The Impact of Pt Concentration on Crystal Growth Mechanism in Pt-Pd Binary Alloy System in the Context of Molecular Dynamics

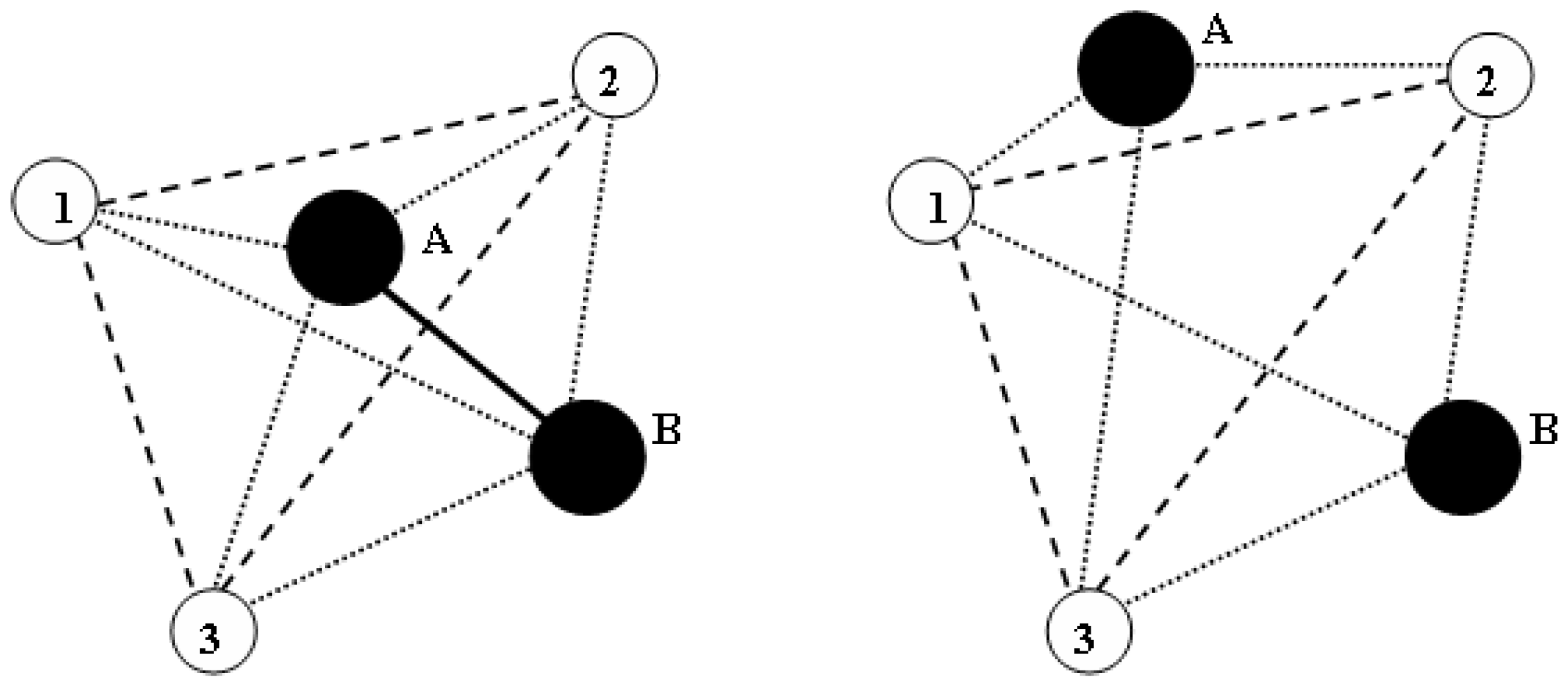{kind=link}
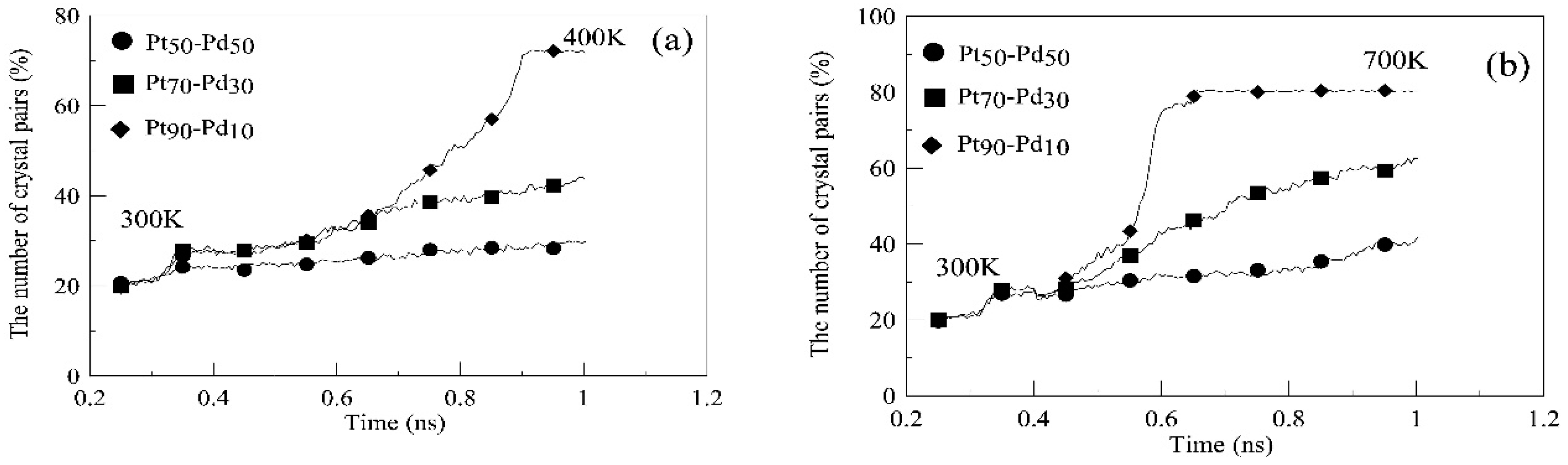{kind=link}
{kind=link}
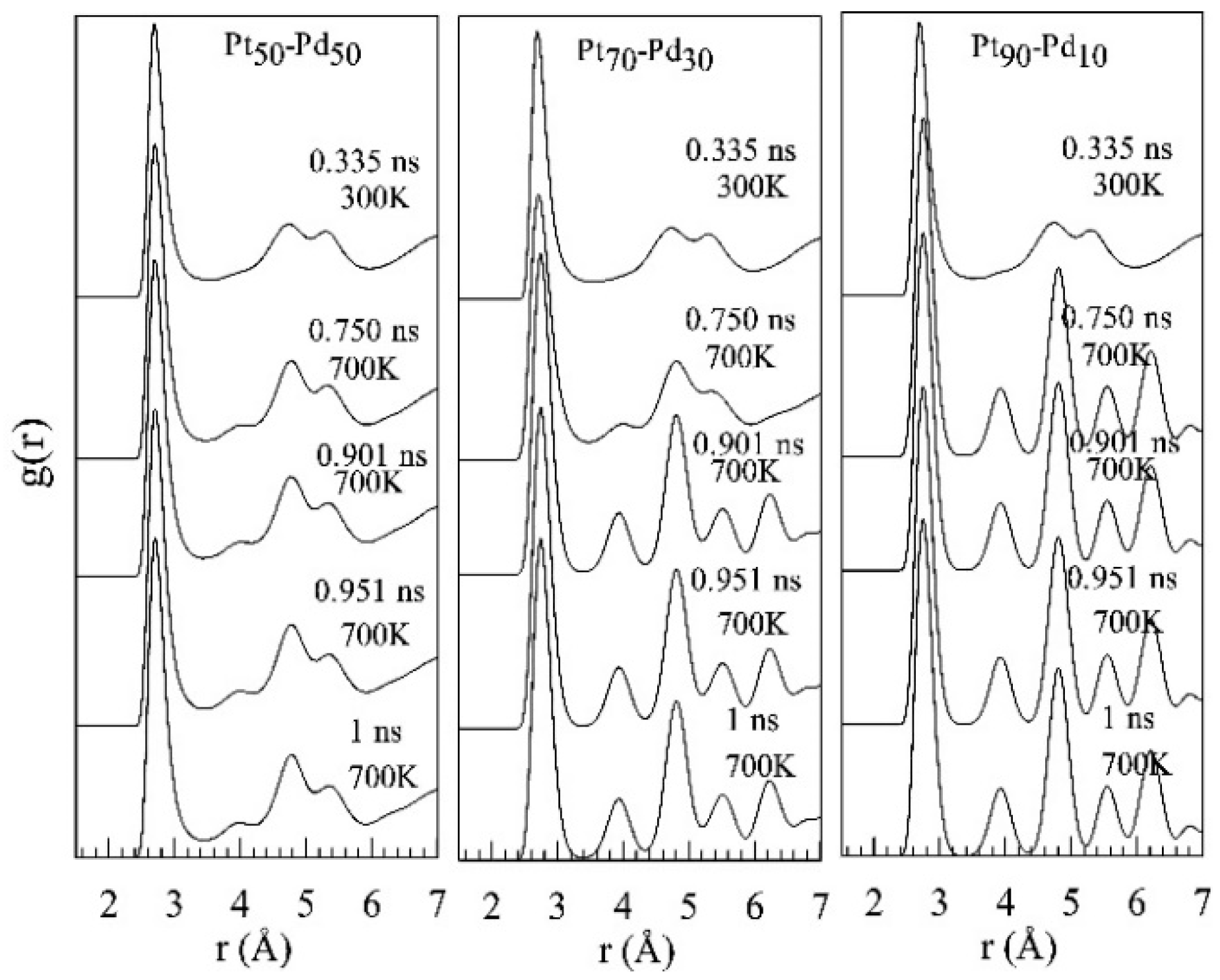{kind=link}
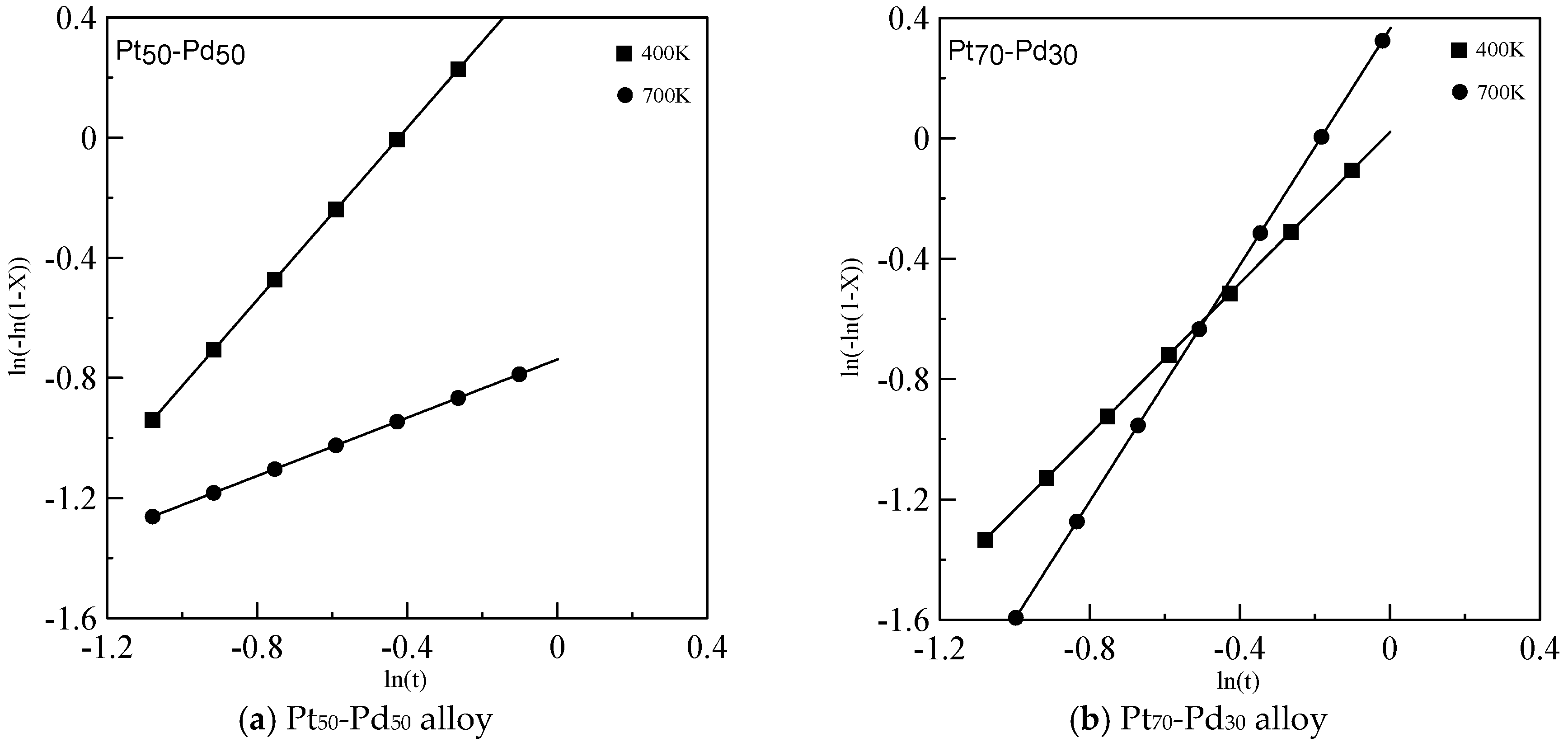{kind=link}
{kind=link}
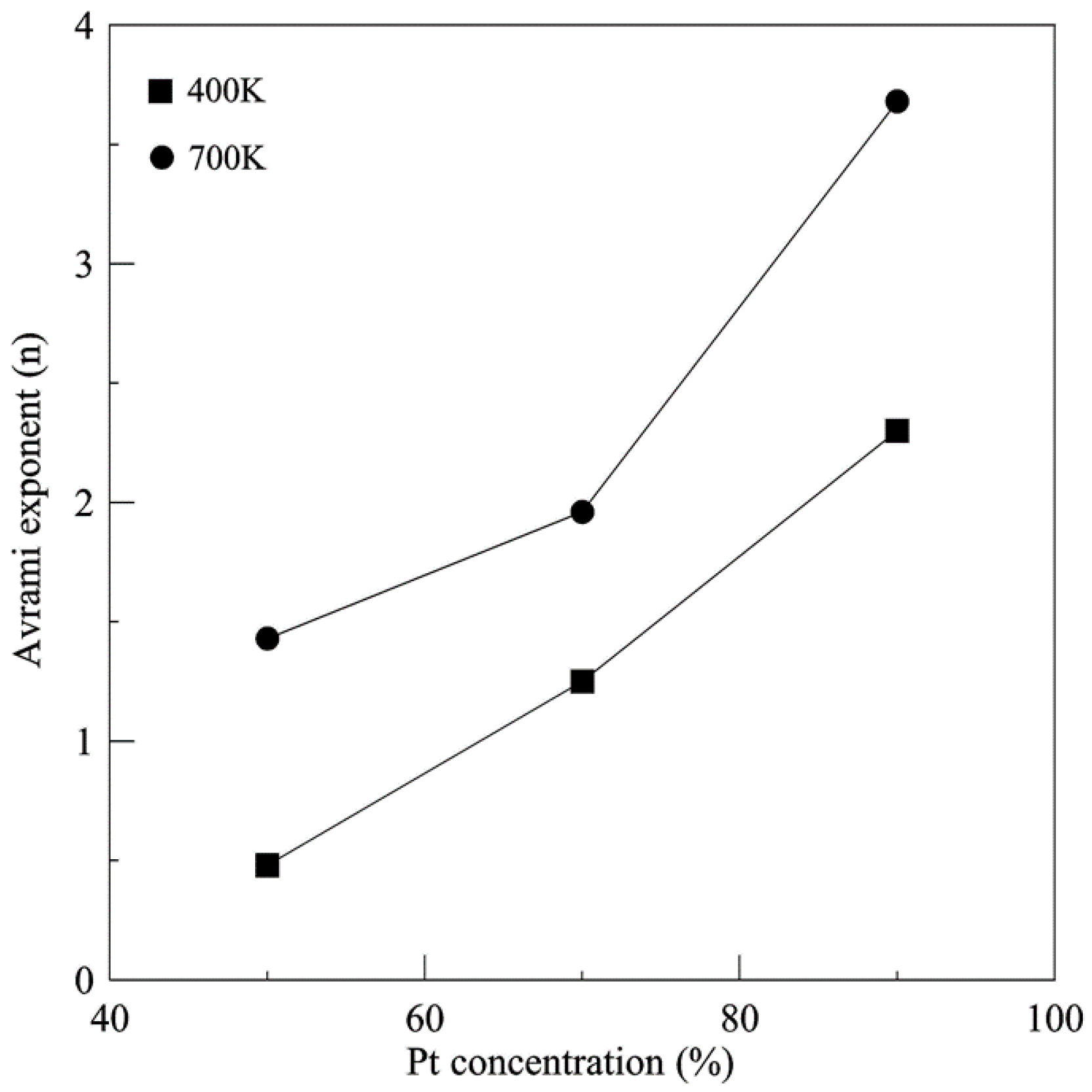{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Theory
2.1. Molecular Dynamic Simulation
2.2. Embedded Atom Method (EAM)
2.3. Honeycutt-Andersen Method (HA)
2.4. Johnson-Mehl-Avrami (JMA) Crystallization Kinetics
3. Results and Discussion
4. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Rdzawski, Z.M.; Stobrawa, J.P. Microstructure and properties of the new Pt-Rh based alloys for hightemperature applications. J. Mater. Process. Technol. 2004, 153, 681–687. [Google Scholar] [CrossRef]
- Ren, D.; Qin, J.; Wang, J.; Tsong, T.T. Oscillatory compositional depth profiles in surface segregation of a Pt-Rh alloy. Phys. Rev. B 1993, 47, 3944. [Google Scholar] [CrossRef]
- Clay, J.P.; Greeley, J.P.; Ribeiro, F.H.; Delgass, W.N.; Schneider, W.F. DFT comparison of intrinsic WGS kinetics over Pd and Pt. J. Catal. 2014, 320, 106–117. [Google Scholar] [CrossRef]
