
Predominance of the SARS-CoV-2 Lineage P.1 and Its Sublineage P.1.2 in Patients from the Metropolitan Region of Porto Alegre, Southern Brazil in March 2021

 , , , , ,
, , , , , 
Abstract
:1. Introduction
2. Results
2.1. Epidemiological Information
2.2. SARS-CoV-2 Mutations and Lineages
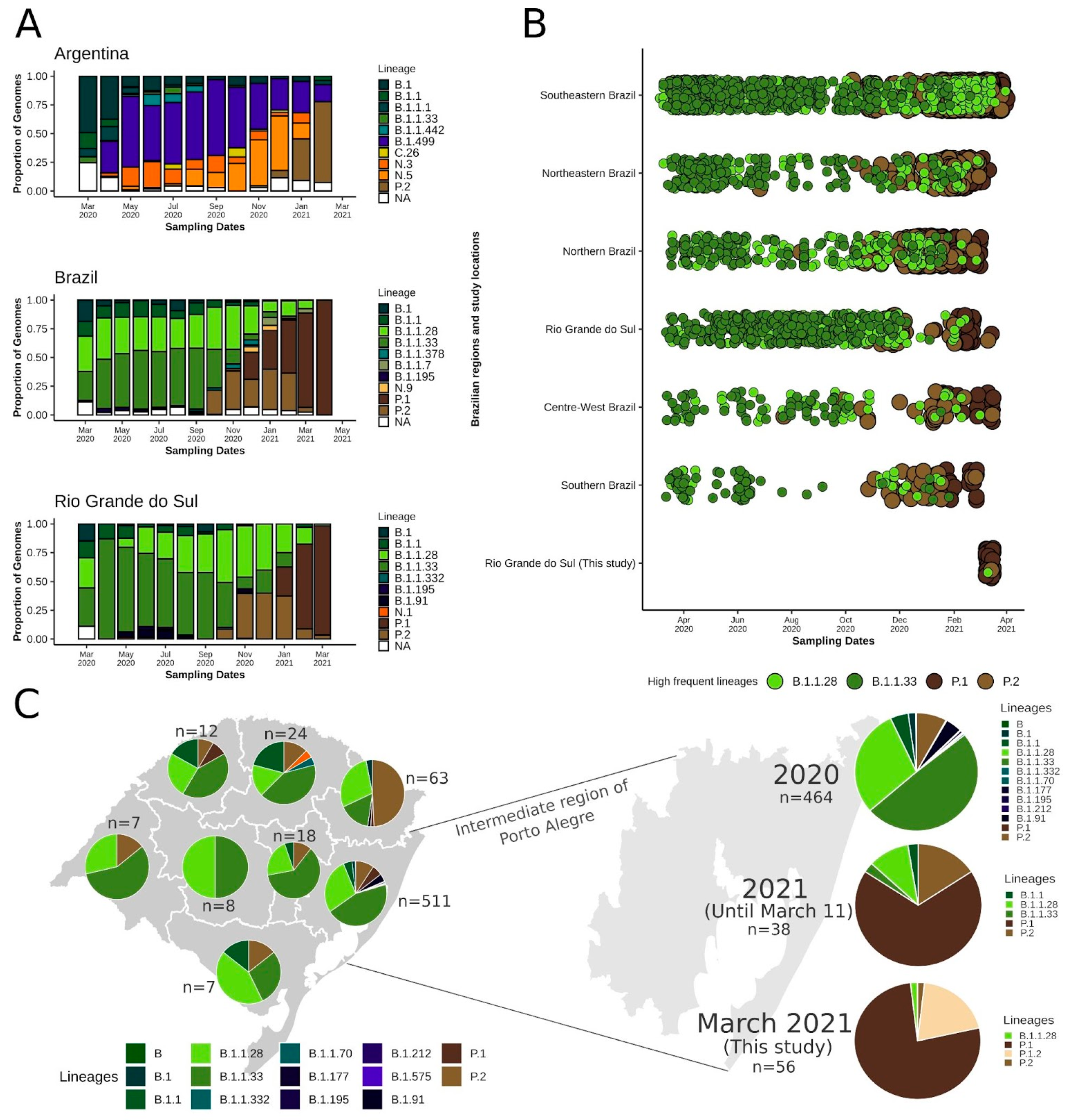
2.3. Lineage Distribution in Neighboring Countries and Brazilian Regions
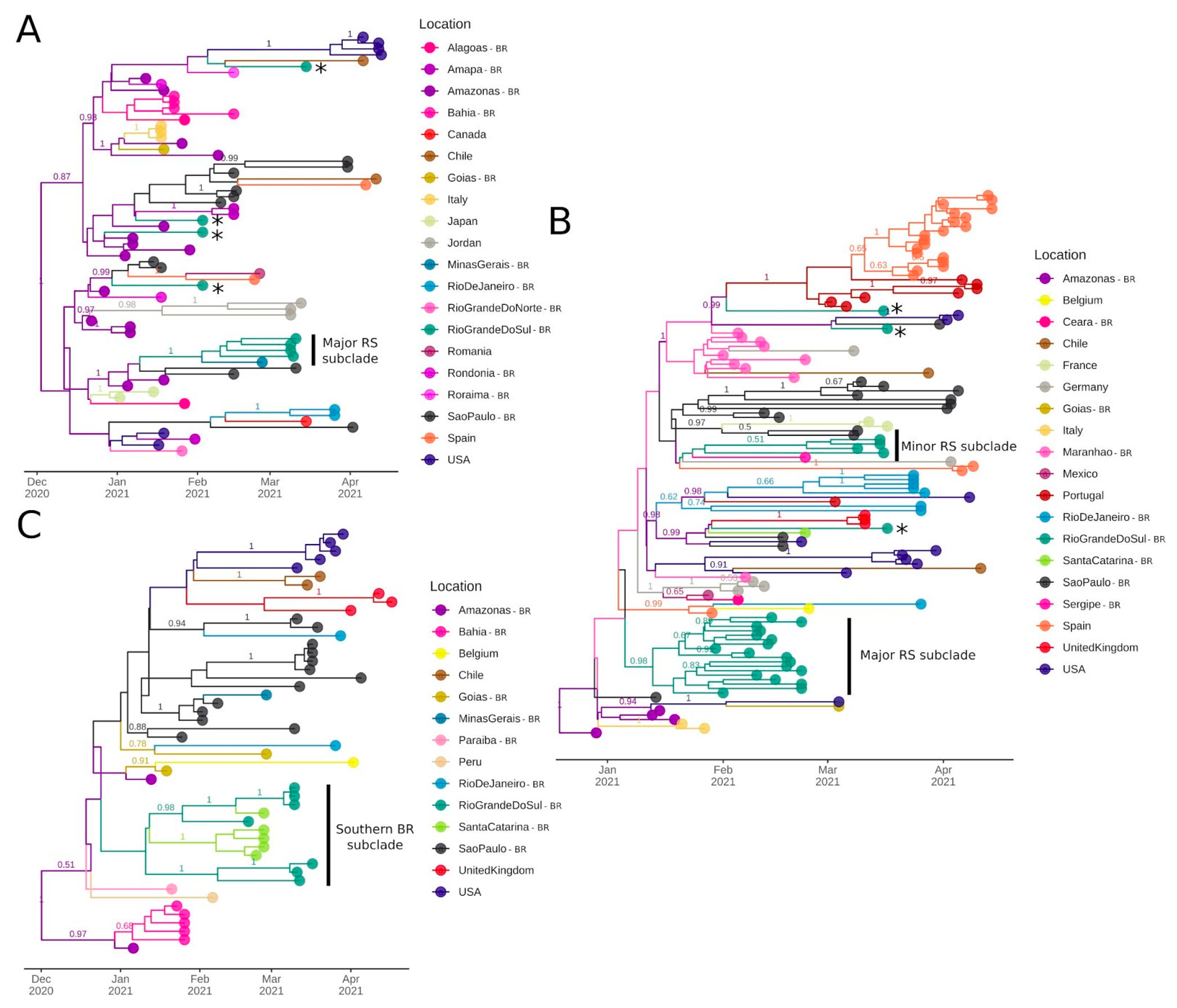
2.4. Maximum Likelihood Phylogenomic Analysis
2.5. Bayesian Molecular Clock and Phylogeographic Analysis
3. Discussion
4. Materials and Methods
4.1. Sample Collection and Clinical Testing
4.2. RNA Extraction, Library Preparation, and Sequencing
4.3. Quality Control and Consensus Calling
4.4. Mutation Analysis
4.5. Maximum Likelihood Phylogenomic Analysis
4.6. Discrete Bayesian Phylogeographic and Phylodynamic Analysis
4.7. Geoplotting
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- World Health Organization. WHO Director-General’s Opening Remarks at the Media Briefing on COVID-19—11 March 2020. Available online: https://www.who.int/director-general/speeches/detail/who-director-general-s-opening-remarks-at-the-media-briefing-on-covid-19---11-march-2020 (accessed on 27 May 2021).
- Candido, D.; Claro, I.M.; de Jesus, J.G.; Souza, W.M.; Moreira, F.R.R.; Dellicour, S.; Mellan, T.A.; du Plessis, L.; Pereira, R.H.M.; Sales, F.C.S.; et al. Evolution and Epidemic Spread of SARS-CoV-2 in Brazil. Science 2020, 369, 1255–1260. [Google Scholar] [CrossRef] [PubMed]
- IBGE (Brazilian Institute of Geography and Statistics) Rio Grande do Sul—Cidades e Estados. Available online: https://www.ibge.gov.br/cidades-e-estados/rs.html (accessed on 17 May 2021).
- IBGE (Brazilian Institute of Geography and Statistics) Regiões Geográficas. Available online: https://www.ibge.gov.br/apps/regioes_geograficas/ (accessed on 17 May 2021).
