
Evidence of a Recent Bottleneck in Plasmodium falciparum Populations on the Honduran–Nicaraguan Border



Abstract
:1. Introduction
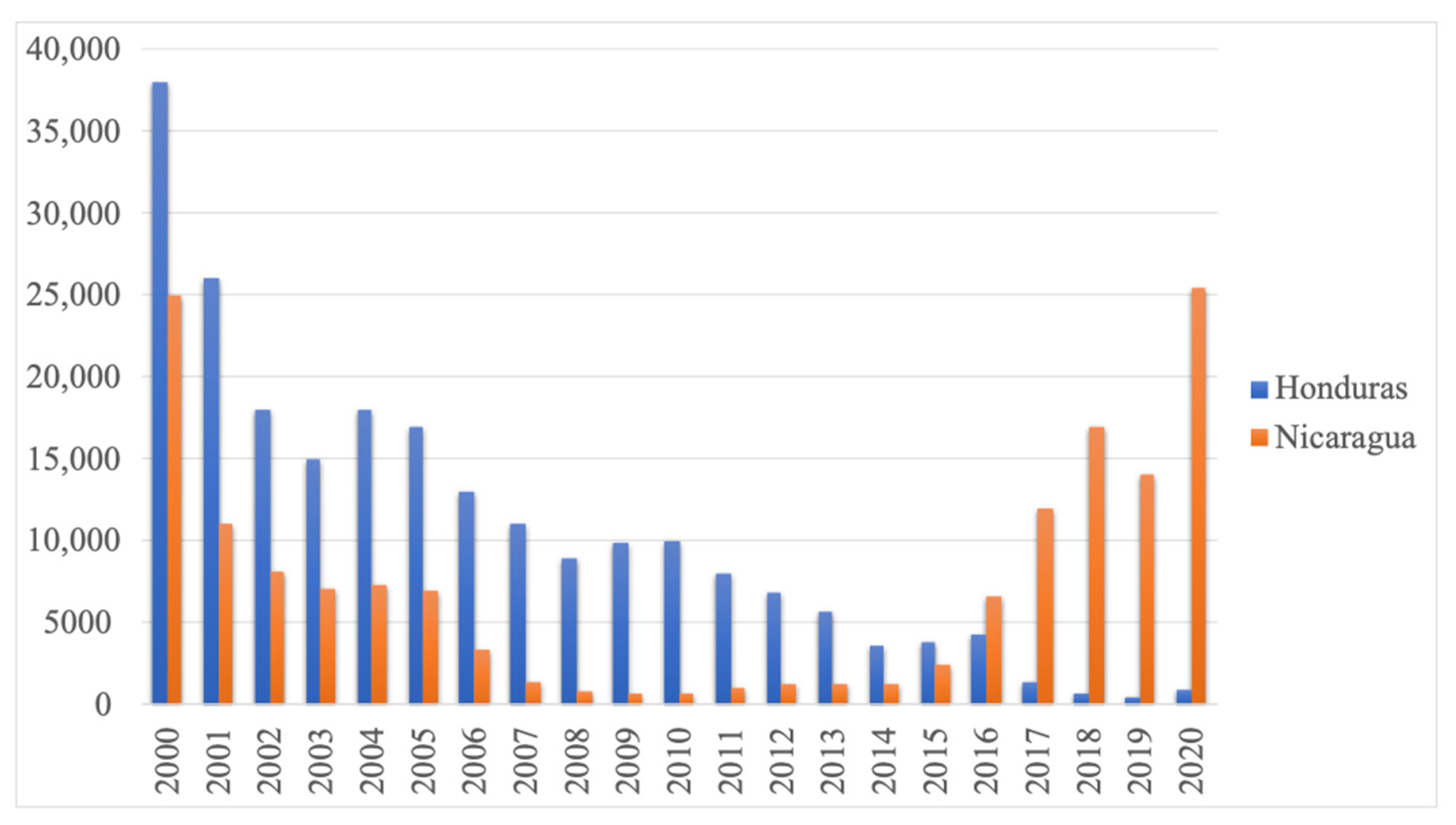
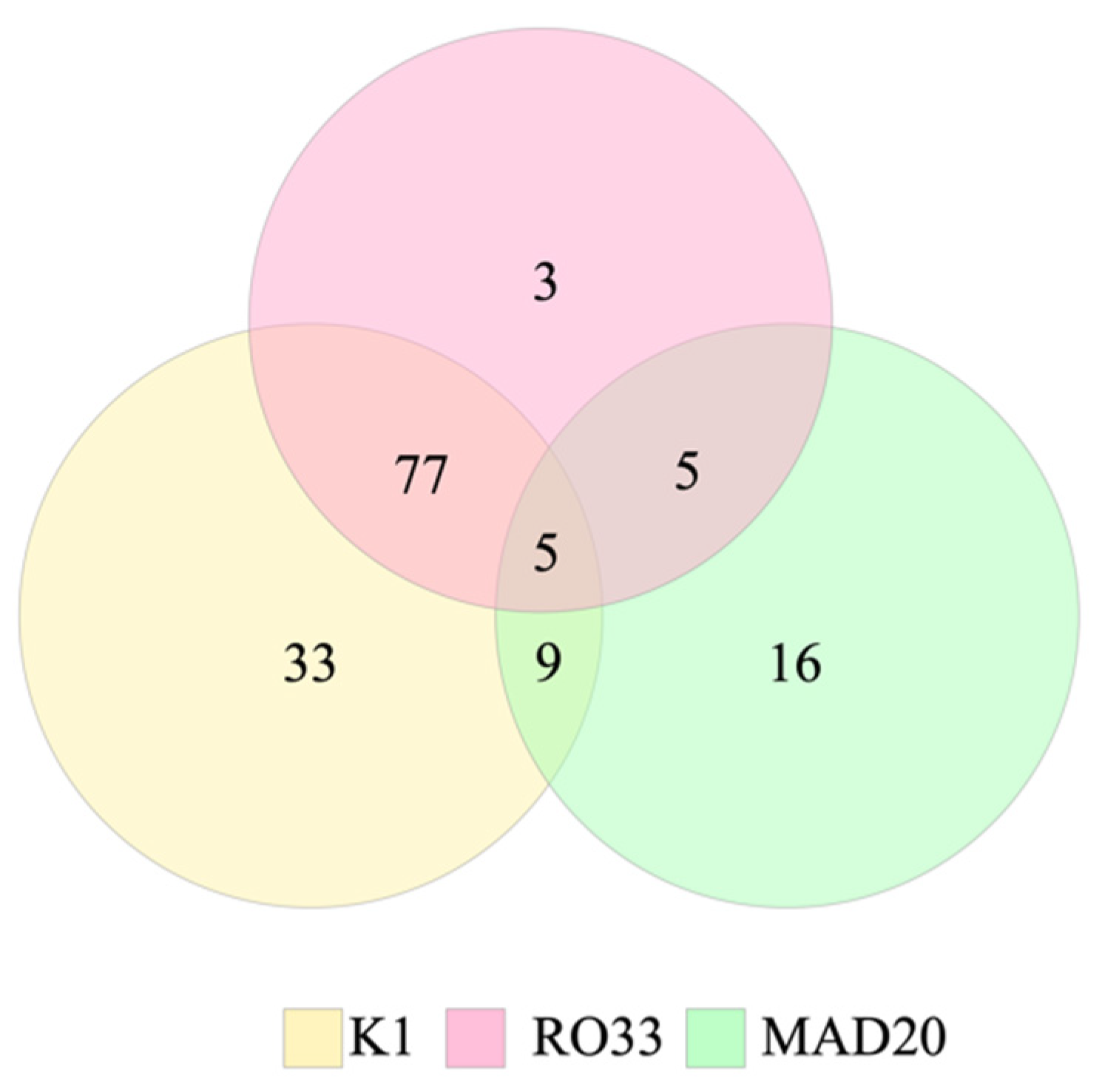
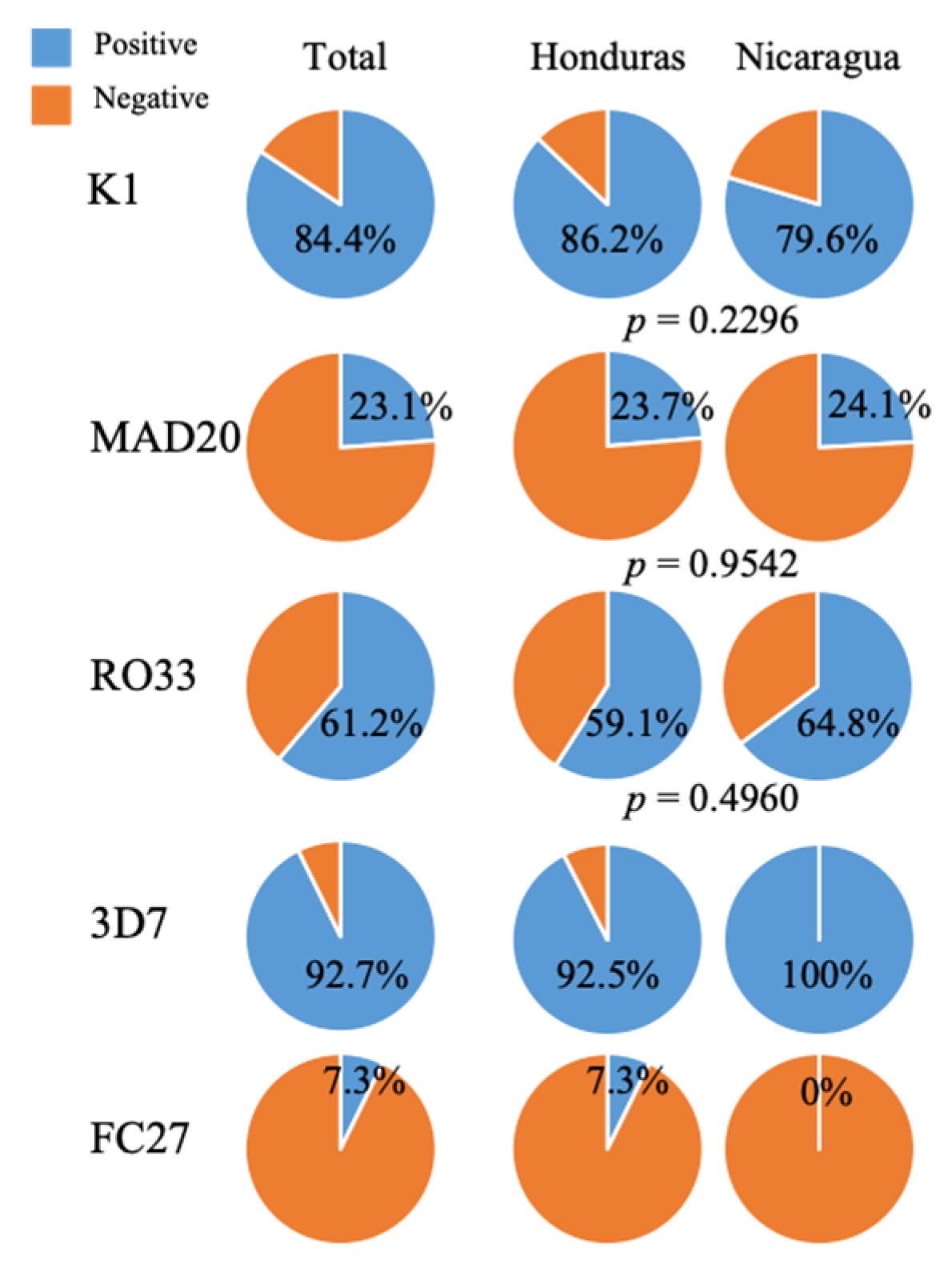
2. Results
3. Discussion
4. Materials and Methods
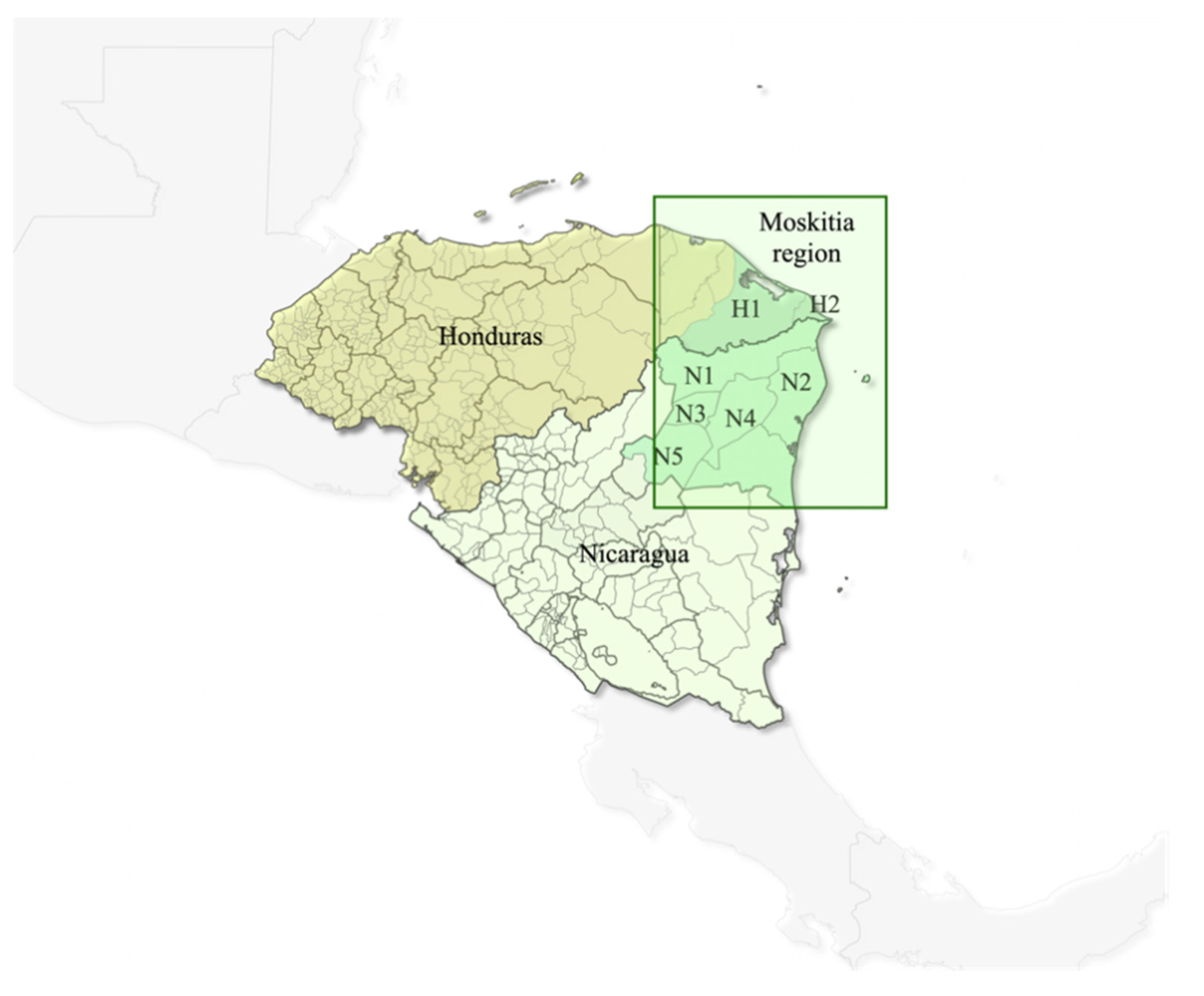
4.1. Sample Collection
4.2. DNA Extraction and Molecular Identification of the Parasite
4.3. Amplification of pfmsp-1 and pfmsp-2
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
Disclaimer
Copyright Statement
References
- World Health Organization. World Malaria Report 2020: 20 Years of Global Progress and Challenges; Licence: CC BY-NC-SA 3.0 IGO; WHO: Geneva, Switzerland, 2020. [Google Scholar]
- World Health Organization. Global Technical Strategy for Malaria 2016–2030; World Health Organization: Geneva, Switzerland, 2021. [Google Scholar]
- Amenga-Etego, L.N.; Asoala, V.; Agongo, G.; Jacob, C.; Goncalves, S.; Awandare, G.A.; Rockett, K.A.; Kwiatkowski, D. Temporal evolution of sulfadoxine-pyrimethamine resistance genotypes and genetic diversity in response to a decade of increased interventions against Plasmodium falciparum in northern Ghana. Malar. J. 2021, 20, 152. [Google Scholar] [CrossRef] [PubMed]
- Da Veiga Leal, S.; Ward, D.; Campino, S.; Benavente, E.D.; Ibrahim, A.; Claret, T.; Isaias, V.; Monteiro, D.; Clark, T.G.; Goncalves, L.; et al. Drug resistance profile and clonality of Plasmodium falciparum parasites in Cape Verde: The 2017 malaria outbreak. Malar. J. 2021, 20, 172. [Google Scholar] [CrossRef] [PubMed]
- Ghansah, A.; Amenga-Etego, L.; Amambua-Ngwa, A.; Andagalu, B.; Apinjoh, T.; Bouyou-Akotet, M.; Cornelius, V.; Golassa, L.; Andrianaranjaka, V.H.; Ishengoma, D.; et al. Monitoring parasite diversity for malaria elimination in sub-Saharan Africa. Science 2014, 345, 1297–1298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montenegro, L.M.; de Las Salas, B.; Neal, A.T.; Tobon-Castano, A.; Fairhurst, R.M.; Lopera-Mesa, T.M. State of Artemisinin and Partner Drug Susceptibility in Plasmodium falciparum Clinical Isolates from Colombia. Am. J. Trop. Med. Hyg. 2021, 104, 263–270. [Google Scholar] [CrossRef]