- Wang, W.; Wang, Z.; Wang, J.; Zhong, C.-J.; Liu, C.-J. Highly active and stable Pt-Pd alloy catalysts synthesized by room-temperature electron reduction for oxygen reduction reaction. Adv. Sci. 2017, 4, 1600486. [Google Scholar] [CrossRef] [PubMed]
- Luyten, J.; De Keyzer, J.; Wollants, P.; Creemers, C. Construction of modified embedded atom method potentials for the study of the bulk phase behaviour in binary Pt-Ph, Pt-Pd, Pd-Ph and ternary Pt-Pd-Rh alloys. Calphad 2009, 33, 370–376. [Google Scholar] [CrossRef]
- Celik, F.A. The investigation of nucleation rate and johnson-mehl-avrami model of Pt-Pd alloy using molecular dynamics simulation during heat treatment processes. J. Alloys Compd. 2015, 632, 116–121. [Google Scholar] [CrossRef]
- Shao, G.; Zhangr, J.; Li, T. Investigation of Potential Parameters Effect on Stable Structures of Pt-Pd Alloy Nanoparticles by Particle Swarm Algorithm. In Proceedings of the 2017 32nd Youth Academic Annual Conference of Chinese Association of Automation (YAC), Hefei, China, 19–21 May 2017. [Google Scholar]
- Garbacz, H.; Mizera, J.; Laskowski, Z.; Gierej, M. Microstructure and mechanical properties of a Pt-Rh alloy produced by powder metallurgy and subjected to plastic working. Powder Technol. 2011, 208, 488–490. [Google Scholar] [CrossRef]