- Rio Grande do Sul Department of Health—SES-RS Confirmado o Primeiro Caso de Novo Coronavírus no Rio Grande do Sul. Available online: https://saude.rs.gov.br/confirmado-o-primeiro-caso-de-novo-coronavirus-no-rio-grande-do-sul (accessed on 24 November 2020).
- Secretaria de Planejamento, Governança e Gestão—Governo do Estado do Rio Grande do Sul Cogestão Regional—Distanciamento Controlado. Available online: https://distanciamentocontrolado.rs.gov.br/ (accessed on 17 May 2021).
- Mullen, J.L.; Tsueng, G.; Latif, A.A.; Alkuzweny, M.; Cano, M.; Haag, E.; Zhou, J.; Zeller, M.; Matteson, N.; Andersen, K.G.; et al. Outbreak.Info. Available online: https://outbreak.info (accessed on 19 July 2021).
- Rambaut, A.; Loman, N.; Pybus, O.; Barclay, W.; Barrett, J.; Carabelli, A.; Connor, T.; Peacock, T.; Robertson, D.; Volz, E.; et al. Preliminary Genomic Characterisation of an Emergent SARS-CoV-2 Lineage in the UK Defined by a Novel Set of Spike Mutations. Available online: https://virological.org/t/preliminary-genomic-characterisation-of-an-emergent-sars-cov-2-lineage-in-the-uk-defined-by-a-novel-set-of-spike-mutations/563 (accessed on 4 January 2021).
- Tegally, H.; Wilkinson, E.; Giovanetti, M.; Iranzadeh, A.; Fonseca, V.; Giandhari, J.; Doolabh, D.; Pillay, S.; San, E.J.; Msomi, N.; et al. Detection of a SARS-CoV-2 Variant of Concern in South Africa. Nature 2021, 592, 438–443. [Google Scholar] [CrossRef]
- Faria, N.; Mellan, T.A.; Whittaker, C.; Claro, I.M.; Candido, D.D.S.; Mishra, S.; Crispim, M.A.E.; Sales, F.C.S.; Hawryluk, I.; McCrone, J.T.; et al. Genomics and Epidemiology of the P.1 SARS-CoV-2 Lineage in Manaus, Brazil. Science 2021, 372, 815–821. [Google Scholar] [CrossRef]
- Naveca, F.; Nascimento, V.; Souza, V.; Corado, A.; Nascimento, F.; Silva, G.; Costa, Á.; Duarte, D.; Pessoa, K.; Mejía, M.; et al. COVID-19 Epidemic in the Brazilian State of Amazonas Was Driven by Long-Term Persistence of Endemic SARS-CoV-2 Lineages and the Recent Emergence of the New Variant of Concern P.1. Available online: https://www.researchsquare.com/article/rs-275494/v1 (accessed on 1 March 2021).
- Dhar, M.S.; Marwal, R.; Radhakrishnan, V.S.; Ponnusamy, K.; Jolly, B.; Bhoyar, R.C.; Sardana, V.; Naushin, S.; Rophina, M.; Mellan, T.A.; et al. Genomic Characterization and Epidemiology of an Emerging SARS-CoV-2 Variant in Delhi, India. medRxiv 2021, 6, 21258076. [Google Scholar] [CrossRef]
- Peacock, T.P.; Sheppard, C.M.; Brown, J.C.; Goonawardane, N.; Zhou, J.; Whiteley, M.; Consortium, P.V.; de Silva, T.I.; Barclay, W.S. The SARS-CoV-2 Variants Associated with Infections in India, B.1.617, Show Enhanced Spike Cleavage by Furin. bioRxiv 2021, 5, 446163. [Google Scholar] [CrossRef]
- Buss, L.F.; Prete, C.A.; Abrahim, C.M.M.; Mendrone, A.; Salomon, T.; de Almeida-Neto, C.; França, R.F.O.; Belotti, M.C.; Carvalho, M.P.S.S.; Costa, A.G.; et al. Three-Quarters Attack Rate of SARS-CoV-2 in the Brazilian Amazon during a Largely Unmitigated Epidemic. Science 2021, 371, 288–292. [Google Scholar] [CrossRef]
- Sabino, E.C.; Buss, L.F.; Carvalho, M.P.S.; Prete, C.A.; Crispim, M.A.E.; Fraiji, N.A.; Pereira, R.H.M.; Parag, K.V.; da Silva Peixoto, P.; Kraemer, M.U.; et al. Resurgence of COVID-19 in Manaus, Brazil, despite High Seroprevalence. Lancet 2021, 397, 452–455. [Google Scholar] [CrossRef]
- Brazilian Ministry of Health Painel Coronavírus Brasil. Available online: https://covid.saude.gov.br/ (accessed on 17 May 2021).