- Lucchi, N.W.; Abdallah, R.; Louzada, J.; Udhayakumar, V.; Oliveira-Ferreira, J. Molecular Surveillance for Polymorphisms Associated with Artemisinin-Based Combination Therapy Resistance in Plasmodium falciparum Isolates Collected in the State of Roraima, Brazil. Am. J. Trop. Med. Hyg. 2020, 102, 310–312. [Google Scholar] [CrossRef]
- Babiker, H.A.; Lines, J.; Hill, W.G.; Walliker, D. Population structure of Plasmodium falciparum in villages with different malaria endemicity in east Africa. Am. J. Trop. Med. Hyg. 1997, 56, 141–147. [Google Scholar] [CrossRef]
- Atroosh, W.M.; Al-Mekhlafi, H.M.; Mahdy, M.A.; Saif-Ali, R.; Al-Mekhlafi, A.M.; Surin, J. Genetic diversity of Plasmodium falciparum isolates from Pahang, Malaysia based on MSP-1 and MSP-2 genes. Parasit. Vectors 2011, 4, 233. [Google Scholar] [CrossRef] [Green Version]
- Ralinoro, F.; Rakotomanga, T.A.; Rakotosaona, R.; Doll Rakoto, D.A.; Menard, D.; Jeannoda, V.; Ratsimbasoa, A. Genetic diversity of Plasmodium falciparum populations in three malaria transmission settings in Madagascar. Malar. J. 2021, 20, 239. [Google Scholar] [CrossRef]
- Gwarinda, H.B.; Tessema, S.K.; Raman, J.; Greenhouse, B.; Birkholtz, L.M. Parasite genetic diversity reflects continued residual malaria transmission in Vhembe District, a hotspot in the Limpopo Province of South Africa. Malar. J. 2021, 20, 96. [Google Scholar] [CrossRef]
- Bosco, A.B.; Anderson, K.; Gresty, K.; Prosser, C.; Smith, D.; Nankabirwa, J.I.; Nsobya, S.; Yeka, A.; Namubiru, R.; Arinaitwe, E.; et al. Genetic diversity and genetic relatedness in Plasmodium falciparum parasite population in individuals with uncomplicated malaria based on microsatellite typing in Eastern and Western regions of Uganda, 2019–2020. Malar. J. 2021, 20, 242. [Google Scholar] [CrossRef]
