
Pan HDACi Valproic Acid and Trichostatin A Show Apparently Contrasting Inflammatory Responses in Cultured J774A.1 Macrophages

 , ,
, , {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results
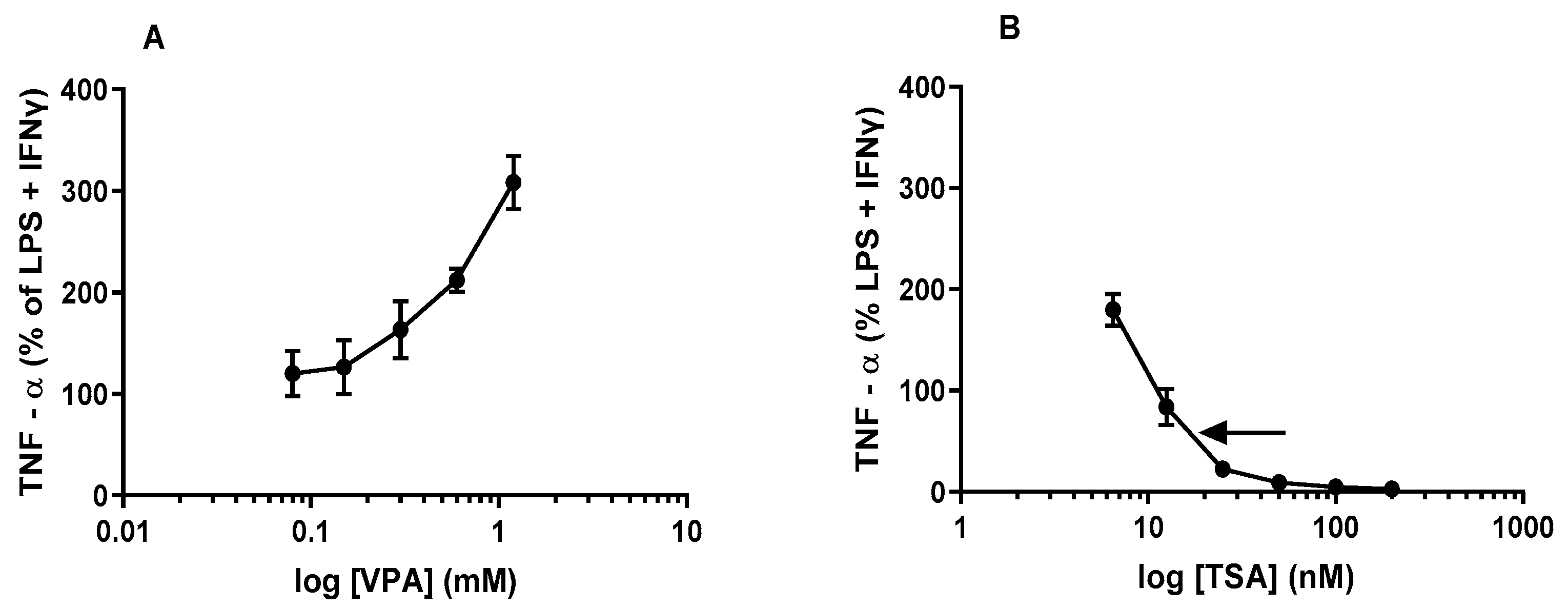
2.1. TNFα Levels Are Differentially Altered by Acute Treatment with HDACis VPA and TSA in Maximally Stimulated J774A.1 Cells
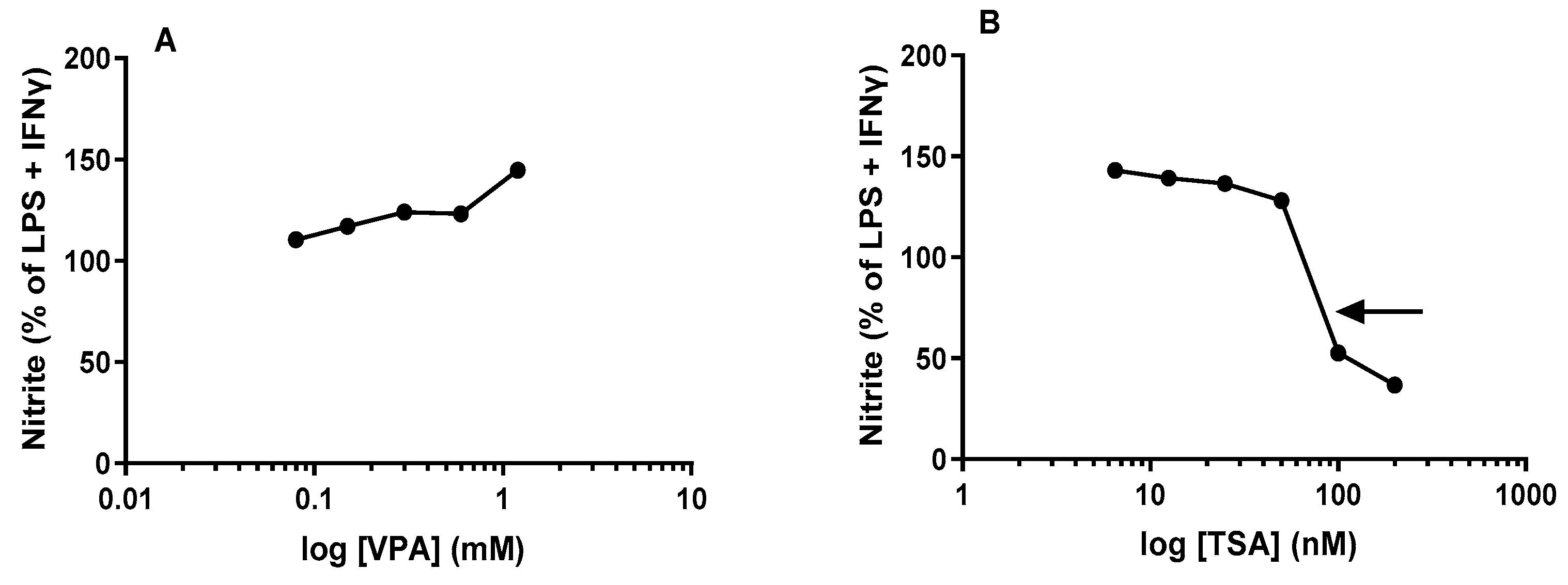
2.2. NO Levels Are Differentially Altered by Acute Treatment with HDACis VPA and TSA in Maximally Stimulated J774A.1 Cells
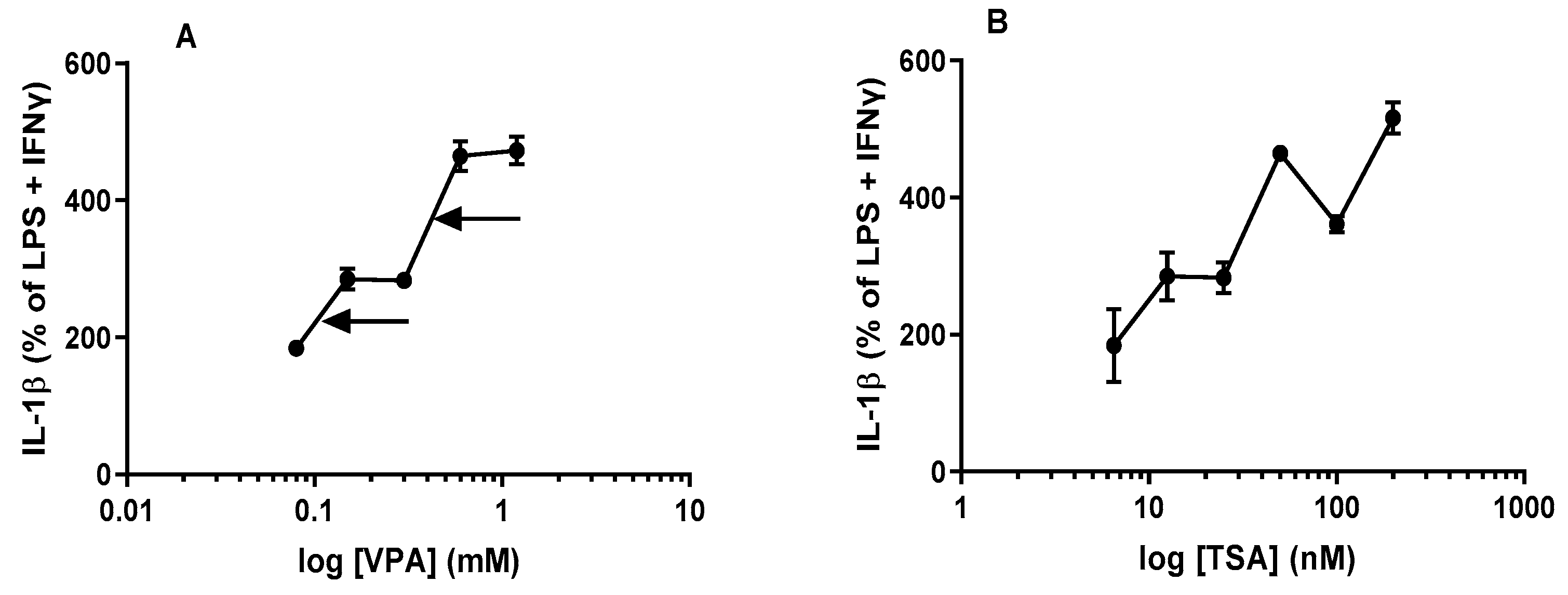
2.3. IL-1β Is Significantly Potentiated by Acute Treatment with HDACis VPA and TSA in Maximally Stimulated J774A.1 Cells
3. Discussion
4. Materials and Methods
4.1. Reagents
4.2. Cell Culture and Treatments
4.3. TNF-Alpha and IL-1βeta ELISAs
4.4. Nitrite Assay
4.5. ED50 and IC50 Calculations
4.6. Statistical Analysis
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Gunawardhana, L.P.; Gibson, P.G.; Simpson, J.L.; Powell, H.; Baines, K.J. Activity and expression of histone acetylases and deacetylases in inflammatory phenotypes of asthma. Clin. Exp. Allergy 2014, 44, 47–57. [Google Scholar] [CrossRef]
- Ito, K.; Caramori, G.; Lim, S.; Oates, T.; Chung, K.F.; Barnes, P.J.; Adcock, I.M. Expression and activity of histone deacetylases in human asthmatic airways. Am. J. Respir. Crit. Care Med. 2002, 166, 392–396. [Google Scholar] [CrossRef]
- Zhang, Z.; Zhang, Z.Y.; Wu, Y.; Schluesener, H.J. Valproic acid ameliorates inflammation in experimental autoimmune encephalomyelitis rats. Neuroscience 2012, 221, 140–150. [Google Scholar] [CrossRef]
- Chen, S.; Ye, J.; Chen, X.; Shi, J.; Wu, W.; Lin, W.; Lin, W.; Li, Y.; Fu, H.; Li, S. Valproic acid attenuates traumatic spinal cord injury-induced inflammation via STAT1 and NF-kappaB pathway dependent of HDAC3. J. Neuroinflamm. 2018, 15, 150. [Google Scholar] [CrossRef]
- Wu, C.; Li, A.; Leng, Y.; Li, Y.; Kang, J. Histone deacetylase inhibition by sodium valproate regulates polarization of macrophage subsets. DNA Cell Biol. 2012, 31, 592–599. [Google Scholar] [CrossRef]