- Yuge, K.; Seko, A.; Kuwabara, A.; Oba, F.; Tanaka, I. First-principles study of bulk ordering and surface segregation in Pt-Rh binary alloys. Phys. Rev. B 2006, 74, 174202. [Google Scholar] [CrossRef]
- Sosa, E.; Liu, H. Oxygen adsorption on Pt-Pd nanoclusters by DFT and ab initio atomistic thermodynamics. J. Alloys Compd. 2018, 735, 643–653. [Google Scholar] [CrossRef]
- Tanaka, H. Relationship among glass-forming ability, fragility, and short-range bond ordering of liquids. J. Non-Cryst. Solids 2005, 351, 678–690. [Google Scholar] [CrossRef]
- Gasser, U.; Schofield, A.; Weitz, D. Local order in a supercooled colloidal fluid observed by confocal microscopy. J. Phys. Condens. Matter 2002, 15, S375. [Google Scholar] [CrossRef]
- Desgranges, C.; Delhommelle, J. Molecular simulation of the nucleation and growth of gold nanoparticles. J. Phys. Chem. C 2009, 113, 3607–3611. [Google Scholar] [CrossRef]
- Celtek, M.; Sengul, S. The characterisation of atomic structure and glass-forming ability of the Zr-Cu-Co metallic glasses studied by molecular dynamics simulations. Philos. Mag. 2018, 98, 783–802. [Google Scholar] [CrossRef]
- Liu, Z.; Zhang, R. AACSD: An atomistic analyzer for crystal structure and defects. Comput. Phys. Commun. 2018, 222, 229–239. [Google Scholar] [CrossRef]