- Franceschi, V.B.; Ferrareze, P.A.G.; Zimerman, R.A.; Cybis, G.B.; Thompson, C.E. Mutation Hotspots, Geographical and Temporal Distribution of SARS-CoV-2 Lineages in Brazil, February 2020 to February 2021: Insights and Limitations from Uneven Sequencing Efforts. medRxiv 2021, 8, 21253152. [Google Scholar] [CrossRef]
- Lamarca, A.P.; de Almeida, L.G.P.; Francisco, R. da S.; Lima, L.F.A.; Scortecci, K.C.; Perez, V.P.; Brustolini, O.J.; Sousa, E.S.S.; Secco, D.A.; Santos, A.M.G.; et al. Genomic Surveillance of SARS-CoV-2 Tracks Early Interstate Transmission of P.1 Lineage and Diversification within P.2 Clade in Brazil. medRxiv 2021, 3, 21253418. [Google Scholar] [CrossRef]
- Rio Grande do Sul Health Surveillance Center Genomic Bulletin 5 (16/04/2021). Available online: https://coronavirus.rs.gov.br/upload/arquivos/202104/16173629-vigilancia-genomica-rs-boletim05-compactado.pdf (accessed on 20 April 2021).
- Shu, Y.; McCauley, J. GISAID: Global Initiative on Sharing All Influenza Data—From Vision to Reality. Eurosurveillance 2017, 22, 30494. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- De Almeira, L.G.; Lamarca, A.P.; Francisco Junior, R.d.S.; Cavalcante, L.; Gerber, A.L.; Guimarães, A.P.d.C.; Machado, D.T.; Alves, C.; Mariani, D.; Cruz, T.F.; et al. Genomic Surveillance of SARS-CoV-2 in the State of Rio de Janeiro, Brazil: Technical Briefing—SARS-CoV-2 Coronavirus/NCoV-2019 Genomic Epidemiology. Available online: https://virological.org/t/genomic-surveillance-of-sars-cov-2-in-the-state-of-rio-de-janeiro-brazil-technical-briefing/683 (accessed on 4 May 2021).
- Voloch, C.M.; Francisco, R.D.S.; de Almeida, L.G.P.; Cardoso, C.C.; Brustolini, O.J.; Gerber, A.L.; Guimarães, A.P.D.C.; Mariani, D.; da Costa, R.M.; Ferreira, O.C.; et al. Genomic Characterization of a Novel SARS-CoV-2 Lineage from Rio de Janeiro, Brazil. J. Virol. 2021, 95. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, V.B.; Caldana, G.D.; de Menezes Mayer, A.; Cybis, G.B.; Neves, C.A.M.; Ferrareze, P.A.G.; Demoliner, M.; de Almeida, P.R.; Gularte, J.S.; Hansen, A.W.; et al. Genomic Epidemiology of SARS-CoV-2 in Esteio, Rio Grande Do Sul, Brazil. BMC Genom. 2021, 22, 371. [Google Scholar] [CrossRef]
- Francisco, R.D.S., Jr.; Benites, L.F.; Lamarca, A.P.; de Almeida, L.G.P.; Hansen, A.W.; Gularte, J.S.; Demoliner, M.; Gerber, A.L.; Guimarães, A.P.D.C.; Antunes, A.K.E.; et al. Pervasive Transmission of E484K and Emergence of VUI-NP13L with Evidence of SARS-CoV-2 Co-Infection Events by Two Different Lineages in Rio Grande Do Sul, Brazil. Virus Res. 2021, 296, 198345. [Google Scholar] [CrossRef] [PubMed]
- Salvato, R.S.; Gregianini, T.S.; Campos, A.A.S.; Crescente, L.V.; Vallandro, M.J.; Ranieri, T.M.S.; Vizeu, S.; Martins, L.G.; da Silva, E.V.; Pedroso, E.R.; et al. Epidemiological Investigation Reveals Local Transmission of SARS-CoV-2 Lineage P.1 in Southern Brazil. Rev. Epidemiol. E Controle Infecção 2021, 1, 1–6. [Google Scholar] [CrossRef]
- Soares da Silva, M.; Demoliner, M.; Hansen, A.; Gularte, J.; Silveira, F.; Heldt, F.; Filippi, M.; Pereira da Silva, F.; Mallmann, L.; Fink, P.; et al. Early Detection of SARS-CoV-2 P.1 Variant in Southern Brazil and Reinfection of the Same Patient by P.2. Available online: https://www.researchsquare.com (accessed on 14 May 2021).
