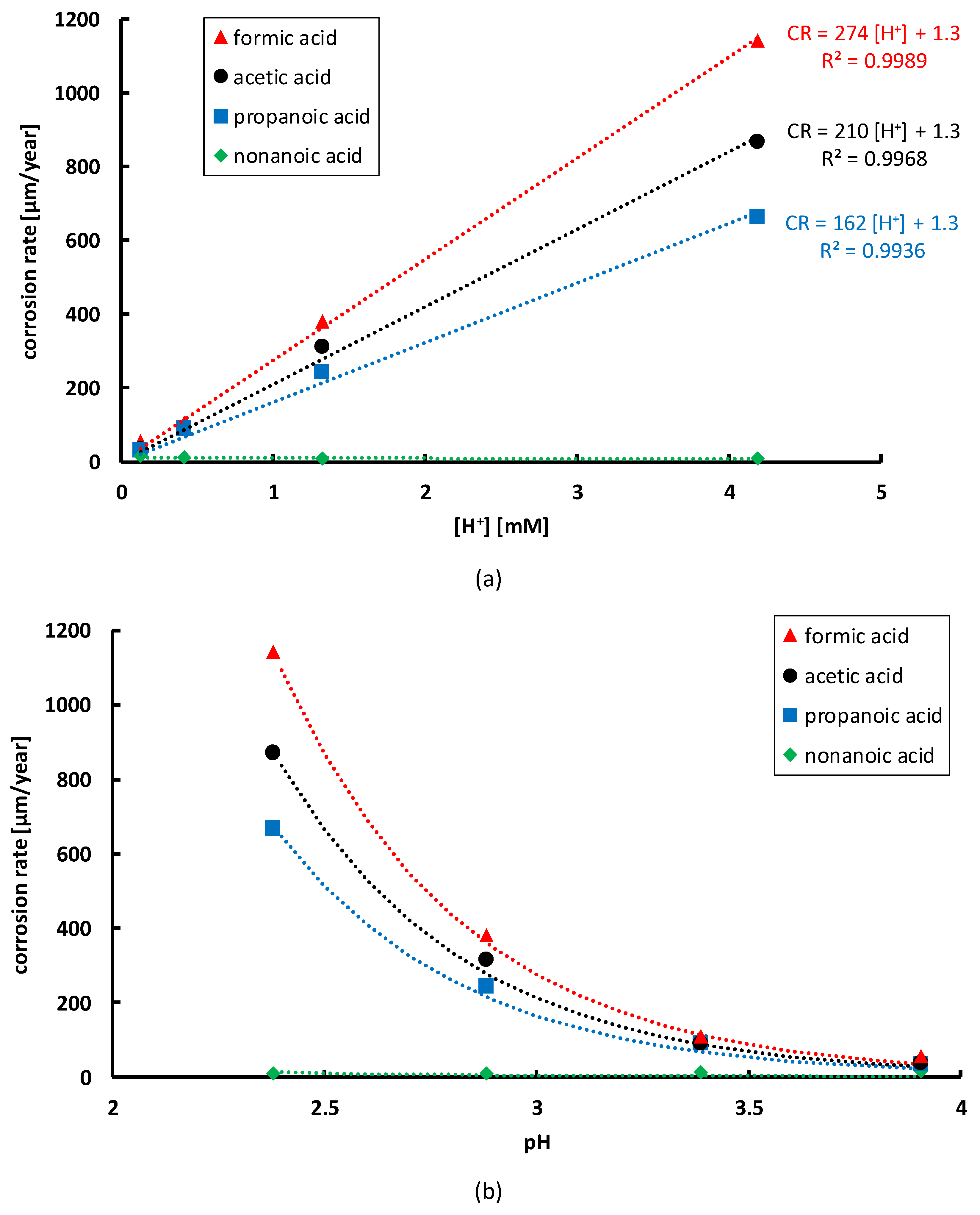
3.2. Influence of Organic Acid Concentration
To investigate the effect of the organic acid concentration on the corrosion rate of NiCrMoV steam turbine steel in the different acids, the corrosion rates are plotted versus the hydrogen proton concentration in
Figure 8a, as well as versus the pH in
Figure 8b. The relation between the organic acid concentration and the hydrogen proton concentration is given by Equation (14), whereas the link with pH is given by Equation (13). Linear relationships between corrosion rate
(µm/year) and hydrogen proton concentration
(mM), according to Equation (18), can be identified [
49,
56,
57].