- Mohammed, H.; Assefa, A.; Chernet, M.; Wuletaw, Y.; Commons, R.J. Genetic polymorphisms of Plasmodium falciparum isolates from Melka-Werer, North East Ethiopia based on the merozoite surface protein-2 (msp-2) gene as a molecular marker. Malar. J. 2021, 20, 85. [Google Scholar] [CrossRef]
- Santamaria, A.M.; Vasquez, V.; Rigg, C.; Moreno, D.; Romero, L.; Justo, C.; Chaves, L.F.; Saldana, A.; Calzada, J.E. Plasmodium falciparum genetic diversity in Panama based on glurp, msp-1 and msp-2 genes: Implications for malaria elimination in Mesoamerica. Life 2020, 10, 319. [Google Scholar] [CrossRef]
- Haddad, D.; Snounou, G.; Mattei, D.; Enamorado, I.G.; Figueroa, J.; Stahl, S.; Berzins, K. Limited genetic diversity of Plasmodium falciparum in field isolates from Honduras. Am. J. Trop. Med. Hyg. 1999, 60, 30–34. [Google Scholar] [CrossRef] [PubMed]
- Samudio, F.; Santamaria, A.M.; Obaldia, N., III; Pascale, J.M.; Bayard, V.; Calzada, J.E. Prevalence of Plasmodium falciparum mutations associated with antimalarial drug resistance during an epidemic in Kuna Yala, Panama, Central America. Am. J. Trop. Med. Hyg. 2005, 73, 839–841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lopez, A.C.; Ortiz, A.; Coello, J.; Sosa-Ochoa, W.; Torres, R.E.; Banegas, E.I.; Jovel, I.; Fontecha, G.A. Genetic diversity of Plasmodium vivax and Plasmodium falciparum in Honduras. Malar. J. 2012, 11, 391. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gutierrez, S.; Gonzalez-Ceron, L.; Montoya, A.; Sandoval, M.A.; Torres, M.E.; Cerritos, R. Genetic structure of Plasmodium vivax in Nicaragua, a country in the control phase, based on the carboxyl terminal region of the merozoite surface protein-1. Infect. Genet. Evol. 2016, 40, 324–330. [Google Scholar] [CrossRef] [PubMed]
- Vallejo, A.F.; Martinez, N.L.; Tobon, A.; Alger, J.; Lacerda, M.V.; Kajava, A.V.; Arevalo-Herrera, M.; Herrera, S. Global genetic diversity of the Plasmodium vivax transmission-blocking vaccine candidate Pvs48/45. Malar. J. 2016, 15, 202. [Google Scholar] [CrossRef] [Green Version]