- Kubik, S.; Marques, A.C.; Xing, X.; Silvery, J.; Bertelli, C.; De Maio, F.; Pournaras, S.; Burr, T.; Duffourd, Y.; Siemens, H.; et al. Recommendations for Accurate Genotyping of SARS-CoV-2 Using Amplicon-Based Sequencing of Clinical Samples. Clin. Microbiol. Infect. 2021, 27, 1036. [Google Scholar] [CrossRef] [PubMed]
- Walls, A.C.; Park, Y.-J.; Tortorici, M.A.; Wall, A.; McGuire, A.T.; Veesler, D. Structure, Function, and Antigenicity of the SARS-CoV-2 Spike Glycoprotein. Cell 2020, 181, 281–292. [Google Scholar] [CrossRef] [PubMed]
- Deng, X.; Garcia-Knight, M.A.; Khalid, M.M.; Servellita, V.; Wang, C.; Morris, M.K.; Sotomayor-González, A.; Glasner, D.R.; Reyes, K.R.; Gliwa, A.S.; et al. Transmission, Infectivity, and Neutralization of a Spike L452R SARS-CoV-2 Variant. Cell 2021, 184, 3426–3437. [Google Scholar] [CrossRef]
- Martin, D.P.; Weaver, S.; Tegally, H.; San, E.J.; Shank, S.D.; Wilkinson, E.; Giandhari, J.; Naidoo, S.; Pillay, Y.; Singh, L.; et al. The Emergence and Ongoing Convergent Evolution of the N501Y Lineages Coincides with a Major Global Shift in the SARS-CoV-2 Selective Landscape. medRxiv 2021, 2, 21252268. [Google Scholar] [CrossRef]
- Resende, P.C.; Delatorre, E.; Gräf, T.; Mir, D.; Motta, F.C.; Appolinario, L.R.; da Paixão, A.C.D.; Mendonça, A.C.D.F.; Ogrzewalska, M.; Caetano, B.; et al. Evolutionary Dynamics and Dissemination Pattern of the SARS-CoV-2 Lineage B.1.1.33 During the Early Pandemic Phase in Brazil. Front. Microbiol. 2021, 11, 615280. [Google Scholar] [CrossRef]
- Mir, D.; Rego, N.; Resende, P.C.; López-Tort, F.; Fernandez-Calero, T.; Noya, V.; Brandes, M.; Possi, T.; Arleo, M.; Reyes, N.; et al. Recurrent Dissemination of SARS-CoV-2 through the Uruguayan-Brazilian Border. medRxiv 2021, 1, 20249026. [Google Scholar] [CrossRef]
- Naveca, F.; Nascimento, V.; Souza, V.; Corado, A.; Nascimento, F.; Silva, G.; Costa, Á.; Duarte, D.; Pessoa, K.; Gonçalves, L.; et al. Phylogenetic Relationship of SARS-CoV-2 Sequences from Amazonas with Emerging Brazilian Variants Harboring Mutations E484K and N501Y in the Spike Protein. Available online: https://virological.org/t/phylogenetic-relationship-of-sars-cov-2-sequences-from-amazonas-with-emerging-brazilian-variants-harboring-mutations-e484k-and-n501y-in-the-spike-protein/585 (accessed on 24 February 2021).
- Choi, B.; Choudhary, M.C.; Regan, J.; Sparks, J.A.; Padera, R.F.; Qiu, X.; Solomon, I.H.; Kuo, H.-H.; Boucau, J.; Bowman, K.; et al. Persistence and Evolution of SARS-CoV-2 in an Immunocompromised Host. N. Engl. J. Med. 2020, 383, 2291–2293. [Google Scholar] [CrossRef]
- Kemp, S.A.; Collier, D.A.; Datir, R.P.; Ferreira, I.A.T.M.; Gayed, S.; Jahun, A.; Hosmillo, M.; Rees-Spear, C.; Mlcochova, P.; Lumb, I.U.; et al. SARS-CoV-2 Evolution during Treatment of Chronic Infection. Nature 2021, 592, 277–282. [Google Scholar] [CrossRef] [PubMed]
- Gräf, T.; Bello, G.; Venas, T.M.M.; Pereira, E.C.; Paixão, A.C.D.; Appolinario, L.R.; Lopes, R.S.; Mendonça, A.C.d.F.; da Rocha, A.S.B.; Motta, F.C.; et al. Identification of SARS-CoV-2 P.1-Related Lineages in Brazil Provides New Insights about the Mechanisms of Emergence of Variants of Concern—SARS-CoV-2 Coronavirus / NCoV-2019 Genomic Epidemiology. Available online: https://virological.org/t/identification-of-sars-cov-2-p-1-related-lineages-in-brazil-provides-new-insights-about-the-mechanisms-of-emergence-of-variants-of-concern/694/1 (accessed on 17 May 2021).
- Plante, J.A.; Mitchell, B.M.; Plante, K.S.; Debbink, K.; Weaver, S.C.; Menachery, V.D. The Variant Gambit: COVID-19′s next Move. Cell Host Microbe 2021, 29, 508–515. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Mallick, D.; Banerjee, K.; Sarkar, S.; Lee, S.T.M.; Basuchowdhuri, P.; Jana, S.S. D155Y Substitution of SARS-CoV-2 ORF3a Weakens Binding with Caveolin-1: An in Silico Study. bioRxiv 2021, 3, 437194. [Google Scholar] [CrossRef]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 Novel Coronavirus (2019-NCoV) by Real-Time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization COVID-19 Clinical Management: Living Guidance. Available online: https://www.who.int/publications-detail-redirect/WHO-2019-nCoV-clinical-2021-1 (accessed on 1 May 2021).