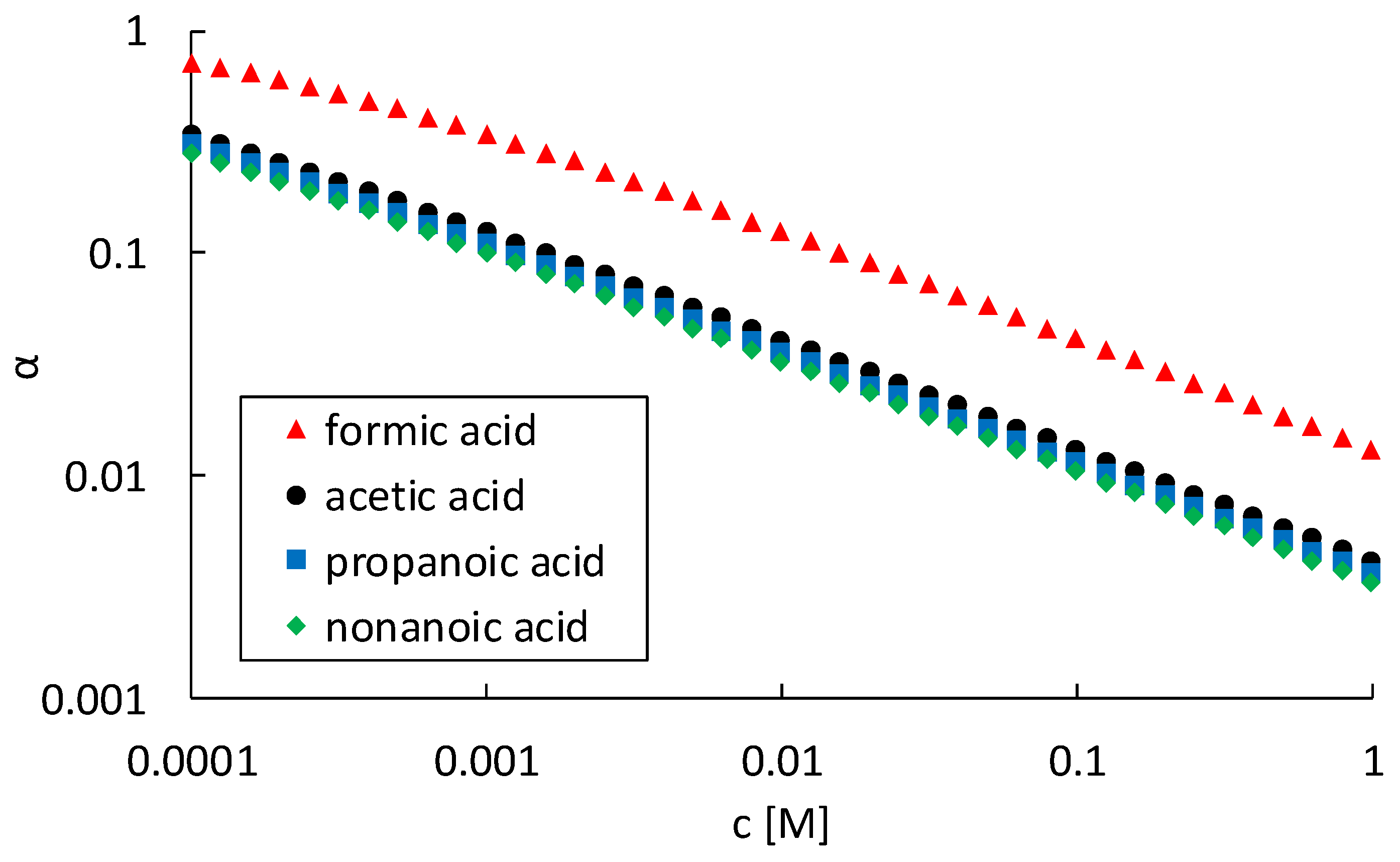
As can be seen in
Figure 8a, the steepest increase in corrosion rate with increasing hydrogen proton concentration is observed for formic acid, followed by acetic and propanoic acid. For nonanoic acid, no increase in corrosion rate was observed. Moreover, even a small decrease in corrosion rate with increasing concentrations was measured for nonanoic acid, as can be seen in
Figure 7. Therefore, no trend line for nonanoic acid is given in
Figure 8. When combining Equations (13) and (18), the corrosion rate can be written as a function of the pH, according to Equation (19).
This correlation is visualised in
Figure 8b. Lowering the pH has a large influence on the corrosion rate of the NiCrMoV steel and it is clear that this influence is dependent on the type of organic acid.
