [27] |
| Elevated Notch signaling associated with treatment resistance and more invasive phenotypes | Melanoma | [28,29,30] | |
| Notch signaling in fibroblasts creates a less favorable microenvironment | Melanoma | [32,33] | |
| G9a levels increase in NICD-induced model of bile duct cancer | Cholangiocarcinoma | [31] | |
| Metastasis | H3K9me2 is increased between non-metastatic mesenchymal-like to metastatic melanoma cells | Melanoma | [34] |
| HOX genes are upregulated in metastatic melanoma | Melanoma | [35] | |
| HOXA1 is a repressed target of G9a | Glioblastoma | [36] | |
| G9a represses CDH10 increasing cellular motility during hypoxia | Breast Cancer | [37] | |
| Autophagy | G9a inhibition activates autophagy | Melanoma | [38] |
| G9a represses MAP1LC3B and blocks autophagy | Glioblastoma | [39,40] | |
| G9a vacates autophagy related gene promoters under starvation in vitro | Cervical and Pancreatic Cancer | [41] | |
| G9a blocks autophagy through activation of mTOR | Gastric Cancer | [42] |
4. Targeting Histone Methyltransferases
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Sigalotti, L.; Covre, A.; Fratta, E.; Parisi, G.; Colizzi, F.; Rizzo, A.; Danielli, R.; Nicolay, H.J.M.; Coral, S.; Maio, M. Epigenetics of human cutaneous melanoma: Setting the stage for new therapeutic strategies. J. Transl. Med. 2010, 8, 56. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, M.A.; Kouzarides, T. Cancer epigenetics: From mechanism to therapy. Cell 2012, 150, 12–27. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-Resolution Profiling of Histone Methylations in the Human Genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schaefer, A.; Sampath, S.C.; Intrator, A.; Min, A.; Gertler, T.S.; Surmeier, D.J.; Tarakhovsky, A.; Greengard, P. Control of Cognition and Adaptive Behavior by the GLP/G9a Epigenetic Suppressor Complex. Neuron 2009, 64, 678–691. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Karimi, M.M.; Goyal, P.; Maksakova, I.A.; Bilenky, M.; Leung, D.; Tang, J.X.; Shinkai, Y.; Mager, D.L.; Jones, S.; Hirst, M.; et al. DNA methylation and SETDB1/H3K9me3 regulate predominantly distinct sets of genes, retroelements, and chimeric transcripts in mescs. Cell Stem Cell 2011, 8, 676–687. [Google Scholar] [CrossRef] [Green Version]
- Peters, A.H.F.M.; O’Carroll, D.; Scherthan, H.; Mechtler, K.; Sauer, S.; Schöfer, C.; Weipoltshammer, K.; Pagani, M.; Lachner, M.; Kohlmaier, A.; et al. Loss of the Suv39h histone methyltransferases impairs mammalian heterochromatin and genome stability. Cell 2001, 107, 323–337. [Google Scholar] [CrossRef] [Green Version]
- Sampath, S.C.; Marazzi, I.; Yap, K.L.; Sampath, S.C.; Krutchinsky, A.N.; Mecklenbräuker, I.; Viale, A.; Rudensky, E.; Zhou, M.M.; Chait, B.T.; et al. Methylation of a Histone Mimic within the Histone Methyltransferase G9a Regulates Protein Complex Assembly. Mol. Cell 2007, 27, 596–608. [Google Scholar] [CrossRef]
- Bao, L.; Chen, Y.; Lai, H.T.; Wu, S.Y.; Wang, J.E.; Hatanpaa, K.J.; Raisanen, J.M.; Fontenot, M.; Lega, B.; Chiang, C.M.; et al. Methylation of hypoxia-inducible factor (HIF)-1α by G9a/GLP inhibits HIF-1 transcriptional activity and cell migration. Nucleic Acids Res. 2018, 46, 6576–6591. [Google Scholar] [CrossRef] [Green Version]