- Chen, P.S.; Wang, C.C.; Bortner, C.D.; Peng, G.S.; Wu, X.; Pang, H.; Lu, R.B.; Gean, P.W.; Chuang, D.M.; Hong, J.S. Valproic acid and other histone deacetylase inhibitors induce microglial apoptosis and attenuate lipopolysaccharide-induced dopaminergic neurotoxicity. Neuroscience 2007, 149, 203–212. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.Y.; Schluesener, H.J. HDAC inhibitor MS-275 attenuates the inflammatory reaction in rat experimental autoimmune prostatitis. Prostate 2012, 72, 90–99. [Google Scholar] [CrossRef]
- Lin, H.S.; Hu, C.Y.; Chan, H.Y.; Liew, Y.Y.; Huang, H.P.; Lepescheux, L.; Bastianelli, E.; Baron, R.; Rawadi, G.; Clement-Lacroix, P. Anti-rheumatic activities of histone deacetylase (HDAC) inhibitors in vivo in collagen-induced arthritis in rodents. Br. J. Pharmacol. 2007, 150, 862–872. [Google Scholar] [CrossRef] [Green Version]
- Choo, Q.Y.; Ho, P.C.; Tanaka, Y.; Lin, H.S. Histone deacetylase inhibitors MS-275 and SAHA induced growth arrest and suppressed lipopolysaccharide-stimulated NF-kappaB p65 nuclear accumulation in human rheumatoid arthritis synovial fibroblastic E11 cells. Rheumatology 2010, 49, 1447–1460. [Google Scholar] [CrossRef]
- Gillespie, J.; Savic, S.; Wong, C.; Hempshall, A.; Inman, M.; Emery, P.; Grigg, R.; McDermott, M.F. Histone deacetylases are dysregulated in rheumatoid arthritis and a novel histone deacetylase 3-selective inhibitor reduces interleukin-6 production by peripheral blood mononuclear cells from rheumatoid arthritis patients. Arthritis Rheum. 2012, 64, 418–422. [Google Scholar] [CrossRef]
- Choi, Y.; Park, S.K.; Kim, H.M.; Kang, J.S.; Yoon, Y.D.; Han, S.B.; Han, J.W.; Yang, J.S.; Han, G. Histone deacetylase inhibitor KBH-A42 inhibits cytokine production in RAW 264.7 macrophage cells and in vivo endotoxemia model. Exp. Mol. Med. 2008, 40, 574–581. [Google Scholar] [CrossRef]
- Morioka, N.; Tomori, M.; Zhang, F.F.; Saeki, M.; Hisaoka-Nakashima, K.; Nakata, Y. Stimulation of nuclear receptor REV-ERBs regulates tumor necrosis factor-induced expression of proinflammatory molecules in C6 astroglial cells. Biochem. Biophys. Res. Commun. 2016, 469, 151–157. [Google Scholar] [CrossRef]