- Eden, J.-S. SARS-CoV-2 Genome Sequencing Using Long Pooled Amplicons on Illumina Platforms. bioRxiv 2020. [Google Scholar] [CrossRef] [Green Version]
- Langmead, B.; Salzberg, S.L. Fast Gapped-Read Alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. The Sequence Alignment/Map Format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Li, H. A Statistical Framework for SNP Calling, Mutation Discovery, Association Mapping and Population Genetical Parameter Estimation from Sequencing Data. Bioinformatics 2011, 27, 2987–2993. [Google Scholar] [CrossRef] [Green Version]
- Gel, B.; Serra, E. KaryoploteR: An R/Bioconductor Package to Plot Customizable Genomes Displaying Arbitrary Data. Bioinforma. Oxf. Engl. 2017, 33, 3088–3090. [Google Scholar] [CrossRef]
- Garrison, E.; Marth, G. Haplotype-Based Variant Detection from Short-Read Sequencing. arXiv 2012, arXiv:1207.3907. Available online: http://arxiv.org/abs/1207.3907 (accessed on 4 May 2021).
- Cingolani, P.; Platts, A.; Wang, L.L.; Coon, M.; Nguyen, T.; Wang, L.; Land, S.J.; Lu, X.; Ruden, D.M. A Program for Annotating and Predicting the Effects of Single Nucleotide Polymorphisms, SnpEff. Fly 2012, 6, 80–92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Du Plessis, L. Laduplessis/SARS-CoV-2_Guangdong_Genomic_Epidemiology; Initial Release; Zenodo. Available online: https://zenodo.org/record/3922606 (accessed on 4 May 2021).
- Rambaut, A.; Holmes, E.C.; O’Toole, Á.; Hill, V.; McCrone, J.T.; Ruis, C.; du Plessis, L.; Pybus, O.G. A Dynamic Nomenclature Proposal for SARS-CoV-2 Lineages to Assist Genomic Epidemiology. Nat. Microbiol. 2020, 5, 1403–1407. [Google Scholar] [CrossRef] [PubMed]
- Argimón, S.; Abudahab, K.; Goater, R.J.E.; Fedosejev, A.; Bhai, J.; Glasner, C.; Feil, E.J.; Holden, M.T.G.; Yeats, C.A.; Grundmann, H.; et al. Microreact: Visualizing and Sharing Data for Genomic Epidemiology and Phylogeography. Microb. Genom. 2016, 2, e000093. [Google Scholar] [CrossRef] [Green Version]
- Hadfield, J.; Megill, C.; Bell, S.M.; Huddleston, J.; Potter, B.; Callender, C.; Sagulenko, P.; Bedford, T.; Neher, R.A. Nextstrain: Real-Time Tracking of Pathogen Evolution. Bioinformatics 2018, 34, 4121–4123. [Google Scholar] [CrossRef]
- Nguyen, L.-T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A Fast and Effective Stochastic Algorithm for Estimating Maximum-Likelihood Phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef]
- Tavare, S. Some Probabilistic and Statistical Problems in the Analysis of DNA Sequences. Lect. Math. Life Sci. 1986, 17, 57–86. [Google Scholar]
- Sagulenko, P.; Puller, V.; Neher, R.A. TreeTime: Maximum-Likelihood Phylodynamic Analysis. Virus Evol. 2018, 4. [Google Scholar] [CrossRef]
- Kalyaanamoorthy, S.; Minh, B.Q.; Wong, T.K.F.; von Haeseler, A.; Jermiin, L.S. ModelFinder: Fast Model Selection for Accurate Phylogenetic Estimates. Nat. Methods 2017, 14, 587–589. [Google Scholar] [CrossRef] [Green Version]
- Guindon, S.; Dufayard, J.-F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New Algorithms and Methods to Estimate Maximum-Likelihood Phylogenies: Assessing the Performance of PhyML 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rambaut, A.; Lam, T.T.; Max Carvalho, L.; Pybus, O.G. Exploring the Temporal Structure of Heterochronous Sequences Using TempEst (Formerly Path-O-Gen). Virus Evol. 2016, 2, vew007. [Google Scholar] [CrossRef] [Green Version]
- Yu, G.; Smith, D.K.; Zhu, H.; Guan, Y.; Lam, T.T.-Y. Ggtree: An r Package for Visualization and Annotation of Phylogenetic Trees with Their Covariates and Other Associated Data. Methods Ecol. Evol. 2017, 8, 28–36. [Google Scholar] [CrossRef]
- Orme, D.; Freckleton, R.; Thomas, G.; Petzoldt, T.; Fritz, S.; Isaac, N.; Pearse, W. Caper: Comparative Analyses of Phylogenetics and Evolution in R (R Package Version 1.0.1). Available online: https://www.scienceopen.com/document?vid=d750bff2-a400-41dd-aa1e-728bb7aaf4d5 (accessed on 10 May 2021).