- Jan, S.; Dar, M.I.; Wani, R.; Sandey, J.; Mushtaq, I.; Lateef, S.; Syed, S.H. Targeting EHMT2/ G9a for cancer therapy: Progress and perspective. Eur. J. Pharmacol. 2021, 893, 173827. [Google Scholar] [CrossRef]
- Rada, M.; Vasileva, E.; Lezina, L.; Marouco, D.; Antonov, A.V.; MacIp, S.; Melino, G.; Barlev, N.A. Human EHMT2/G9a activates p53 through methylation-independent mechanism. Oncogene 2017, 36, 922–932. [Google Scholar] [CrossRef] [Green Version]
- Zeng, T.B.; Han, L.; Pierce, N.; Pfeifer, G.P.; Szabó, P.E. EHMT2 and SETDB1 protect the maternal pronucleus from 5mC oxidation. Proc. Natl. Acad. Sci. USA 2019, 166, 10834–10841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wen, B.; Wu, H.; Shinkai, Y.; Irizarry, R.A.; Feinberg, A.P. Large histone H3 lysine 9 dimethylated chromatin blocks distinguish differentiated from embryonic stem cells. Nat. Genet. 2009, 41, 246–250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Filion, G.J.; Van Steensel, B. Reassessing the abundance of H3K9me2 chromatin domains in embryonic stem cells. Nat. Genet. 2010, 42, 4. [Google Scholar] [CrossRef] [PubMed]
- Antignano, F.; Braam, M.; Hughes, M.R.; Chenery, A.L.; Burrows, K.; Gold, M.J.; Oudhoff, M.J.; Rattray, D.; Halim, T.Y.; Cait, A.; et al. G9a regulates group 2 innate lymphoid cell development by repressing the group 3 innate lymphoid cell program. J. Exp. Med. 2016, 213, 1153–1162. [Google Scholar] [CrossRef]
- Gaballa, J.M.; Braga Neto, M.B.; Ramos, G.P.; Bamidele, A.O.; Gonzalez, M.M.; Sagstetter, M.R.; Sarmento, O.F.; Faubion, W.A. The Role of Histone Methyltransferases and Long Non-coding RNAs in the Regulation of T Cell Fate Decisions. Front. Immunol. 2018, 9, 2955. [Google Scholar] [CrossRef]
- Verbaro, D.J.; Sakurai, N.; Kim, B.; Shinkai, Y.; Egawa, T. Cutting Edge: The Histone Methyltransferase G9a Is Required for Silencing of Helper T Lineage–Associated Genes in Proliferating CD8 T Cells. J. Immunol. 2018, 200, 3891–3896. [Google Scholar] [CrossRef]
- Shin, H.M.; Kapoor, V.N.; Guan, T.; Kaech, S.M.; Welsh, R.M.; Berg, L.J. Epigenetic modifications induced by blimp-1 regulate CD8+ T cell memory progression during acute virus infection. Immunity 2013, 39, 661–675. [Google Scholar] [CrossRef] [Green Version]
- Thomas, L.R.; Miyashita, H.; Cobb, R.M.; Pierce, S.; Tachibana, M.; Hobeika, E.; Reth, M.; Shinkai, Y.; Oltz, E.M. Functional Analysis of Histone Methyltransferase G9a in B and T Lymphocytes. J. Immunol. 2008, 181, 485–493. [Google Scholar] [CrossRef] [Green Version]
- Nakajima, N.I.; Niimi, A.; Isono, M.; Oike, T.; Sato, H.; Nakano, T.; Shibata, A. Inhibition of the HDAC/Suv39/G9a pathway restores the expression of DNA damage-dependent major histocompatibility complex class I-related chain A and B in cancer cells. Oncol. Rep. 2017, 38, 693–702. [Google Scholar] [CrossRef] [Green Version]