- Prajapati, S.K.; Joshi, H.; Shalini, S.; Patarroyo, M.A.; Suwanarusk, R.; Kumar, A.; Sharma, S.K.; Eapen, A.; Dev, V.; Bhatt, R.M.; et al. Plasmodium vivax lineages: Geographical distribution, tandem repeat polymorphism, and phylogenetic relationship. Malar. J. 2011, 10, 374. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larranaga, N.; Mejia, R.E.; Hormaza, J.I.; Montoya, A.; Soto, A.; Fontecha, G.A. Genetic structure of Plasmodium falciparum populations across the Honduras-Nicaragua border. Malar. J. 2013, 12, 354. [Google Scholar] [CrossRef] [Green Version]
- Valdivia, H.O.; Villena, F.E.; Lizewski, S.E.; Garcia, J.; Alger, J.; Bishop, D.K. Genomic surveillance of Plasmodium falciparum and Plasmodium vivax cases at the University Hospital in Tegucigalpa, Honduras. Sci. Rep. 2020, 10, 20975. [Google Scholar] [CrossRef] [PubMed]
- Ajogbasile, F.V.; Kayode, A.T.; Oluniyi, P.E.; Akano, K.O.; Uwanibe, J.N.; Adegboyega, B.B.; Philip, C.; John, O.G.; Eromon, P.J.; Emechebe, G.; et al. Genetic diversity and population structure of Plasmodium falciparum in Nigeria: Insights from microsatellite loci analysis. Malar. J. 2021, 20, 236. [Google Scholar] [CrossRef]
- Knudson, A.; Gonzalez-Casabianca, F.; Feged-Rivadeneira, A.; Pedreros, M.F.; Aponte, S.; Olaya, A.; Castillo, C.F.; Mancilla, E.; Piamba-Dorado, A.; Sanchez-Pedraza, R.; et al. Spatio-temporal dynamics of Plasmodium falciparum transmission within a spatial unit on the Colombian Pacific Coast. Sci. Rep. 2020, 10, 3756. [Google Scholar] [CrossRef] [PubMed]
- Vo, T.C.; Le, H.G.; Kang, J.M.; Naw, H.; Fan, C.K.; Trinh, N.T.M.; Quang, H.H.; Na, B.K. Molecular surveillance of malaria in the Central Highlands, Vietnam. Parasitol. Int. 2021, 83, 102374. [Google Scholar] [CrossRef] [PubMed]
- Goh, X.T.; Lim, Y.A.L.; Lee, P.C.; Nissapatorn, V.; Chua, K.H. Diversity and natural selection of merozoite surface protein-1 in three species of human malaria parasites: Contribution from South-East Asian isolates. Mol. Biochem. Parasitol. 2021, 244, 111390. [Google Scholar] [CrossRef] [PubMed]