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian Phylogenetic and Phylodynamic Data Integration Using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef] [Green Version]
- Ayres, D.L.; Darling, A.; Zwickl, D.J.; Beerli, P.; Holder, M.T.; Lewis, P.O.; Huelsenbeck, J.P.; Ronquist, F.; Swofford, D.L.; Cummings, M.P.; et al. BEAGLE: An Application Programming Interface and High-Performance Computing Library for Statistical Phylogenetics. Syst. Biol. 2012, 61, 170–173. [Google Scholar] [CrossRef] [PubMed]
- Hasegawa, M.; Kishino, H.; Yano, T. Dating of the Human-Ape Splitting by a Molecular Clock of Mitochondrial DNA. J. Mol. Evol. 1985, 22, 160–174. [Google Scholar] [CrossRef]
- Ferreira, M.A.R.; Suchard, M.A. Bayesian Analysis of Elapsed Times in Continuous-Time Markov Chains. Can. J. Stat. 2008, 36, 355–368. [Google Scholar] [CrossRef]
- Gill, M.S.; Lemey, P.; Faria, N.R.; Rambaut, A.; Shapiro, B.; Suchard, M.A. Improving Bayesian Population Dynamics Inference: A Coalescent-Based Model for Multiple Loci. Mol. Biol. Evol. 2013, 30, 713–724. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A.; Drummond, A.J.; Xie, D.; Baele, G.; Suchard, M.A. Posterior Summarization in Bayesian Phylogenetics Using Tracer 1.7. Syst. Biol. 2018, 67, 901–904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lemey, P.; Rambaut, A.; Drummond, A.J.; Suchard, M.A. Bayesian Phylogeography Finds Its Roots. PLoS Comput. Biol. 2009, 5, e1000520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- R Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2020. [Google Scholar]
- Wickham, H. Ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2009; ISBN 978-0-387-98141-3. [Google Scholar]
- Pereira, R.; Gonçalves, C.; De Araujo, P.; Carvalho, G.; De Arruda, R.; Nascimento, I.; Da Costa, B.; Cavedo, W.; Andrade, P.; Da Silva, A.; et al. Geobr: Loads Shapefiles of Official Spatial Data Sets of Brazil. Available online: https://github.com/ipeaGIT/geobr (accessed on 4 May 2021).
- Pebesma, E. Simple Features for R: Standardized Support for Spatial Vector Data. R. J. 2018, 10, 439–446. [Google Scholar] [CrossRef] [Green Version]
- Bielejec, F.; Baele, G.; Vrancken, B.; Suchard, M.A.; Rambaut, A.; Lemey, P. SpreaD3: Interactive Visualization of Spatiotemporal History and Trait Evolutionary Processes. Mol. Biol. Evol. 2016, 33, 2167–2169. [Google Scholar] [CrossRef] [PubMed] [Green Version]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Study ID | GISAID ID | Cycle Threshold | Municipality of Residence | Gender | Age Group | Lineage | Contact with Confirmed Case |
|---|---|---|---|---|---|---|---|
| (HBM-RS) | (EPI_ISL_) | ||||||
| 39468 | 2139494 | 16 | São Leopoldo | Male | 30–39 | P.1 | Yes |
| 39469 | 2139495 | 19 | Porto Alegre | Female | 20–29 | P.1.2 | Yes |
| 39470 | 2139496 | 19 | Porto Alegre | Male | 60–69 | P.1 | No |
| 39471 | 2139497 | 18 | Porto Alegre | Male | 20–29 | P.1 | Yes |
| 39472 | 2139498 | 17 | Gravataí | Male | 30–39 | P.1 | No |
| 39473 | 2139499 | 26 | Cachoeira do Sul | Female | 20–29 | P.1 | Yes |
| 39474 | 2139500 | 18 | Gravataí | Male | 30–39 | P.1 | Yes |
| 39475 | 2139501 | 18 | Porto Alegre | Female | 20–29 | P.1 | No |
| 39476 | 2139502 | 15 | Porto Alegre | Male | 20–29 | P.1 | Yes |
| 39477 | 2139503 | 21 | Porto Alegre | Male | 30–39 | P.1 | Yes |
| 39478 | 2139504 | 15 | Cachoeira do Sul | Male | 30–39 | P.1 | Yes |
| 39479 | 2139505 | 22 | Porto Alegre | Male | 50–59 | P.1 | Yes |
| 39480 | 2139506 | 17 | Novo Hamburgo | Male | 40–49 | P.1 | Yes |
| 39481 | 2139507 | 14 | Porto Alegre | Female | 70–79 | P.1 | Yes |
| 39482 | 2139508 | 14 | Porto Alegre | Female | 80–89 | P.1.2 | No |