- Pereira, R.M.; Martinez, G.J.; Engel, I.; Cruz-Guilloty, F.; Barboza, B.A.; Tsagaratou, A.; Lio, C.W.J.; Berg, L.J.; Lee, Y.; Kronenberg, M.; et al. Jarid2 is induced by TCR signalling and controls iNKT cell maturation. Nat. Commun. 2014, 5, 4540. [Google Scholar] [CrossRef] [Green Version]
- Kato, S.; Weng, Q.Y.; Insco, M.L.; Chen, K.Y.; Muralidhar, S.; Pozniak, J.; Diaz, J.M.S.; Drier, Y.; Nguyen, N.; Lo, J.A.; et al. Gain-of-function genetic alterations of g9a drive oncogenesis. Cancer Discov. 2020, 10, 980–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Serre, C.; Busuttil, V.; Botto, J.-M. Intrinsic and extrinsic regulation of human skin melanogenesis and pigmentation. Int. J. Cosmet. Sci. 2018, 40, 328–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damsky, W.E.; Curley, D.P.; Santhanakrishnan, M.; Rosenbaum, L.E.; Platt, J.T.; Gould Rothberg, B.E.; Taketo, M.M.; Dankort, D.; Rimm, D.L.; McMahon, M.; et al. β-Catenin Signaling Controls Metastasis in Braf-Activated Pten-Deficient Melanomas. Cancer Cell 2011, 20, 741–754. [Google Scholar] [CrossRef] [Green Version]
- Spranger, S.; Bao, R.; Gajewski, T.F. Melanoma-intrinsic β-catenin signalling prevents anti-tumour immunity. Nature 2015, 523, 231–235. [Google Scholar] [CrossRef]
- Kim, J.T.; Li, J.; Jang, E.R.; Gulhati, P.; Rychahou, P.G.; Napier, D.L.; Wang, C.; Weiss, H.L.; Lee, E.Y.; Anthony, L.; et al. Deregulation of Wnt/β-catenin signaling through genetic or epigenetic alterations in human neuroendocrine tumors. Carcinogenesis 2013, 34, 953–961. [Google Scholar] [CrossRef] [Green Version]
- Bergin, C.J.; Zouggar, A.; Haebe, J.R.; Masibag, A.N.; Desrochers, F.M.; Reilley, S.Y.; Agrawal, G.; Benoit, Y.D. G9a controls pluripotent-like identity and tumor-initiating function in human colorectal cancer. Oncogene 2021, 40, 1191–1202. [Google Scholar] [CrossRef]
- Dang, N.N.; Jiao, J.; Meng, X.; An, Y.; Han, C.; Huang, S. Abnormal overexpression of G9a in melanoma cells promotes cancer progression via upregulation of the Notch1 signaling pathway. Aging 2020, 12, 2393–2407. [Google Scholar] [CrossRef]
- Yang, Z.; Qi, Y.; Lai, N.; Zhang, J.; Chen, Z.; Liu, M.; Zhang, W.; Luo, R.; Kang, S. Notch1 signaling in melanoma cells promoted tumor-induced immunosuppression via upregulation of TGF-β1. J. Exp. Clin. Cancer Res. 2018, 37, 1–13. [Google Scholar] [CrossRef]
- Porcelli, L.; Mazzotta, A.; Garofoli, M.; Di Fonte, R.; Guida, G.; Guida, M.; Tommasi, S.; Azzariti, A. Active notch protects MAPK activated melanoma cell lines from MEK inhibitor cobimetinib. Biomed. Pharmacother. 2021, 133, 111006. [Google Scholar] [CrossRef]
- Golan, T.; Levy, C. Negative regulatory loop between microphthalmia-associated transcription factor (MITF) and notch signaling. Int. J. Mol. Sci. 2019, 20, 576. [Google Scholar] [CrossRef] [Green Version]
- Ma, W.; Han, C.; Zhang, J.; Song, K.; Chen, W.; Kwon, H.; Wu, T. The Histone Methyltransferase G9a Promotes Cholangiocarcinogenesis Through Regulation of the Hippo Pathway Kinase LATS2 and YAP Signaling Pathway. Hepatology 2020, 72, 1283–1297. [Google Scholar] [CrossRef]