- Metoh, T.N.; Chen, J.H.; Fon-Gah, P.; Zhou, X.; Moyou-Somo, R.; Zhou, X.N. Genetic diversity of Plasmodium falciparum and genetic profile in children affected by uncomplicated malaria in Cameroon. Malar. J. 2020, 19, 115. [Google Scholar] [CrossRef] [Green Version]
- Patgiri, S.J.; Sarma, K.; Sarmah, N.; Bhattacharyya, N.; Sarma, D.K.; Nirmolia, T.; Bhattacharyya, D.R.; Mohapatra, P.K.; Bansal, D.; Bharti, P.K.; et al. Characterization of drug resistance and genetic diversity of Plasmodium falciparum parasites from Tripura, Northeast India. Sci. Rep. 2019, 9, 13704. [Google Scholar] [CrossRef]
- Le, H.G.; Kang, J.M.; Jun, H.; Lee, J.; Thai, T.L.; Myint, M.K.; Aye, K.S.; Sohn, W.M.; Shin, H.J.; Kim, T.S.; et al. Changing pattern of the genetic diversities of Plasmodium falciparum merozoite surface protein-1 and merozoite surface protein-2 in Myanmar isolates. Malar. J. 2019, 18, 241. [Google Scholar] [CrossRef]
- Ghoshal, S.; Gajendra, P.; Kanjilal, S.D.; Mitra, M.; Sengupta, S. Diversity analysis of MSP1 identifies conserved epitope organization in block 2 amidst high sequence variability in Indian Plasmodium falciparum isolates. Malar. J. 2018, 17, 447. [Google Scholar] [CrossRef]
- Miller, L.H.; Roberts, T.; Shahabuddin, M.; McCutchan, T.F. Analysis of sequence diversity in the Plasmodium falciparum merozoite surface protein-1 (MSP-1). Mol. Biochem. Parasitol. 1993, 59, 1–14. [Google Scholar] [CrossRef]
- Ferreira, M.U.; Liu, Q.; Zhou, M.; Kimura, M.; Kaneko, O.; Van Thien, H.; Isomura, S.; Tanabe, K.; Kawamoto, F. Stable patterns of allelic diversity at the Merozoite surface protein-1 locus of Plasmodium falciparum in clinical isolates from southern Vietnam. J. Eukaryot. Microbiol. 1998, 45, 131–136. [Google Scholar] [CrossRef]
- Ngomo, J.M.N.; M’Bondoukwe, N.P.; Yavo, W.; Mavoungou, L.C.B.; Bouyou-Akotet, M.K.; Mawili-Mboumba, D.P. Spatial and temporal distribution of Pfmsp1 and Pfmsp2 alleles and genetic profile change of Plasmodium falciparum populations in Gabon. Acta Trop. 2018, 178, 27–33. [Google Scholar] [CrossRef]
