
A Remarkable Genetic Diversity of Rotavirus A Circulating in Red Fox Population in Croatia



Abstract
:1. Introduction
2. Results
2.1. The Results of VP2 Real-Time RT-PCR
2.2. The Results of VP7 and VP4 Genotyping and Phylogenetic Analysis
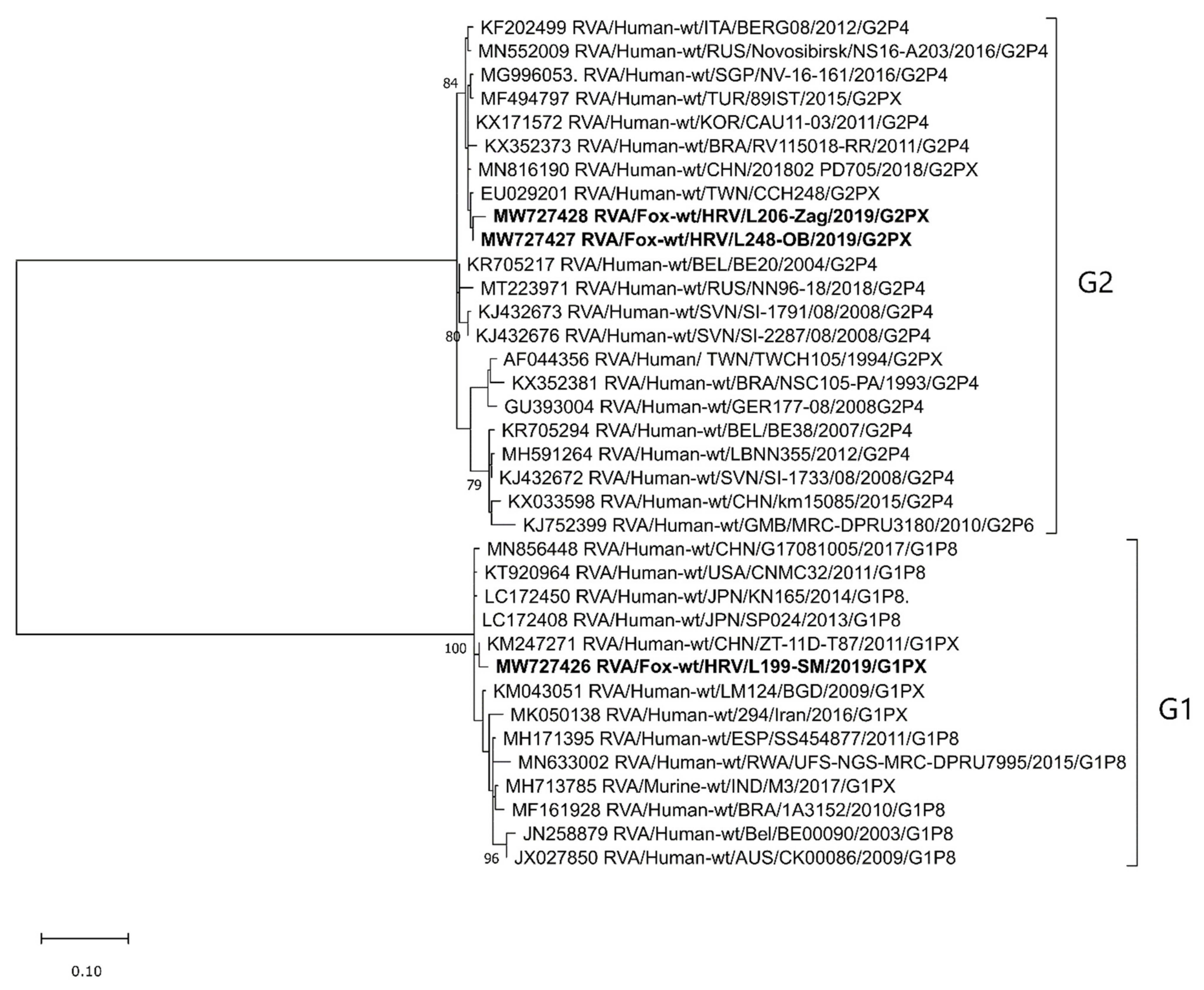
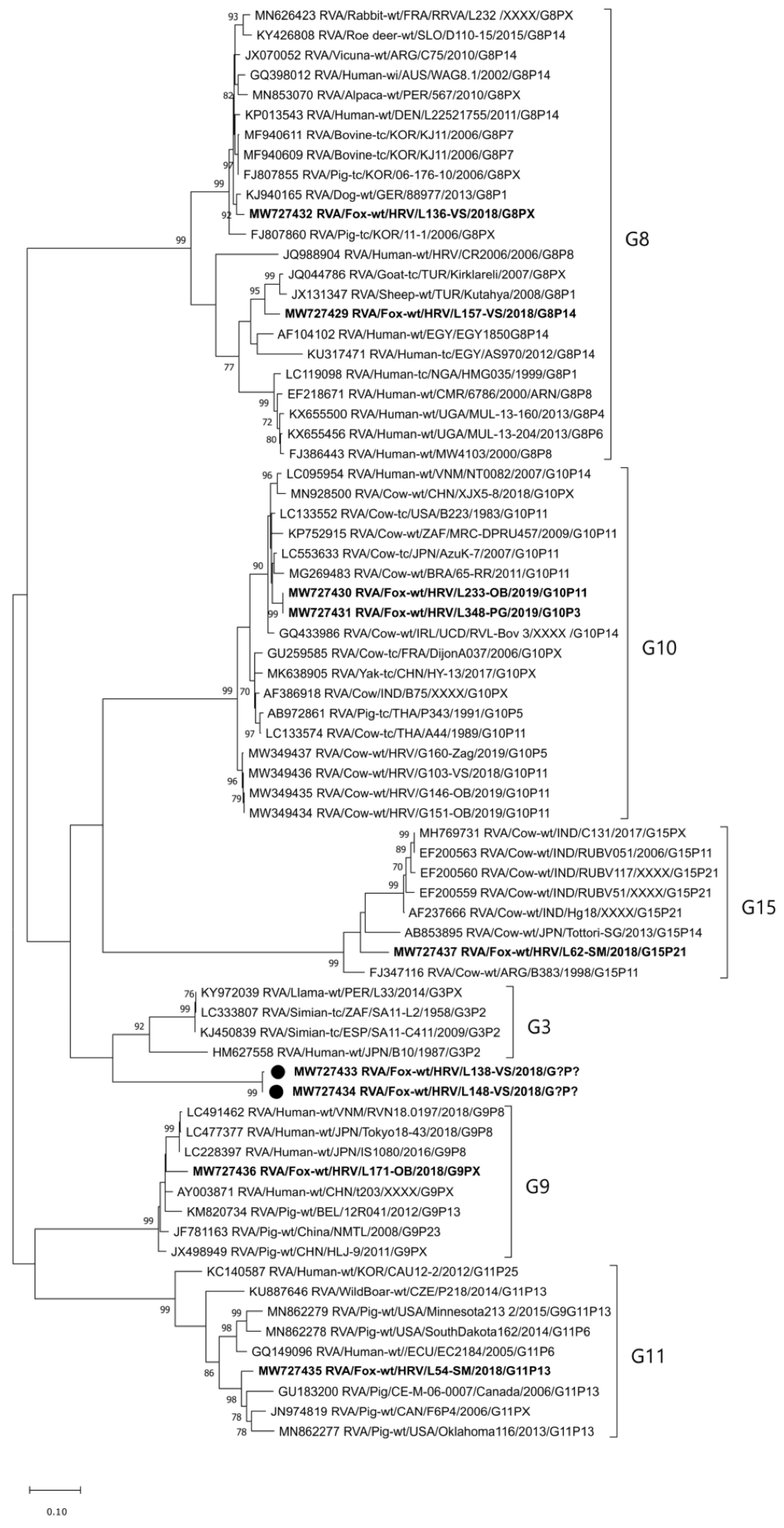
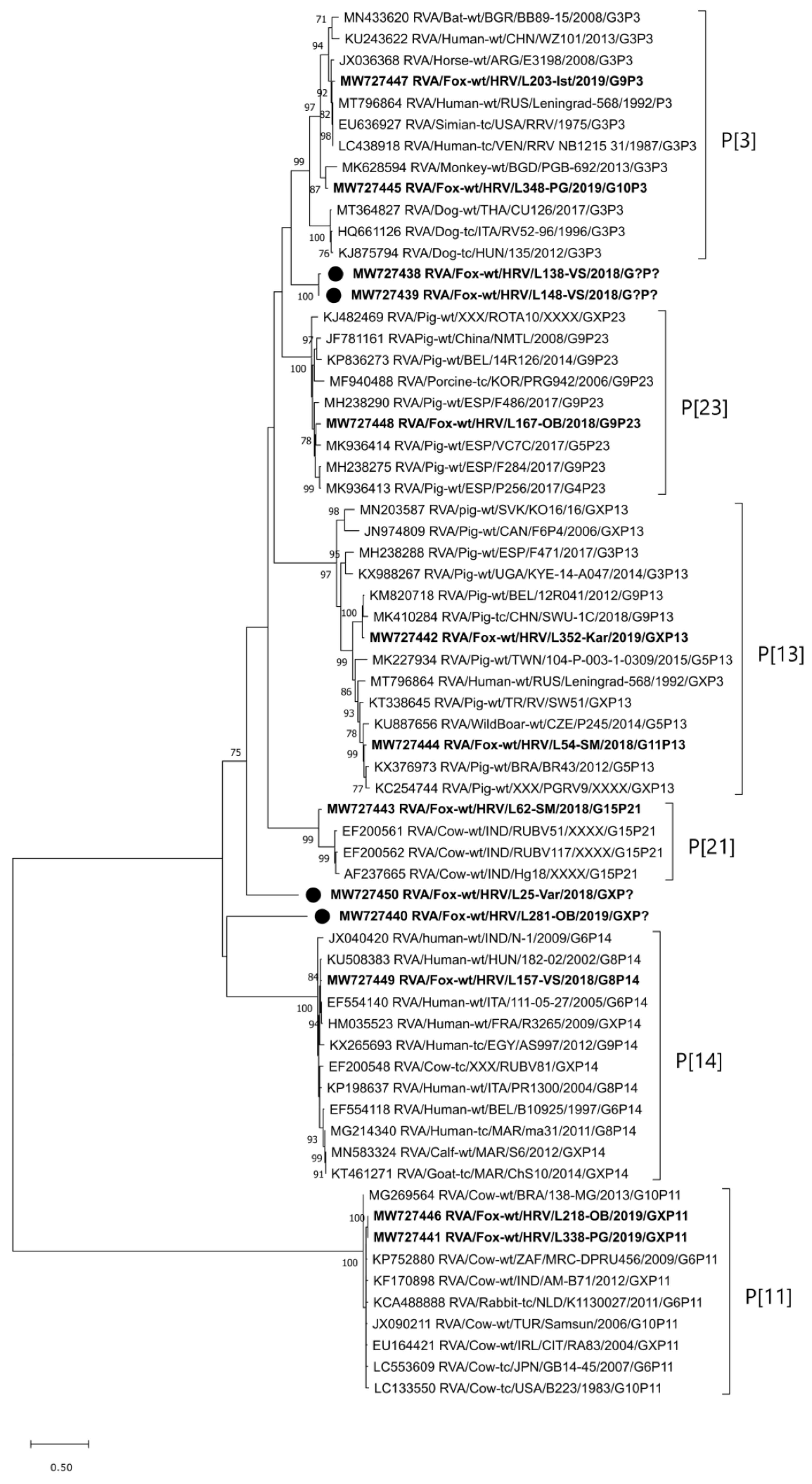
2.2.1. VP7 Genotypes and Phylogenetic Analysis
2.2.2. VP4 Genotypes and Phylogenetic Analysis
3. Discussion
4. Materials and Methods
4.1. Samples
4.2. RNA Extraction and Real-Time RT-PCR
4.3. VP7 and VP4 Genotyping of RVA Strains
4.4. Sequencing and Phylogenetic Analysis
4.5. Statistical Analysis