- Shao, H.; Moller, M.; Cai, L.; Prokupets, R.; Yang, C.; Costa, C.; Yu, K.; Le, N.; Liu, Z.J. Converting melanoma-associated fibroblasts into a tumor-suppressive phenotype by increasing intracellular Notch1 pathway activity. PLoS ONE 2021, 16, e0248260. [Google Scholar] [CrossRef]
- Du, Y.; Shao, H.; Moller, M.; Prokupets, R.; Tse, Y.T.; Liu, Z.J. Intracellular Notch1 Signaling in Cancer-Associated Fibroblasts Dictates the Plasticity and Stemness of Melanoma Stem/Initiating Cells. Stem Cells 2019, 37, 865–875. [Google Scholar] [CrossRef] [Green Version]
- Azevedo, H.; Pessoa, G.C.; De Luna Vitorino, F.N.; Nsengimana, J.; Newton-Bishop, J.; Reis, E.M.; Da Cunha, J.P.C.; Jasiulionis, M.G. Gene co-expression and histone modification signatures are associated with melanoma progression, epithelial-to-mesenchymal transition, and metastasis. Clin. Epigenetics 2020, 12, 127. [Google Scholar] [CrossRef]
- Maeda, K.; Hamada, J.I.; Takahashi, Y.; Tada, M.; Yamamoto, Y.; Sugihara, T.; Moriuchi, T. Altered expressions of HOX genes in human cutaneous malignant melanoma. Int. J. Cancer 2005, 114, 436–441. [Google Scholar] [CrossRef]
- Li, Q.; Dong, C.; Cui, J.; Wang, Y.; Hong, X. Over-expressed lncRNA HOTAIRM1 promotes tumor growth and invasion through up-regulating HOXA1 and sequestering G9a/EZH2/Dnmts away from the HOXA1 gene in glioblastoma multiforme. J. Exp. Clin. Cancer Res. 2018, 37, 265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Casciello, F.; Al-Ejeh, F.; Miranda, M.; Kelly, G.; Baxter, E.; Windloch, K.; Gannon, F.; Lee, J.S. G9a-mediated repression of CDH10 in hypoxia enhances breast tumour cell motility and associates with poor survival outcome. Theranostics 2020, 10, 4515–4529. [Google Scholar] [CrossRef] [PubMed]
- Kelly, G.M.; Al-Ejeh, F.; McCuaig, R.; Casciello, F.; Kamal, N.A.; Ferguson, B.; Pritchard, A.L.; Ali, S.; Silva, I.P.; Wilmott, J.S.; et al. G9a Inhibition Enhances Checkpoint Inhibitor Blockade Response in Melanoma. Clin. Cancer Res. 2021, 27, 2624–2635. [Google Scholar] [CrossRef] [PubMed]
- Ciechomska, I.A.; Przanowski, P.; Jackl, J.; Wojtas, B.; Kaminska, B. BIX01294, an inhibitor of histone methyltransferase, induces autophagy-dependent differentiation of glioma stem-like cells. Sci. Rep. 2016, 6, 38723. [Google Scholar] [CrossRef] [Green Version]
- Ke, X.X.; Zhang, R.; Zhong, X.; Zhang, L.; Cui, H. Deficiency of G9a Inhibits Cell Proliferation and Activates Autophagy via Transcriptionally Regulating c-Myc Expression in Glioblastoma. Front. Cell Dev. Biol. 2020, 8. [Google Scholar] [CrossRef] [PubMed]
- Artal-Martinez de Narvajas, A.; Gomez, T.S.; Zhang, J.-S.; Mann, A.O.; Taoda, Y.; Gorman, J.A.; Herreros-Villanueva, M.; Gress, T.M.; Ellenrieder, V.; Bujanda, L.; et al. Epigenetic Regulation of Autophagy by the Methyltransferase G9a. Mol. Cell. Biol. 2013, 33, 3983–3993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yin, C.; Ke, X.; Zhang, R.; Hou, J.; Dong, Z.; Wang, F.; Zhang, K.; Zhong, X.; Yang, L.; Cui, H. G9a promotes cell proliferation and suppresses autophagy in gastric cancer by directly activating mTOR. FASEB J. 2019, 33, 14036–14050. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mullard, A. FDA approves an inhibitor of a novel “epigenetic writer”. Nat. Rev. Drug Discov. 2020, 19, 156. [Google Scholar] [CrossRef] [PubMed]