- Khatoon, L.; Jan, M.I.; Khan, I.U.; Ullah, F.; Malik, S.A. Genetic diversity in human malarial parasites of Khyber Agency Pakistan. Acta Parasitol. 2013, 58, 564–569. [Google Scholar] [CrossRef] [Green Version]
- Anderson, T.J.; Haubold, B.; Williams, J.T.; Estrada-Franco, J.G.; Richardson, L.; Mollinedo, R.; Bockarie, M.; Mokili, J.; Mharakurwa, S.; French, N.; et al. Microsatellite markers reveal a spectrum of population structures in the malaria parasite Plasmodium falciparum. Mol. Biol. Evol. 2000, 17, 1467–1482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ndiaye, T.; Sy, M.; Gaye, A.; Siddle, K.J.; Park, D.J.; Bei, A.K.; Deme, A.B.; Mbaye, A.; Dieye, B.; Ndiaye, Y.D.; et al. Molecular epidemiology of Plasmodium falciparum by multiplexed amplicon deep sequencing in Senegal. Malar. J. 2020, 19, 403. [Google Scholar] [CrossRef] [PubMed]
- Jimenez, J.N.; Snounou, G.; Letourneur, F.; Renia, L.; Velez, I.D.; Muskus, C.E. Near-fixation of a Pfmsp1 block 2 allelic variant in genetically diverse Plasmodium falciparum populations across Western Colombia. Acta Trop. 2010, 114, 67–70. [Google Scholar] [CrossRef] [PubMed]
- Al-abd, N.M.; Mahdy, M.A.; Al-Mekhlafi, A.M.; Snounou, G.; Abdul-Majid, N.B.; Al-Mekhlafi, H.M.; Fong, M.Y. The suitability of P. falciparum merozoite surface proteins 1 and 2 as genetic markers for in vivo drug trials in Yemen. PLoS ONE 2013, 8, e67853. [Google Scholar] [CrossRef]
- Sallenave-Sales, S.; Ferreira-da-Cruz, M.F.; Faria, C.P.; Cerruti, C., Jr.; Daniel-Ribeiro, C.T.; Zalis, M.G. Plasmodium falciparum: Limited genetic diversity of MSP-2 in isolates circulating in Brazilian endemic areas. Exp. Parasitol. 2003, 103, 127–135. [Google Scholar] [CrossRef]
- Sutton, P.L.; Neyra, V.; Hernandez, J.N.; Branch, O.H. Plasmodium falciparum and Plasmodium vivax infections in the Peruvian Amazon: Propagation of complex, multiple allele-type infections without super-infection. Am. J. Trop. Med. Hyg. 2009, 81, 950–960. [Google Scholar] [CrossRef] [Green Version]
- de Lamballerie, X.; Zandotti, C.; Vignoli, C.; Bollet, C.; de Micco, P. A one-step microbial DNA extraction method using “Chelex 100” suitable for gene amplification. Res. Microbiol. 1992, 143, 785–790. [Google Scholar] [CrossRef]