- Leus, N.G.; van der Wouden, P.E.; van den Bosch, T.; Hooghiemstra, W.T.R.; Ourailidou, M.E.; Kistemaker, L.E.; Bischoff, R.; Gosens, R.; Haisma, H.J.; Dekker, F.J. HDAC 3-selective inhibitor RGFP966 demonstrates anti-inflammatory properties in RAW 264.7 macrophages and mouse precision-cut lung slices by attenuating NF-kappaB p65 transcriptional activity. Biochem. Pharmacol. 2016, 108, 58–74. [Google Scholar] [CrossRef] [Green Version]
- Sharif, O.; Bolshakov, V.N.; Raines, S.; Newham, P.; Perkins, N.D. Transcriptional profiling of the LPS induced NF-kappaB response in macrophages. BMC Immunol. 2007, 8, 1. [Google Scholar] [CrossRef] [Green Version]
- Dorrington, M.G.; Fraser, I.D.C. NF-kappaB Signaling in Macrophages: Dynamics, Crosstalk, and Signal Integration. Front. Immunol. 2019, 10, 705. [Google Scholar] [CrossRef]
- Bosisio, D.; Polentarutti, N.; Sironi, M.; Bernasconi, S.; Miyake, K.; Webb, G.R.; Martin, M.U.; Mantovani, A.; Muzio, M. Stimulation of toll-like receptor 4 expression in human mononuclear phagocytes by interferon-gamma: A molecular basis for priming and synergism with bacterial lipopolysaccharide. Blood 2002, 99, 3427–3431. [Google Scholar] [CrossRef] [Green Version]
- Schroder, K.; Hertzog, P.J.; Ravasi, T.; Hume, D.A. Interferon-gamma: An overview of signals, mechanisms and functions. J. Leukoc. Biol. 2004, 75, 163–189. [Google Scholar] [CrossRef] [Green Version]
- Christensen, J.; Gronborg, T.K.; Sorensen, M.J.; Schendel, D.; Parner, E.T.; Pedersen, L.H.; Vestergaard, M. Prenatal valproate exposure and risk of autism spectrum disorders and childhood autism. JAMA 2013, 309, 1696–1703. [Google Scholar] [CrossRef] [Green Version]
- Triyasakorn, K.; Ubah, U.D.B.; Roan, B.; Conlin, M.; Aho, K.; Awale, P.S. The Antiepileptic Drug and Toxic Teratogen Valproic Acid Alters Microglia in an Environmental Mouse Model of Autism. Toxics 2022, 10, 379. [Google Scholar] [CrossRef]
- Agbanoma, G.; Li, C.; Ennis, D.; Palfreeman, A.C.; Williams, L.M.; Brennan, F.M. Production of TNF-alpha in macrophages activated by T cells, compared with lipopolysaccharide, uses distinct IL-10-dependent regulatory mechanism. J. Immunol. 2012, 188, 1307–1317. [Google Scholar] [CrossRef] [Green Version]