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Troeger, C.; Khalil, I.A.; Rao, P.C.; Cao, S.; Blacker, B.F.; Ahmed, T.; Armah, G.; Bines, J.E.; Brewer, T.G.; Colombara, D.V.; et al. Rotavirus Vaccination and the Global Burden of Rotavirus Diarrhea Among Children Younger Than 5 Years. JAMA Pediatr. 2018, 172, 958–965. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hungerford, D.; Vivancos, R.; Read, J.M.; Pitzer, V.E.; Cunliffe, N.; French, N.; Iturriza-Gomara, M. In-season and out-of-season variation of rotavirus genotype distribution and age of infection across 12 European countries before the introduction of routine vaccination, 2007/08 to 2012/13. Eurosurveillance 2016, 21, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martella, V.; Bányai, K.; Matthijnssens, J.; Buonavoglia, C.; Ciarlet, M. Zoonotic aspects of rotaviruses. Vet. Microbiol. 2010, 140, 246–255. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palmarini, M. Reoviridae. In Fenner’s Veterinary Virology, 5th ed.; MacLachlan, N.J., Dubovi, E.J., Eds.; Academic Press: Boston, MA, USA, 2017; pp. 299–317. [Google Scholar]
- Ghosh, S.; Kobayashi, N. Exotic rotaviruses in animals and rotaviruses in exotic animals. Virusdisease 2014, 25, 158–172. [Google Scholar] [CrossRef] [Green Version]
- ICTV. Rotavirus Taxonomy. 2019. Available online: https://talk.ictvonline.org/taxonomy/ (accessed on 22 February 2021).
- Dóró, R.; Farkas, S.L.; Martella, V.; Bányai, K. Zoonotic transmission of rotavirus: Surveillance and control. Expert Rev. Anti Infect. Ther. 2015, 13, 1337–1350. [Google Scholar] [CrossRef] [PubMed]
- Crawford, S.E.; Ramani, S.; Tate, J.E.; Parashar, U.D.; Svensson, L.; Hagbom, M.; Franco, M.A.; Greenberg, H.B.; O’Ryan, M.; Kang, G.; et al. Rotavirus infection. Nat. Rev. Dis. Prim. 2017, 3, 17083. [Google Scholar] [CrossRef] [Green Version]
- Matthijnssens, J.; Ciarlet, M.; Rahman, M.; Attoui, H.; Bányai, K.; Estes, M.K.; Gentsch, J.R.; Iturriza-Gómara, M.; Kirkwood, C.D.; Martella, V.; et al. Recommendations for the classification of group A rotaviruses using all 11 genomic RNA segments. Arch. Virol. 2008, 153, 1621–1629. [Google Scholar] [CrossRef] [Green Version]
- RCWG. List of Accepted Genotypes by Rotavirus Classification Working Group. 2018. Available online: https://rega.kuleuven.be/cev/viralmetagenomics/virus-classification/rcwg (accessed on 1 March 2021).