- Singh, B.; Bobogare, A.; Cox-Singh, J.; Snounou, G.; Abdullah, M.S.; Rahman, H.A. A genus- and species-specific nested polymerase chain reaction malaria detection assay for epidemiologic studies. Am. J. Trop. Med. Hyg. 1999, 60, 687–692. [Google Scholar] [CrossRef]
- Schoepflin, S.; Valsangiacomo, F.; Lin, E.; Kiniboro, B.; Mueller, I.; Felger, I. Comparison of Plasmodium falciparum allelic frequency distribution in different endemic settings by high-resolution genotyping. Malar. J. 2009, 8, 250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mwingira, F.; Nkwengulila, G.; Schoepflin, S.; Sumari, D.; Beck, H.P.; Snounou, G.; Felger, I.; Olliaro, P.; Mugittu, K. Plasmodium falciparum msp1, msp2 and glurp allele frequency and diversity in sub-Saharan Africa. Malar. J. 2011, 10, 79. [Google Scholar] [CrossRef] [PubMed] [Green Version]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| pfmsp-1 Subfamilies | pfmsp-2 Subfamilies | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| K1 (bp) | n (%) | MAD20 (bp) | n (%) | RO33 (bp) | n (%) | 3D7 (bp) | n (%) | FC27 (bp) | n (%) |
| 235 | 124 (84.4) | 146 | 6 (4.01) | 335 | 90 (61.2) | 276 | 51 (92.7) | 327 | 4 (7.3) |
| Negative | 23 (15.6) | 183 | 16 (10.9) | Negative | 57 (38.8) | Negative | 4 (7.3) | Negative | 51 (92.7) |
| 202 | 13 (8.8) | ||||||||
| Negative | 112 (76.2) | ||||||||
| Total | 147 | Total | 147 | Total | 147 | Total | 55 | Total | 55 |
| pfmsp-1 Subfamilies | pfmsp-2 Subfamilies | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| K1 | MAD20 | RO33 | K1/MAD20 | MAD20/RO33 | K1/RO33 | K1/MAD20/RO33 | 3D7 | FC27 | |
| Honduras | 81 | 22 | 55 | 8 | 0 | 48 | 4 | 49 | 4 |
| Nicaragua | 43 | 13 | 35 | 1 | 5 | 29 | 1 | 2 | 0 |
| Total | 124 (84.4%) | 35 (23.8%) | 90 (61.2%) | 9 (6.1%) | 5 (3.4%) | 77 (52.4%) | 5 (3.4%) | 51 (92.7%) | 4 (7.3%) |
| PCR/Allelic Family | Primer Name | Primer Sequence 5′-3′ | Annealing Temperature | Approximate Amplicon Size (bp) | |