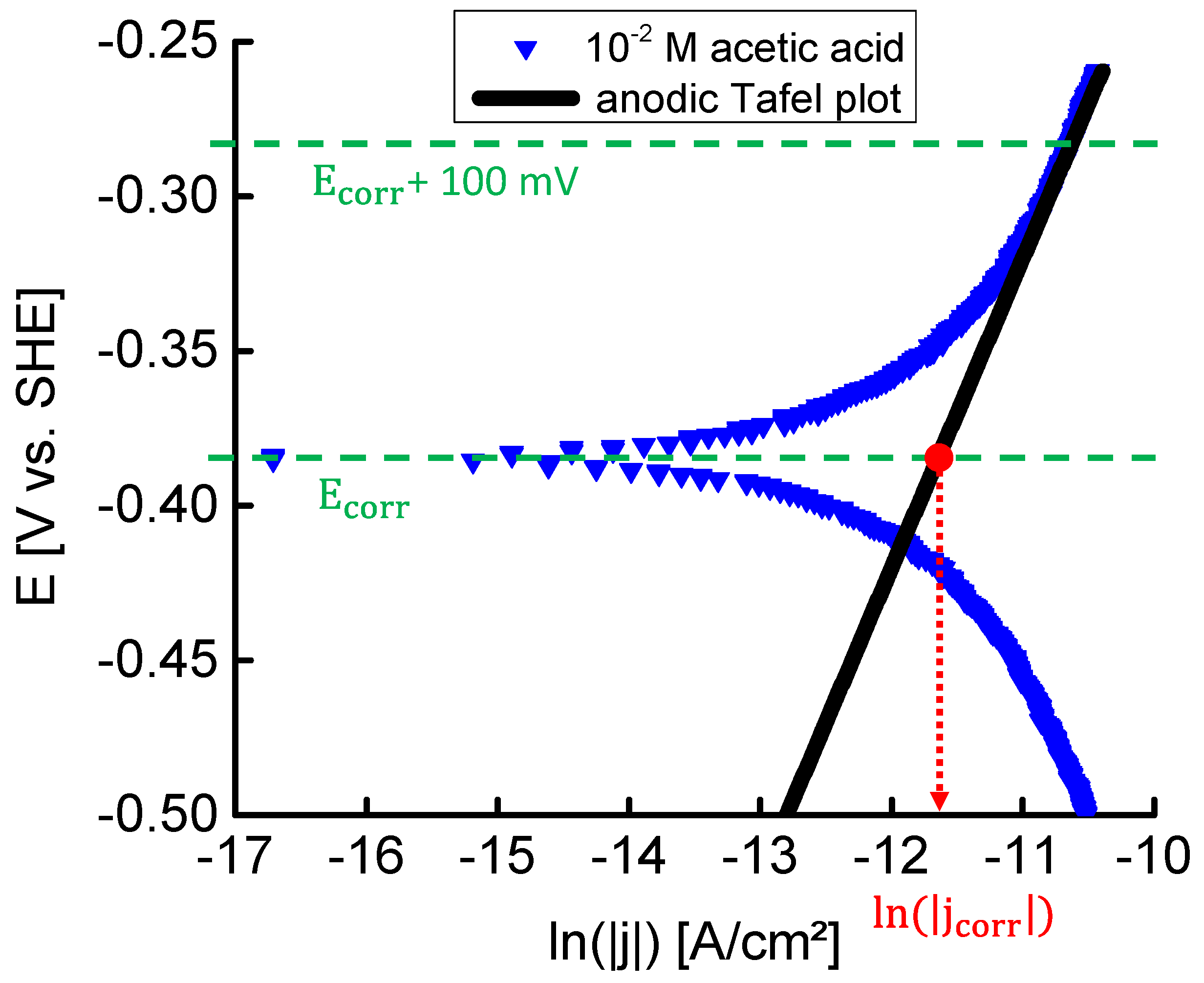
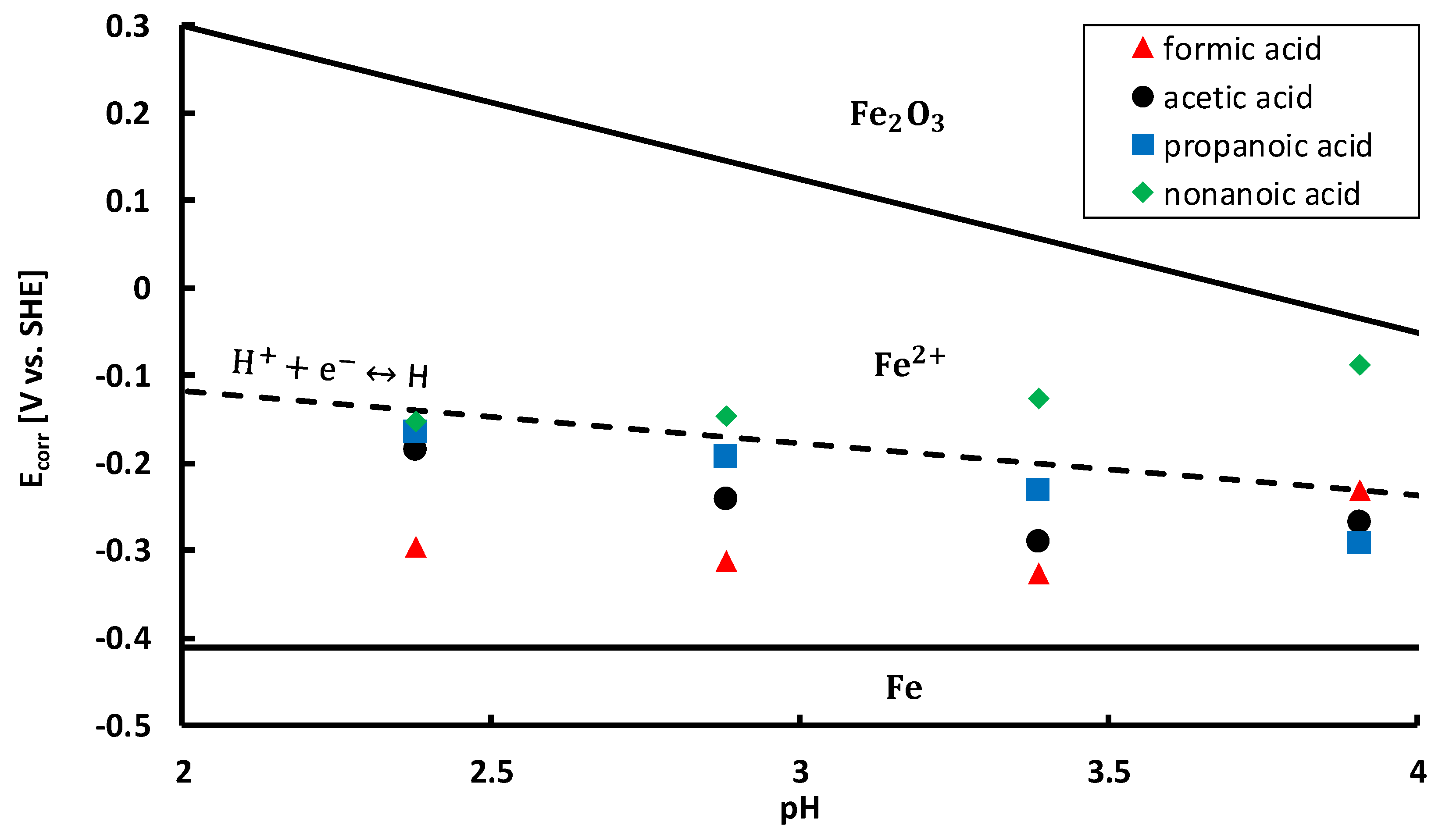
To gain more insight in the active corrosion mechanisms, corrosion potentials
(V) were measured and plotted on a Pourbaix diagram [
63,
64,
65,
66,
67,
68,
69]. Corrosion potentials of the acidic aqueous solutions, given in
Table 5, were determined from the polarisation curves and included on a Pourbaix diagram in
Figure 9. The Pourbaix diagram was constructed based on thermodynamic data from FactSage
©. The diagram was calculated for iron at 25 °C with an iron molality of 1. Thermodynamically stable forms of iron are delimited by solid black lines. All corrosion potentials are situated in the
region, i.e., above the iron oxidation reaction line, cf. Equation (20).
As such, anodic dissolution of the steel takes place in all conditions. The anodic reaction is coupled with at least one reduction reaction in a redox system. All corrosion potentials are situated below the oxygen reduction reaction line, cf. Equation (21).
This line is not shown in
Figure 9, since it is situated at higher potentials, according to the Nernst equation for the oxygen reduction reaction at 25 °C, cf. Equation (22),
where,
(V vs. SHE) is the equilibrium potential. Since all corrosion potentials are situated below the oxygen reduction reaction line, the reduction of oxygen takes place in all tested conditions. Furthermore, almost all corrosion potentials, except for most of the nonanoic acid solutions, are also located below the hydrogen proton reduction reaction line, cf. Equation (23) (Volmer reaction) [
65,
66].