- Ozgen, S.; Duruk, E. Molecular dynamics simulation of solidification kinetics of aluminium using Sutton–Chen version of EAM. Mater. Lett. 2004, 58, 1071–1075. [Google Scholar] [CrossRef]
- Yildiz, A.; Celik, F. Atomic concentration effect on thermal properties during solidification of Pt-Rh alloy: A molecular dynamics simulation. J. Cryst. Growth 2017, 463, 194–200. [Google Scholar] [CrossRef]
- Zhang, Y.; Jiang, S. Atomistic mechanisms for temperature-induced crystallization of amorphous copper based on molecular dynamics simulation. Comput. Mater. Sci. 2018, 151, 25–33. [Google Scholar] [CrossRef]
- Zhu, X.; Chen, K. Molecular dynamics simulation of homogeneous nucleation of K-Br cluster. J. Phys. Chem. Solids 2005, 66, 1732–1738. [Google Scholar] [CrossRef]
- Yu, W.; Wang, X.; Wang, H. Atomic simulation on evolution of nano-crystallizaion in amorphous metals. Trans. Nonferrous Met. Soc. China 2006, 16, s327–s331. [Google Scholar]
- Kazanc, S.; Ozgen, S.; Adiguzel, O. Pressure effects on martensitic transformation under quenching process in a molecular dynamics model of Ni-Al alloy. Phys. B 2003, 334, 375–381. [Google Scholar] [CrossRef]
- Kazanc, S. Molecular dynamics study of pressure effect on crystallization behaviour of amorphous Cu-Ni alloy during isothermal annealing. Phys. Lett. A 2007, 365, 473–477. [Google Scholar] [CrossRef]