|---|---|---|---|---|---|
| pfmsp-1 | 1st | MI-OF | CTAGAAGCTTTAGAAGATGCAGTATTG | 54 °C | >1000 |
| MI-OR | CTTAAATAGATTCTAATTCAAGTGGATCA | ||||
| 2nd K1 | K1F | AAATGAAGAAGAAATTACTACAAAAGGTGC | 59 °C | 235 | |
| K1R | GCTTGCATCAGCTGGAGGGCTTGCACCAGA | ||||
| 2nd RO33 | RO33F | TAAAGGATGGAGCAAATACTCAAGTTGTTG | 58 °C | 335 | |
| RO33R | CAAGTAATTTTGAACTCTATGTTTTAAATCAGCGTA | ||||
| 2nd MAD20 | MAD20F | AAATGAAGGAACAAGTGGAACAGCTGTTAC | 59 °C | 146–202 | |
| MAD20R | ATCTGAAGGATTTGTACGTCTTGAATTACC | ||||
| pfmsp-2 | 1st | MSP2 S2 | GAAGGTAATTAAAACATTGTC | 60 °C | 905 |
| MSP2 S3 | GAGGGATGTTGCTGCTCCACAG | ||||
| 2nd 3D7 | RvNS 3D7 | CTGAAGAGGTACTGGTAGA | 50 °C | 276 | |
| MSP-2 S1 | GCTTATAATATGAGTATAAGGAGAA | ||||
| 2nd FC27 | RvMS FC27 | GCATTGCCAGAACTTGAA | 50 °C | 327 | |
| MSP-2 S1 | GCTTATAATATGAGTATAAGGAGAA |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pinto, A.; Archaga, O.; Mejía, Á.; Escober, L.; Henríquez, J.; Montoya, A.; Valdivia, H.O.; Fontecha, G. Evidence of a Recent Bottleneck in Plasmodium falciparum Populations on the Honduran–Nicaraguan Border. Pathogens 2021, 10, 1432. https://doi.org/10.3390/pathogens10111432
Pinto A, Archaga O, Mejía Á, Escober L, Henríquez J, Montoya A, Valdivia HO, Fontecha G. Evidence of a Recent Bottleneck in Plasmodium falciparum Populations on the Honduran–Nicaraguan Border. Pathogens. 2021; 10(11):1432. https://doi.org/10.3390/pathogens10111432
Chicago/Turabian StylePinto, Alejandra, Osman Archaga, Ángel Mejía, Lenin Escober, Jessica Henríquez, Alberto Montoya, Hugo O. Valdivia, and Gustavo Fontecha. 2021. "Evidence of a Recent Bottleneck in Plasmodium falciparum Populations on the Honduran–Nicaraguan Border" Pathogens 10, no. 11: 1432. https://doi.org/10.3390/pathogens10111432
APA StylePinto, A., Archaga, O., Mejía, Á., Escober, L., Henríquez, J., Montoya, A., Valdivia, H. O., & Fontecha, G. (2021). Evidence of a Recent Bottleneck in Plasmodium falciparum Populations on the Honduran–Nicaraguan Border. Pathogens, 10(11), 1432. https://doi.org/10.3390/pathogens10111432