- Marletta, M.A. Nitric oxide synthase: Aspects concerning structure and catalysis. Cell 1994, 78, 927–930. [Google Scholar] [CrossRef] [Green Version]
- Moncada, S. Nitric oxide: Discovery and impact on clinical medicine. J. R. Soc. Med. 1999, 92, 164–169. [Google Scholar] [CrossRef] [Green Version]
- Geller, D.A.; Billiar, T.R. Molecular biology of nitric oxide synthases. Cancer Metastasis Rev. 1998, 17, 7–23. [Google Scholar] [CrossRef]
- Taylor, B.S.; Geller, D.A. Molecular regulation of the human inducible nitric oxide synthase (iNOS) gene. Shock 2000, 13, 413–424. [Google Scholar] [CrossRef]
- Heba, G.; Krzeminski, T.; Porc, M.; Grzyb, J.; Dembinska-Kiec, A. Relation between expression of TNF alpha, iNOS, VEGF mRNA and development of heart failure after experimental myocardial infarction in rats. J. Physiol. Pharmacol. 2001, 52, 39–52. [Google Scholar]
- Stuehr, D.J.; Marletta, M.A. Induction of nitrite/nitrate synthesis in murine macrophages by BCG infection, lymphokines, or interferon-gamma. J. Immunol. 1987, 139, 518–525. [Google Scholar]
- Morgan, M.M.; Clayton, C.C.; Heinricher, M.M. Dissociation of hyperalgesia from fever following intracerebroventricular administration of interleukin-1beta in the rat. Brain Res. 2004, 1022, 96–100. [Google Scholar] [CrossRef]
- Suuronen, T.; Huuskonen, J.; Pihlaja, R.; Kyrylenko, S.; Salminen, A. Regulation of microglial inflammatory response by histone deacetylase inhibitors. J. Neurochem. 2003, 87, 407–416. [Google Scholar] [CrossRef] [Green Version]
- Meda, L.; Cassatella, M.A.; Szendrei, G.I.; Otvos, L., Jr.; Baron, P.; Villalba, M.; Ferrari, D.; Rossi, F. Activation of microglial cells by beta-amyloid protein and interferon-gamma. Nature 1995, 374, 647–650. [Google Scholar] [CrossRef]
- Lee, J.Y.; Kim, H.S.; Choi, H.Y.; Oh, T.H.; Ju, B.G.; Yune, T.Y. Valproic acid attenuates blood-spinal cord barrier disruption by inhibiting matrix metalloprotease-9 activity and improves functional recovery after spinal cord injury. J. Neurochem. 2012, 121, 818–829. [Google Scholar] [CrossRef] [PubMed]
- Han, S.B.; Lee, J.K. Anti-inflammatory effect of Trichostatin-A on murine bone marrow-derived macrophages. Arch. Pharm. Res. 2009, 32, 613–624. [Google Scholar] [CrossRef] [PubMed]