- Parrinello, M.; Rahman, A. Crystal structure and pair potentials: A molecular-dynamics study. Phys. Rev. Lett. 1980, 45, 1196. [Google Scholar] [CrossRef]
- Haile, J.M. Molecular Dynamics Simulation: Elementary Methods; Wiley: New York, NY, USA, 1992. [Google Scholar]
- Rapaport, D.C.; Rapaport, D.C.R. The Art of Molecular Dynamics Simulation; Cambridge University Press: Cambridge, UK, 2004. [Google Scholar]
- Sutton, A.; Chen, J. Long-range finnis-sinclair potentials. Philos. Mag. Lett. 1990, 61, 139–146. [Google Scholar] [CrossRef]
- Dereli, G.; Cagin, T.; Uludogan, M.; Tomak, M. Thermal and mechanical properties of Pt-Rh alloys. Philos. Mag. Lett. 1997, 75, 209–218. [Google Scholar] [CrossRef] [Green Version]
- Honeycutt, J.D.; Andersen, H.C. Molecular dynamics study of melting and freezing of small Lennard Jones clusters. J. Phys. Chem. 1987, 91, 4950–4963. [Google Scholar] [CrossRef]
- Wang, L.; Liu, H.; Chen, K.; Hu, Z. The local orientational orders and structures of liquid and amorphous metals Au and Ni during rapid solidification. Phys. B 1997, 239, 267–273. [Google Scholar] [CrossRef]
- Jakse, N.; Pasturel, A. Ab initio molecular dynamics simulations of local structure of supercooled Ni. Chin. J. Chem. Phys. 2004, 120, 6124–6127. [Google Scholar] [CrossRef] [PubMed]