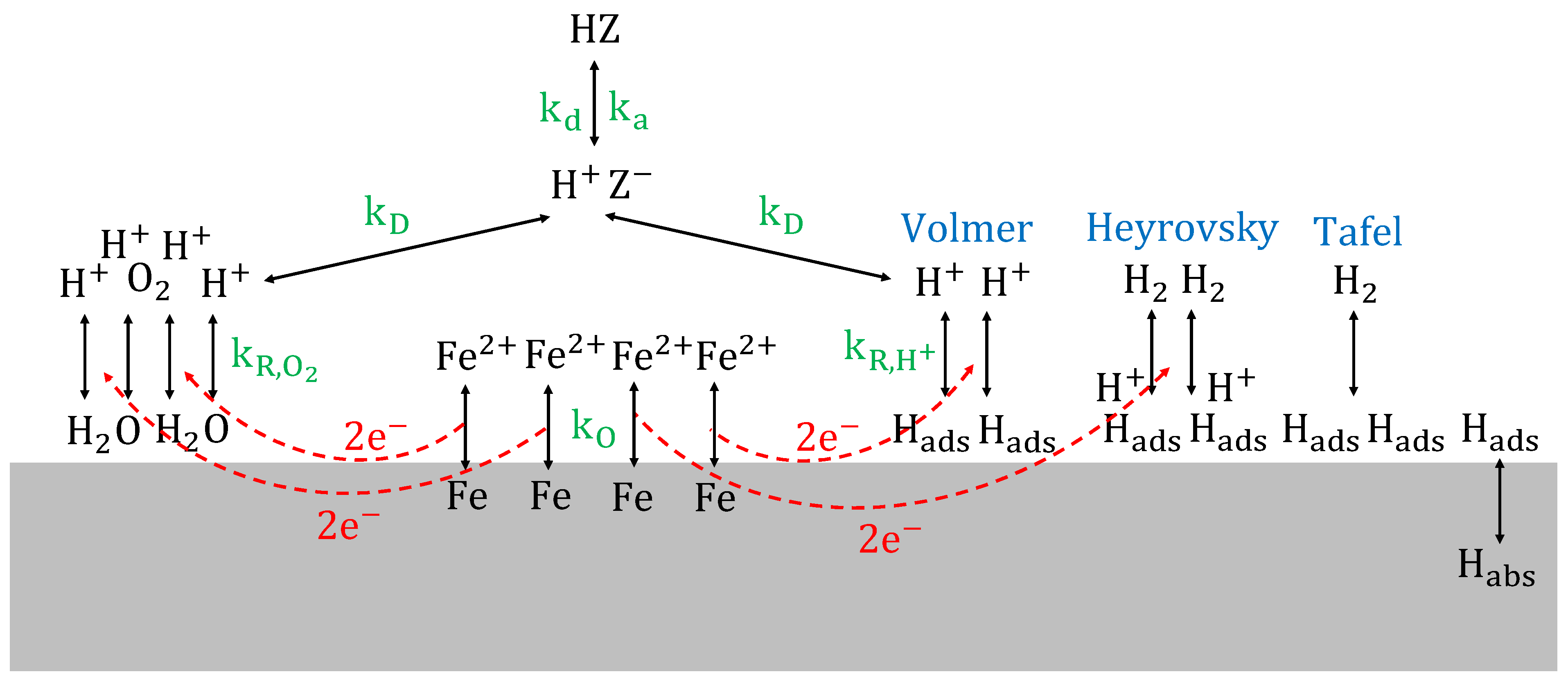
This means that in almost every situation, protons are reduced to atomic hydrogen, as a second reduction reaction in the redox system. Hydrogen atoms adsorbed on the metal surface can recombine by chemical desorption (Tafel reaction), cf. Equation (24), recombine by electrochemical desorption (Heyrovsky reaction), cf. Equation (25), or absorb into the steel, cf. Equation (26) [
65,
66].
Hydrogen absorption into steel, followed by hydrogen diffusion through the lattice, can cause hydrogen embrittlement [
70,
71,
72,
73]. Based on the positioning of the corrosion potentials in
Figure 9 and the corrosion rates in
Figure 7, the most significant hydrogen embrittlement would be expected in formic acid, followed by acetic acid, propanoic acid and nonanoic acid. Moreover, in most conditions of nonanoic acid, no hydrogen embrittlement of the steam turbine steel would be expected.