- Yu, Z.; Zhang, W.; Kone, B.C. Histone deacetylases augment cytokine induction of the iNOS gene. J. Am. Soc. Nephrol. 2002, 13, 2009–2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Weiss, U.; Moller, M.; Husseini, S.A.; Manderscheid, C.; Hausler, J.; Geisslinger, G.; Niederberger, E. Inhibition of HDAC Enzymes Contributes to Differential Expression of Pro-Inflammatory Proteins in the TLR-4 Signaling Cascade. Int. J. Mol. Sci. 2020, 21, 8943. [Google Scholar] [CrossRef]
- Blanchette, J.; Jaramillo, M.; Olivier, M. Signalling events involved in interferon-gamma-inducible macrophage nitric oxide generation. Immunology 2003, 108, 513–522. [Google Scholar] [CrossRef]
- Yagi, K.; Ishii, M.; Namkoong, H.; Fujii, H.; Asami, T.; Suzuki, S.; Asakura, T.; Mizoguchi, K.; Kamo, T.; Tasaka, S.; et al. Histone Deacetylase Inhibition Protects Mice Against Lethal Postinfluenza Pneumococcal Infection. Crit. Care Med. 2016, 44, e980–e987. [Google Scholar] [CrossRef]
- Andrei, C.; Margiocco, P.; Poggi, A.; Lotti, L.V.; Torrisi, M.R.; Rubartelli, A. Phospholipases C and A2 control lysosome-mediated IL-1 beta secretion: Implications for inflammatory processes. Proc. Natl. Acad. Sci. USA 2004, 101, 9745–9750. [Google Scholar] [CrossRef] [Green Version]
- Andrei, C.; Dazzi, C.; Lotti, L.; Torrisi, M.R.; Chimini, G.; Rubartelli, A. The secretory route of the leaderless protein interleukin 1beta involves exocytosis of endolysosome-related vesicles. Mol. Biol. Cell 1999, 10, 1463–1475. [Google Scholar] [CrossRef]
- Di Virgilio, F.; Chiozzi, P.; Ferrari, D.; Falzoni, S.; Sanz, J.M.; Morelli, A.; Torboli, M.; Bolognesi, G.; Baricordi, O.R. Nucleotide receptors: An emerging family of regulatory molecules in blood cells. Blood 2001, 97, 587–600. [Google Scholar] [CrossRef] [Green Version]
- Carta, S.; Tassi, S.; Semino, C.; Fossati, G.; Mascagni, P.; Dinarello, C.A.; Rubartelli, A. Histone deacetylase inhibitors prevent exocytosis of interleukin-1beta-containing secretory lysosomes: Role of microtubules. Blood 2006, 108, 1618–1626. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Y.; Li, N.; Caron, C.; Matthias, G.; Hess, D.; Khochbin, S.; Matthias, P. HDAC-6 interacts with and deacetylates tubulin and microtubules in vivo. EMBO J. 2003, 22, 1168–1179. [Google Scholar] [CrossRef] [Green Version]