- Preston, N.D.; Daszak, P.; Colwell, R.R. The Human Environment Interface: Applying Ecosystem Concepts to Health. Curr. Top. Microbiol. Immunol. 2013, 365, 83–100. [Google Scholar] [CrossRef]
- Zecchin, B.; De Nardi, M.; Nouvellet, P.; Vernesi, C.; Babbucci, M.; Crestanello, B.; Bagó, Z.; Bedeković, T.; Hostnik, P.; Milani, A.; et al. Genetic and spatial characterization of the red fox (Vulpes vulpes) population in the area stretching between the Eastern and Dinaric Alps and its relationship with rabies and canine distemper dynamics. PLoS ONE 2019, 14, e0213515. [Google Scholar] [CrossRef] [Green Version]
- Evans, R.H. Rotavirus-associated diarrhea in young raccoons (procyon lotor), striped skunks (mephitis mephitis) and red foxes (vulpes vulpes). J. Wildl. Dis. 1984, 20, 79–85. [Google Scholar] [CrossRef]
- Busi, C.; Martella, V.; Papetti, A.; Sabelli, C.; Lelli, D.; Alborali, G.L.; Gibelli, L.; Gelmetti, D.; Lavazza, A.; Cordioli, P.; et al. Group A Rotavirus Associated with Encephalitis in Red Fox. Emerg. Infect. Dis. 2017, 23, 1535–1538. [Google Scholar] [CrossRef] [Green Version]
- Mijatovic-Rustempasic, S.; Esona, M.D.; Williams, A.L.; Bowen, M.D. Sensitive and specific nested PCR assay for detection of rotavirus A in samples with a low viral load. J. Virol. Methods 2016, 236, 41–46. [Google Scholar] [CrossRef] [Green Version]
- Vilibic-Cavlek, T.; Barbic, L.; Mrzljak, A.; Brnic, D.; Klobucar, A.; Ilic, M.; Janev-Holcer, N.; Bogdanic, M.; Jemersic, L.; Stevanovic, V.; et al. Emerging and Neglected Viruses of Zoonotic Importance in Croatia. Pathogens 2021, 10, 73. [Google Scholar] [CrossRef]
- Bányai, K.; László, B.; Duque, J.; Steele, A.D.; Nelson, E.A.S.; Gentsch, J.R.; Parashar, U.D. Systematic review of regional and temporal trends in global rotavirus strain diversity in the pre rotavirus vaccine era: Insights for understanding the impact of rotavirus vaccination programs. Vaccine 2012, 30 (Suppl. 1), A122–A130. [Google Scholar] [CrossRef] [PubMed]
- Papp, H.; László, B.; Jakab, F.; Ganesh, B.; De Grazia, S.; Matthijnssens, J.; Ciarlet, M.; Martella, V.; Bányai, K. Review of group A rotavirus strains reported in swine and cattle. Vet. Microbiol. 2013, 165, 190–199. [Google Scholar] [CrossRef]
- Moutelíková, R.; Dufková, L.; Kamler, J.; Drimaj, J.; Plhal, R.; Prodělalová, J. Epidemiological survey of enteric viruses in wild boars in the Czech Republic: First evidence of close relationship between wild boar and human rotavirus A strains. Vet. Microbiol. 2016, 193, 28–35. [Google Scholar] [CrossRef] [PubMed]
- Jamnikar-Ciglenecki, U.; Kuhar, U.; Sturm, S.; Kirbis, A.; Racki, N.; Steyer, A. The first detection and whole genome characterization of the G6P[15] group A rotavirus strain from roe deer. Vet. Microbiol. 2016, 191, 52–59. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Aguirre, I.; Steyer, A.; Boben, J.; Gruden, K.; Poljsak-Prijatelj, M.; Ravnikar, M. Sensitive detection of multiple rota-virus genotypes with a single reverse transcription-real-time quantitative PCR assay. J. Clin. Microbiol. 2008, 46, 2547–2554. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bwogi, J.; Jere, K.C.; Karamagi, C.; Byarugaba, D.K.; Namuwulya, P.; Baliraine, F.N.; Desselberger, U.; Iturriza-Gomara, M. Whole genome analysis of selected human and animal rotaviruses identified in Uganda from 2012 to 2014 reveals complex genome reassortment events between human, bovine, caprine and porcine strains. PLoS ONE 2017, 12, e0178855. [Google Scholar] [CrossRef] [Green Version]
- Messenger, A.M.; Barnes, A.N.; Gray, G.C. Reverse Zoonotic Disease Transmission (Zooanthroponosis): A Systematic Review of Seldom-Documented Human Biological Threats to Animals. PLoS ONE 2014, 9, e89055. [Google Scholar] [CrossRef] [Green Version]