Knowing that protons participate in the reduction reactions of the corrosion process, cf. Equations (21) and (23), the linear correlation between corrosion rate and hydrogen proton concentration, cf. Equation (18) and
Figure 8a, can be substantiated with formulas. According to Faraday’s law, cf. Equation (16), the corrosion rate is proportional to the corrosion current density
(A/m²). The current density
(A/m²) is related to the reaction rate
(mol/m²s) via Equation (27).
Furthermore, the rate equation for an electrochemical reaction can be written as Equation (28),
where,
(m/s) and
(m/s) are the rate constants for oxidation and reduction, respectively, whereas
(M) and
(M) are the concentrations of reductant and oxidant at the electrode surface, respectively. Therefore, concentrations of reacting species have a large influence on the reaction rate
, current density
, as well as on the corrosion rate. Although the kinetics of the oxygen reduction reaction and hydrogen proton reduction reaction are different, the hydrogen proton concentration plays an important role in both reactions.
The kinetics of the oxygen reduction reaction in these conditions are determined by mass transfer [
59,
74]. The limiting current density
(A/m²) can be written as Equation (29),
where,
is the concentration of oxidant in the bulk of the aqueous solution and
(m/s) is the mass transfer coefficient, equal to Equation (30),
where,
(m) is the thickness of the diffusion layer and
(m²/s) is the diffusion coefficient of the reacting species, according to Equation (31),
where,
(m²/s) is a temperature-independent pre-exponential and
(J/mol) is the activation energy for diffusion. The kinetics of the oxygen reduction reaction, cf. Equation (21), can be limited by oxygen diffusion, but also by hydrogen proton diffusion [
75,
76]. In the latter case, the limiting current density increases when increasing the hydrogen proton concentration in the bulk solution, cf. Equation (29).
In contrast, the kinetics of the hydrogen proton reduction reaction are determined by the charge transfer rate. For this situation, the current density can be written according to the Butler–Volmer equation, cf. Equation (32),
where,
and
are the anodic and cathodic charge transfer coefficients, respectively;
(m/s) and
(m/s) are equilibrium rate constants for oxidation and reduction, respectively. The reaction rate constants
(m/s) typically follow an Arrhenius equation, cf. Equation (33),
where,
(m/s) is a pre-exponential factor and
(J/mol) is the activation energy for the reaction. Note the similarity of Equation (31) and Equation (33). The concentration of oxidant
in the Butler–Volmer Equation of Equation (32) can be identified with the hydrogen proton concentration in the hydrogen proton reduction reaction (Volmer reaction), cf. Equation (23). As such, increasing the hydrogen proton concentration leads to an increase in current density. In addition, as can be seen in
Figure 9, it must be taken into account that also the equilibrium potential
of the hydrogen proton reduction reaction is dependent on the hydrogen proton concentration, cf. Equation (13) and the Nernst equation of the hydrogen proton reduction reaction at 25 °C, given in Equation (34) [
49].
The concentration dependent equilibrium potential is present in the Butler–Volmer equation of Equation (32) since the potential (V) is equal to (V), with (V) the overpotential.
Therefore, when increasing the hydrogen proton concentration, the current densities of both the oxygen reduction reaction and the hydrogen proton reduction reaction, according to Equations (29) and (32), respectively, roughly increase linearly. When taking into account Faraday’s law, cf. Equation (16), this also leads to a linear increase in corrosion rate with increasing hydrogen proton concentration, as such explaining the linear correlation observed in
Figure 8.
3.3. Influence of Organic Acid Type
Another remarkable observation is the difference in corrosion rate depending on the type of organic acid, for solutions with the same pH, as can be seen in
Figure 7 and
Figure 8. The rate of metal dissolution is determined by the kinetics of the reduction reactions. Based on the location of the corrosion potentials, cf.
Figure 9, protons are consumed by both reduction reactions, cf. Equations (21) and (23). When protons are consumed, new protons are produced by organic acid dissociation, according to the principle of Le Chatelier. Hence, protons are continuously consumed and produced during the corrosion process. Therefore, the rate of acid dissociation, which depends on the type of organic acid, plays an important role. The equilibrium dissociation constant
can be written as the quotient of the dissociation rate constant
(s
−1) and the association rate constant
(M
−1 s
−1), cf. Equation (35).
Values of
and
for formic, acetic and propanoic acid are given in
Table 7 [
77,
78,
79]. It can be seen that a larger equilibrium dissociation constant
corresponds with a larger dissociation rate constant
for these organic acids. The organic acid with the largest dissociation rate constant
, i.e., formic acid, can produce protons the fastest, which means that protons are again the earliest available for the reduction reactions, as such explaining the larger corrosion rate in formic acid, followed by acetic acid and propanoic acid, cf.