- Hubbert, C.; Guardiola, A.; Shao, R.; Kawaguchi, Y.; Ito, A.; Nixon, A.; Yoshida, M.; Wang, X.F.; Yao, T.P. HDAC6 is a microtubule-associated deacetylase. Nature 2002, 417, 455–458. [Google Scholar] [CrossRef]
- North, B.J.; Marshall, B.L.; Borra, M.T.; Denu, J.M.; Verdin, E. The human Sir2 ortholog, SIRT2, is an NAD+-dependent tubulin deacetylase. Mol. Cell 2003, 11, 437–444. [Google Scholar] [CrossRef]
- Chi, Z.; Chen, S.; Xu, T.; Zhen, W.; Yu, W.; Jiang, D.; Guo, X.; Wang, Z.; Zhang, K.; Li, M.; et al. Histone Deacetylase 3 Couples Mitochondria to Drive IL-1beta-Dependent Inflammation by Configuring Fatty Acid Oxidation. Mol. Cell 2020, 80, 43–58.e7. [Google Scholar] [CrossRef]
- Deverman, B.E.; Patterson, P.H. Cytokines and CNS development. Neuron 2009, 64, 61–78. [Google Scholar] [CrossRef] [Green Version]
- Meltzer, A.; Van de Water, J. The Role of the Immune System in Autism Spectrum Disorder. Neuropsychopharmacology 2017, 42, 284–298. [Google Scholar] [CrossRef] [Green Version]
- Gassowska-Dobrowolska, M.; Cieslik, M.; Czapski, G.A.; Jesko, H.; Frontczak-Baniewicz, M.; Gewartowska, M.; Dominiak, A.; Polowy, R.; Filipkowski, R.K.; Babiec, L.; et al. Prenatal Exposure to Valproic Acid Affects Microglia and Synaptic Ultrastructure in a Brain-Region-Specific Manner in Young-Adult Male Rats: Relevance to Autism Spectrum Disorders. Int. J. Mol. Sci. 2020, 21, 3576. [Google Scholar] [CrossRef]
- Nau, H. Teratogenic valproic acid concentrations: Infusion by implanted minipumps vs conventional injection regimen in the mouse. Toxicol. Appl. Pharmacol. 1985, 80, 243–250. [Google Scholar] [CrossRef]
- Anderton, H.; Wicks, I.P.; Silke, J. Cell death in chronic inflammation: Breaking the cycle to treat rheumatic disease. Nat. Rev. Rheumatol. 2020, 16, 496–513. [Google Scholar] [CrossRef]
- Chen, P.S.; Peng, G.S.; Li, G.; Yang, S.; Wu, X.; Wang, C.C.; Wilson, B.; Lu, R.B.; Gean, P.W.; Chuang, D.M.; et al. Valproate protects dopaminergic neurons in midbrain neuron/glia cultures by stimulating the release of neurotrophic factors from astrocytes. Mol. Psychiatry 2006, 11, 1116–1125. [Google Scholar] [CrossRef] [Green Version]