- Okadera, K.; Abe, M.; Ito, N.; Morikawa, S.; Yamasaki, A.; Masatani, T.; Nakagawa, K.; Yamaoka, S.; Sugiyama, M. Evidence of natural transmission of group A rotavirus between domestic pigs and wild boars (Sus scrofa) in Japan. Infect. Genet. Evol. 2013, 20, 54–60. [Google Scholar] [CrossRef] [PubMed]
- Matthijnssens, J.; Potgieter, C.A.; Ciarlet, M.; Parreño, V.; Martella, V.; Bányai, K.; Garaicoechea, L.; Palombo, E.A.; Novo, L.; Zeller, M.; et al. Are Human P[14] Rotavirus Strains the Result of Interspecies Transmissions from Sheep or Other Ungulates That Belong to the Mammalian Order Artiodactyla? J. Virol. 2009, 83, 2917–2929. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rao, C.D.; Gowda, K.; Reddy, B.S.Y. Sequence Analysis of VP4 and VP7 Genes of Nontypeable Strains Identifies a New Pair of Outer Capsid Proteins Representing Novel P and G Genotypes in Bovine Rotaviruses. Virology 2000, 276, 104–113. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Masuda, T.; Nagai, M.; Yamasato, H.; Tsuchiaka, S.; Okazaki, S.; Katayama, Y.; Oba, M.; Nishiura, N.; Sassa, Y.; Omatsu, T.; et al. Identification of novel bovine group A rotavirus G15P[14] strain from epizootic diarrhea of adult cows by de novo sequencing using a next-generation sequencer. Vet. Microbiol. 2014, 171, 66–73. [Google Scholar] [CrossRef] [PubMed]
- Matthijnssens, J.; Ciarlet, M.; Heiman, E.; Arijs, I.; Delbeke, T.; McDonald, S.M.; Palombo, E.A.; Iturriza-Gómara, M.; Maes, P.; Patton, J.T.; et al. Full Genome-Based Classification of Rotaviruses Reveals a Common Origin between Human Wa-Like and Porcine Rotavirus Strains and Human DS-1-Like and Bovine Rotavirus Strains. J. Virol. 2008, 82, 3204–3219. [Google Scholar] [CrossRef] [Green Version]
- Eurorotanet. Rotavirus Detection and Typing. 2009, p. 25. Available online: https://www.eurorotanet.com/project-information/documents-and-methods/ (accessed on 2 April 2018).
- Abe, M.; Ito, N.; Morikawa, S.; Takasu, M.; Murase, T.; Kawashima, T.; Kawai, Y.; Kohara, J.; Sugiyama, M. Molecular epidemiology of rotaviruses among healthy calves in Japan: Isolation of a novel bovine rotavirus bearing new P and G genotypes. Virus Res. 2009, 144, 250–257. [Google Scholar] [CrossRef]
- Schumann, T.; Hotzel, H.; Otto, P.; Johne, R. Evidence of interspecies transmission and reassortment among avian group A rotaviruses. Virology 2009, 386, 334–343. [Google Scholar] [CrossRef] [Green Version]
- Gouvea, V.; Glass, R.I.; Woods, P.; Taniguchi, K.; Clark, H.F.; Forrester, B.; Fang, Z.Y. Polymerase chain reaction amplification and typing of rotavirus nucleic acid from stool specimens. J. Clin. Microbiol. 1990, 28, 276–282. [Google Scholar] [CrossRef] [Green Version]
- Theuns, S.; Desmarets, L.M.B.; Heylen, E.; Zeller, M.; Dedeurwaerder, A.; Roukaerts, I.D.M.; Van Ranst, M.; Matthijnssens, J.; Nauwynck, H.J. Porcine group a rotaviruses with heterogeneous VP7 and VP4 genotype combinations can be found together with enteric bacteria on Belgian swine farms. Vet. Microbiol. 2014, 172, 23–34. [Google Scholar] [CrossRef]
- Robardet, E.; Demerson, J.-M.; Andrieu, S.; Cliquet, F. First European Interlaboratory Comparison of Tetracycline and Age Determination with Red Fox Teeth Following Oral Rabies Vaccination Programs. J. Wildl. Dis. 2012, 48, 858–868. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular evolutionary genetics analysis across computing platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2019; Available online: https://www.r-project.org/ (accessed on 12 January 2021).