Figure 7 and
Figure 8. Although the solutions of different types of organic acids were prepared in such a way that their concentrations were balanced taking into account their different equilibrium dissociation constants, cf. Equation (14), as such incorporating thermodynamics, their difference in kinetics of acid dissociation still has a significant impact on the corrosion process. In
Figure 10, the acid dissociation reaction, acting as the rate determining step in the corrosion of steam turbine steel in organic acid solutions, is schematically represented.
The influence of the dissociation constant
of the different organic acids on the overall kinetics of the corrosion process can be represented in a similar way as the Brønsted catalysis equation. The Brønsted catalysis equation constitutes the first linear free-energy relationship, i.e., Gibbs energy relation [
80]. It gives a relationship between acid strength (dissociation constant
) and catalytic activity (reaction rate constant
) in general acid catalysis, cf. Equation (36) [
81,
82],
where,
is the slope and
is the intercept. This equation implies that the strongest acids are the most effective in producing protons, as such contributing the most to reaction rate acceleration. In this work, the reaction rate constant
of Equation (36) can be represented by an Arrhenius relation, cf. Equation (37),
where,
(m/s),
(m/s) and
(J/mol) are the reaction rate constant, the pre-exponential factor and the activation energy of the total reaction, respectively. When combining Equations (2), (36) and (37), it can be seen that the Gibbs free energy for proton dissociation
is proportional to the activation energy of the overall reaction
, as such resulting in a relationship according to the Evans–Polanyi principle, cf. Equation (38) [
83,
84],
where,
is the slope and
is the intercept. Therefore, a larger (i.e., more negative) Gibbs free energy for proton dissociation
, i.e., favoured thermodynamics, corresponds with a decrease in activation energy
, i.e., enhanced kinetics, which is in accordance with the experimental observations for the different organic acids, as such confirming
Figure 7 and
Figure 8.
Furthermore, the hydrocarbon chains of the organic acids can also influence the corrosion behaviour. Especially for nonanoic acid a lot of long chains were present in the test solutions due to its large molar mass, cf.
Table 2, and its small dissociation constant, cf.
Table 3, as such requiring large concentrations of nonanoic acid to obtain a certain low pH, according to Equations (13) and (14). Nonanoic acid, also known as pelargonic acid, has been reported to form an emulsion in water [
85]. Without emulsifiers, such as surfactants, an unstable nonanoic acid emulsion progressively separates, and hydrocarbon chains can gather at the metal surface. Increased van der Waals interactions between long hydrocarbon chains and the charge density of the surface film can induce a change in local pH at the surface compared to bulk values [
86,
87,
88]. As a result of the gathering at the metal surface, hydrocarbon chains can block the surface active sites for corrosion, as such acting as a corrosion inhibitor [
89,
90,
91,
92]. This can explain the low corrosion rates observed in
Figure 7 and
Figure 8. Moreover, when lowering the pH of the nonanoic solution, a decrease in corrosion rate can be noticed. When decreasing the pH, the effect becomes more pronounced due to the increasing nonanoic acid concentration, as such even more shielding the metal surface from its environment.
It must be mentioned that the oxygen concentration in the first condensate of steam–water cycles is much lower than in the tested solutions because of the partitioning of oxygen between the steam and liquid phases in the low-pressure steam turbine section [
2]. The International Association for the Properties of Water and Steam (IAPWS) [
2] reported that oxygen levels in the first condensate might be lower than 1 ppb for steam inlet oxygen levels of 150 ppb. However, the solutions were not deaerated in this study. The main goal of this study was to evaluate whether, next to the role of the concentration of organic acid, the type of organic acid also matters in terms of corrosion behaviour for steam turbine steel in an acidic aqueous environment with a certain pH. To investigate this, no deaeration of the solutions was required. Nevertheless, electrochemical measurements in deaerated conditions are recommended for more industrially relevant quantitative corrosion rates.