- Dragunow, M.; Greenwood, J.M.; Cameron, R.E.; Narayan, P.J.; O’Carroll, S.J.; Pearson, A.G.; Gibbons, H.M. Valproic acid induces caspase 3-mediated apoptosis in microglial cells. Neuroscience 2006, 140, 1149–1156. [Google Scholar] [CrossRef]
- Silva, M.F.B.; Aires, C.C.P.; Luis, P.B.M.; Ruiter, J.P.N.; Ijlst, L.; Duran, M.; Wanders, R.J.A.; de Almeida, I.T. Valproic acid metabolism and its effects on mitochondrial fatty acid oxidation: A review. J. Inherit. Metab. Dis. 2008, 31, 205–216. [Google Scholar] [CrossRef]
- Carroll, R.T.; Galatsis, P.; Borosky, S.; Kopec, K.K.; Kumar, V.; Althaus, J.S.; Hall, E.D. 4-Hydroxy-2,2,6,6-tetramethylpiperidine-1-oxyl (Tempol) inhibits peroxynitrite-mediated phenol nitration. Chem. Res. Toxicol. 2000, 13, 294–300. [Google Scholar] [CrossRef]





Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ubah, U.D.B.; Triyasakorn, K.; Roan, B.; Conlin, M.; Lai, J.C.K.; Awale, P.S. Pan HDACi Valproic Acid and Trichostatin A Show Apparently Contrasting Inflammatory Responses in Cultured J774A.1 Macrophages. Epigenomes 2022, 6, 38. https://doi.org/10.3390/epigenomes6040038
Ubah UDB, Triyasakorn K, Roan B, Conlin M, Lai JCK, Awale PS. Pan HDACi Valproic Acid and Trichostatin A Show Apparently Contrasting Inflammatory Responses in Cultured J774A.1 Macrophages. Epigenomes. 2022; 6(4):38. https://doi.org/10.3390/epigenomes6040038
Chicago/Turabian StyleUbah, Ubah Dominic Babah, Korawin Triyasakorn, Brandon Roan, Minsyusheen Conlin, James C. K. Lai, and Prabha S. Awale. 2022. "Pan HDACi Valproic Acid and Trichostatin A Show Apparently Contrasting Inflammatory Responses in Cultured J774A.1 Macrophages" Epigenomes 6, no. 4: 38. https://doi.org/10.3390/epigenomes6040038
APA StyleUbah, U. D. B., Triyasakorn, K., Roan, B., Conlin, M., Lai, J. C. K., & Awale, P. S. (2022). Pan HDACi Valproic Acid and Trichostatin A Show Apparently Contrasting Inflammatory Responses in Cultured J774A.1 Macrophages. Epigenomes, 6(4), 38. https://doi.org/10.3390/epigenomes6040038