- Grant, B.J.; Rodrigues, A.P.C.; ElSawy, K.M.; McCammon, J.A.; Caves, L.S.D. Bio3d: An R package for the comparative analysis of protein structures. Bioinformatics 2006, 22, 2695–2696. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| County | The Number of RVA-Positive/Sampled Foxes | G Genotype | P Genotype | ||
|---|---|---|---|---|---|
| 2018 | 2019 | Total | |||
| Bjelovar-Bilogora | 0/0 | 1/2 | 1/2 | G9 | |
| Brod-Posavina | 0/1 | 2/7 | 2/8 | G9 | |
| Istria | 4/18 | 3/11 | 7/29 | G6, G9 | P[3] |
| Karlovac | 0/15 | 3/19 | 3/34 | G9 | P[13] |
| Krapina-Zagorje | 1/15 | 1/2 | 2/17 | ||
| Lika-Senj | 0/1 | 0/5 | 0/6 | ||
| Međimurje | 0/1 | 1/6 | 1/7 | ||
| Osijek-Baranja | 5/21 | 8/41 | 13/62 | G2, G5, G8, G9, G10 | P[11], P[23], P[?] * |
| Požega-Slavonia | 0/4 | 1/2 | 1/6 | ||
| Primorje-Gorski kotar | 1/14 | 2/19 | 3/33 | G10 | P[3] |
| Sisak-Moslavina | 3/19 | 2/9 | 5/28 | G1, G11, G15 | P[13], P[21] |
| Varaždin | 1/22 | 1/6 | 2/28 | P[?] * | |
| Virovitica-Podravina | 1/4 | 0/5 | 1/9 | ||
| Vukovar-Srijem | 7/34 | 1/28 | 8/62 | G1, G3, G8, G9, G? * | P[14], P[?] * |
| Zagreb County | 0/19 | 4/11 | 4/30 | G2 | P[11] |
| City of Zagreb | 1/4 | 1/5 | 2/9 | ||
| Total | 24/192 | 31/178 | 55/370 | ||
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Čolić, D.; Krešić, N.; Mihaljević, Ž.; Andreanszky, T.; Balić, D.; Lolić, M.; Brnić, D. A Remarkable Genetic Diversity of Rotavirus A Circulating in Red Fox Population in Croatia. Pathogens 2021, 10, 485. https://doi.org/10.3390/pathogens10040485
Čolić D, Krešić N, Mihaljević Ž, Andreanszky T, Balić D, Lolić M, Brnić D. A Remarkable Genetic Diversity of Rotavirus A Circulating in Red Fox Population in Croatia. Pathogens. 2021; 10(4):485. https://doi.org/10.3390/pathogens10040485
Chicago/Turabian StyleČolić, Daniel, Nina Krešić, Željko Mihaljević, Tibor Andreanszky, Davor Balić, Marica Lolić, and Dragan Brnić. 2021. "A Remarkable Genetic Diversity of Rotavirus A Circulating in Red Fox Population in Croatia" Pathogens 10, no. 4: 485. https://doi.org/10.3390/pathogens10040485
APA StyleČolić, D., Krešić, N., Mihaljević, Ž., Andreanszky, T., Balić, D., Lolić, M., & Brnić, D. (2021). A Remarkable Genetic Diversity of Rotavirus A Circulating in Red Fox Population in Croatia. Pathogens, 10(4), 485. https://doi.org/10.3390/pathogens10040485