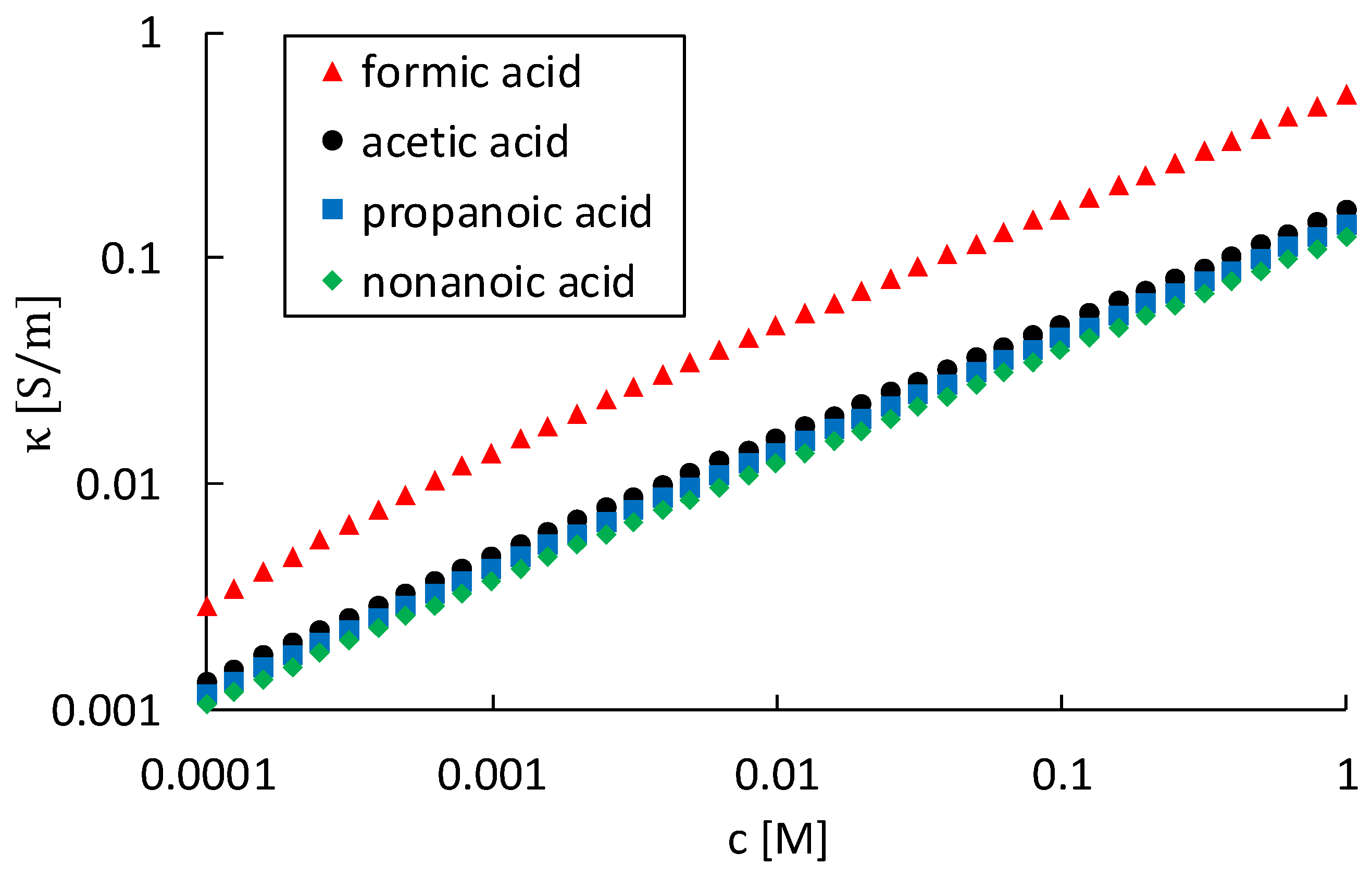
Conclusively, the experiments were designed to compare solutions of organic acids with the same pH, i.e., similar conductivity, according to
Table 5. Based on the findings in this work, it can be stated that using (cation) conductivity as a guide value for general corrosion behaviour in steam–water cycles is only partially correct, cf. the increase in corrosion rate when increasing the organic acid concentration, cf.
Figure 7 and
Figure 8. However, based on these graphs, it can be seen that it is also important to know the type(s) of organic acids present in the aqueous solution, especially at low pH, i.e., at high organic acid concentrations. Therefore, drawing conclusions for corrosion monitoring on (cation) conductivity measurements alone fall short to some extent. By only measuring the conductivity, no information is gained on the composition of the organic acids, thus lacking information about the active dissociation kinetics, which play a crucial role. An inherent problem in practice is that organic acids with long chains are less stable at high temperatures, as such leading to a shift towards small, more corrosive, organic acids [
40]. Unfortunately, knowing the exact composition of the aqueous environment is not at all straightforward in industry. Nevertheless, screening which compounds are detrimental on a fundamental level, combined with goal-oriented measurements of their concentrations, seems feasible. In this way, industrial guidelines should be re-assessed, and appropriate water treatments can be designed, as such aiming for a better corrosion prevention and control in steam–water cycles and beyond.


 ,
, 















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}