- William, J.; Mehl, R. Reaction kinetics in processes of nucleation and growth. Trans. Metall. Soc. AIME 1939, 135, 416–442. [Google Scholar]
- Avrami, M. Kinetics of phase change. J. Chem. Phys. 1939, 7, 1103–1112. [Google Scholar] [CrossRef]
- Li, X.; Yan, M.; Wang, J.; Huang, H.; Kong, C.; Schaffer, G.; Qian, M. Non-isothermal crystallization kinetics and mechanical properties of Al86Ni6Y4.5Co2La1.5 metallic glass powder. J. Alloys Compd. 2012, 530, 127–131. [Google Scholar] [CrossRef]
- Xie, X.; Gao, H. Calorimetric studies on the crystallization of Li2S–B2O3 glasses. J. Non-Cryst. Solids 1998, 240, 166–176. [Google Scholar] [CrossRef]
- Qi, Y.; Çağın, T.; Kimura, Y.; Goddard, W.A., III. Molecular-dynamics simulations of glass formation and crystallization in binary liquid metals: Cu-Ag and Cu-Ni. Phys. Rev. B 1999, 59, 3527. [Google Scholar] [CrossRef]
- Zhang, P.; Hui, W.; Zhang, Y.; Ren, X.; Zhang, D. Molecular dynamics simulation for the rapid solidification process of MgO–Al2O3–SiO2 glass-ceramics. J. Non-Cryst. Solids 2012, 358, 1465–1473. [Google Scholar] [CrossRef]
- Lu, K. Nanocrystalline metals crystallized from amorphous solids: Nanocrystallization, structure, and properties. Mater. Sci. Eng. 1996, 16, 161–221. [Google Scholar] [CrossRef]