- Varambally, S.; Dhanasekaran, S.M.; Zhou, M.; Barrette, T.R.; Kumar-Sinha, C.; Sanda, M.G.; Ghosh, D.; Pienta, K.J.; Sewalt, R.G.A.B.; Rubin, M.A.; et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002, 419, 624–629. [Google Scholar] [CrossRef]
- Visser, H.P.J.; Gunster, M.J.; Kluin-Nelemans, H.C.; Manders, E.M.M.; Raaphorst, F.M.; Meijer, C.J.L.M.; Willemze, R.; Otte, A.P. The Polycomb group protein EZH2 is upregulated in proliferating, cultured human mantle cell lymphoma. Br. J. Haematol. 2001, 112, 950–958. [Google Scholar] [CrossRef]
- Kleer, C.G.; Cao, Q.; Varambally, S.; Shen, R.; Ota, I.; Tomlins, S.A.; Ghosh, D.; Sewalt, R.G.A.B.; Otte, A.P.; Hayes, D.F.; et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc. Natl. Acad. Sci. USA. 2003, 100, 11606–11611. [Google Scholar] [CrossRef] [Green Version]
- Bitler, B.G.; Aird, K.M.; Garipov, A.; Li, H.; Amatangelo, M.; Kossenkov, A.V.; Schultz, D.C.; Liu, Q.; Shih, I.M.; Conejo-Garcia, J.R.; et al. Synthetic lethality by targeting EZH2 methyltransferase activity in ARID1A-mutated cancers. Nat. Med. 2015, 21, 231–238. [Google Scholar] [CrossRef] [Green Version]
- Ashizawa, M.; Saito, M.; Min, A.K.T.; Ujiie, D.; Saito, K.; Sato, T.; Kikuchi, T.; Okayama, H.; Fujita, S.; Endo, H.; et al. Prognostic role of ARID1A negative expression in gastric cancer. Sci. Rep. 2019, 9, 6769. [Google Scholar] [CrossRef] [Green Version]
- Deogharkar, A.; Singh, S.V.; Bharambe, H.S.; Paul, R.; Moiyadi, A.; Goel, A.; Shetty, P.; Sridhar, E.; Gupta, T.; Jalali, R.; et al. Downregulation of ARID1B, a tumor suppressor in the WNT subgroup medulloblastoma, activates multiple oncogenic signaling pathways. Hum. Mol. Genet. 2021, 30, 1721–1733. [Google Scholar] [CrossRef]
- Helming, K.C.; Wang, X.; Wilson, B.G.; Vazquez, F.; Haswell, J.R.; Manchester, H.E.; Kim, Y.; Kryukov, G.V.; Ghandi, M.; Aguirre, A.J.; et al. ARID1B is a specific vulnerability in ARID1A-mutant cancers. Nat. Med. 2014, 20, 251–254. [Google Scholar] [CrossRef] [Green Version]
- Garczyk, S.; Schneider, U.; Lurje, I.; Becker, K.; Vögeli, T.A.; Gaisa, N.T.; Knüchel, R. ARID1A-deficiency in urothelial bladder cancer: No predictive biomarker for EZH2-inhibitor treatment response? PLoS ONE 2018, 13, e0202965. [Google Scholar] [CrossRef] [PubMed]

Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Flesher, J.L.; Fisher, D.E. G9a: An Emerging Epigenetic Target for Melanoma Therapy. Epigenomes 2021, 5, 23. https://doi.org/10.3390/epigenomes5040023
Flesher JL, Fisher DE. G9a: An Emerging Epigenetic Target for Melanoma Therapy. Epigenomes. 2021; 5(4):23. https://doi.org/10.3390/epigenomes5040023
Chicago/Turabian StyleFlesher, Jessica L., and David E. Fisher. 2021. "G9a: An Emerging Epigenetic Target for Melanoma Therapy" Epigenomes 5, no. 4: 23. https://doi.org/10.3390/epigenomes5040023
APA StyleFlesher, J. L., & Fisher, D. E. (2021). G9a: An Emerging Epigenetic Target for Melanoma Therapy. Epigenomes, 5(4), 23. https://doi.org/10.3390/epigenomes5040023