- Çelik, F.A.; Kazanc, S.; Yildiz, A.; Ozgen, S. Pressure effect on the structural properties of amorphous Ag during isothermal annealing. Intermetallics 2008, 16, 793–800. [Google Scholar] [CrossRef]









© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kizilagac, S.; Celik, F.A.; Koksal, K. The Impact of Pt Concentration on Crystal Growth Mechanism in Pt-Pd Binary Alloy System in the Context of Molecular Dynamics. Metals 2018, 8, 926. https://doi.org/10.3390/met8110926
Kizilagac S, Celik FA, Koksal K. The Impact of Pt Concentration on Crystal Growth Mechanism in Pt-Pd Binary Alloy System in the Context of Molecular Dynamics. Metals. 2018; 8(11):926. https://doi.org/10.3390/met8110926
Chicago/Turabian StyleKizilagac, Servet, Fatih Ahmet Celik, and Koray Koksal. 2018. "The Impact of Pt Concentration on Crystal Growth Mechanism in Pt-Pd Binary Alloy System in the Context of Molecular Dynamics" Metals 8, no. 11: 926. https://doi.org/10.3390/met8110926
APA StyleKizilagac, S., Celik, F. A., & Koksal, K. (2018). The Impact of Pt Concentration on Crystal Growth Mechanism in Pt-Pd Binary Alloy System in the Context of Molecular Dynamics. Metals, 8(11), 926. https://doi.org/10.3390/met8110926