| 39483 | 2139509 | 13 | Gravataí | Male | 30–39 | P.1 | Yes |
| 39484 | 2139510 | 20 | Porto Alegre | Male | 20–29 | P.1 | Yes |
| 39485 | 2139511 | 16 | Porto Alegre | Male | 50–59 | P.1 | Yes |
| 39486 | 2139512 | 27 | Porto Alegre | Male | 30–39 | P.2 | No |
| 39487 | 2139513 | 14 | São Sebastião do Caí | Male | 40–49 | P.1.2 | Yes |
| 39488 | 2139514 | 28 | Santo Antônio da Patrulha | Male | 70–79 | P.1 | Yes |
| 39489 | 2139515 | 27 | Porto Alegre | Female | 20–29 | P.1.2 | Yes |
| 39490 | 2139516 | 18 | Porto Alegre | Male | 20–29 | P.1 | Yes |
| 39491 | 2139517 | 15 | Alvorada | Female | 20–29 | B.1.1.28 | Yes |
| 39492 | 2139518 | 17 | Gravataí | Female | 30–39 | P.1 | Yes |
| 39493 | 2139519 | 22 | Canoas | Male | 30–39 | P.1.2 | Yes |
| 39494 | 2139520 | 17 | Porto Alegre | Female | 30–39 | P.1 | No |
| 39495 | 2139521 | 17 | Porto Alegre | Male | 30–39 | P.1 | Yes |
| 39496 | 2139522 | 17 | Canoas | Female | 30–39 | P.1 | Yes |
| 39497 | 2139523 | 21 | Porto Alegre | Male | 40–49 | P.1.2 | Yes |
| 39498 | 2139524 | 20 | Porto Alegre | Female | 40–49 | P.1 | Yes |
| 39499 | 2139525 | 22 | Portão | Male | 30–39 | P.1 | Yes |
| 39500 | 2139526 | 11 | Porto Alegre | Male | 20–29 | P.1.2 | Yes |
| 39501 | 2139527 | 14 | Santa Maria | Male | 20–29 | P.1.2 | Yes |
| 39502 | 2139528 | 21 | Porto Alegre | Male | 30–39 | P.1 | Yes |
| 39503 | 2139529 | 16 | Porto Alegre | Male | 30–39 | P.1 | No |
| 39504 | 2139530 | 21 | Gravataí | Male | 40–49 | P.1 | Yes |
| 39505 | 2139531 | 13 | Porto Alegre | Male | 30–39 | P.1.2 | Yes |
| 39506 | 2139532 | 23 | Porto Alegre | Female | 40–49 | P.1 | Yes |
| 39507 | 2139533 | 28 | Canoas | Female | 30–39 | P.1 | Yes |
| 39508 | 2139534 | 22 | Porto Alegre | Male | 20–29 | P.1 | Yes |
| 39509 | 2139535 | 23 | Alvorada | Male | 20–29 | P.1 | Yes |
| 39510 | 2139536 | 19 | Canoas | Male | 50–59 | P.1.2 | Yes |
| 39511 | 2139537 | 22 | Porto Alegre | Male | 30–39 | P.1 | No |
| 39512 | 2139538 | 25 | Cachoeira do Sul | Female | 40–49 | P.1 | Yes |
| 39513 | 2139539 | 23 | Santa Maria | Male | 40–49 | P.1 | Yes |
| 39514 | 2139540 | 15 | Porto Alegre | Male | 30–39 | P.1 | No |
| 39515 | 2139541 | 21 | Porto Alegre | Male | 20–29 | P.1 | Yes |
| 39516 | 2139542 | 28 | Porto Alegre | Male | 50–59 | P.1 | Yes |
| 39517 | 2139543 | 17 | Sapiranga | Male | 30–39 | P.1 | Yes |
| 39518 | 2139544 | 17 | Porto Alegre | Male | 30–39 | P.1.2 | Yes |
| 39519 | 2139545 | 23 | Porto Alegre | Male | 20–29 | P.1 | Yes |
| 39520 | 2139546 | 15 | Campo Bom | Male | 20–29 | P.1 | Yes |
| 39521 | 2139547 | 15 | Porto Alegre | Male | 20–29 | P.1 | Yes |
| 39522 | 2139548 | 21 | Porto Alegre | Male | 50–59 | P.1 | Yes |
| 39523 | 2139549 | 18 | Porto Alegre | Male | 20–29 | P.1 | Yes |
| Genomic Position | Effect | Amino Acid Change | Gene/Region | Product | Frequency Our Study (%) | Frequency in Brazilian’s P.1 (%) |
|---|---|---|---|---|---|---|
| C241T | Intergenic | NA | 5′ UTR | NA | 100.0 | 97.2 |
| T733C | Synonymous | D156D | ORF1ab | Leader Protein | 96.4 | 99.9 |
| C1912T | Synonymous | S549S | nsp2 | 19.6 | 1.4 | |
| A2550G | Missense | D762G | 19.6 | 1.5 | ||
| C2749T | Synonymous | D828D | nsp3 | 94.6 | 99.7 | |
| C3037T | Synonymous | F924F | 100.0 | 99.9 | ||
| C3828T | Missense | S1188L | 96.4 | 95.3 | ||
| A5648C | Missense | K1795Q | 85.7 | 100.0 | ||
| C5724T | Missense | T1820I | 17.9 | 2.3 | ||
| A6319G | Synonymous | P2018P | 87.5 | 99.5 | ||
| A6613G | Synonymous | V2116V | 96.4 | 99.8 | ||
| T11296G | Missense | F3677L | nsp6 | 30.4 | 8.2 | |
| C12778T | Synonymous | Y4171Y | nsp9 | 94.6 | 98.9 | |
| C13860T | Missense | T4532I | RdRp | 94.6 | 99.8 | |
| C14408T | Synonymous | L4715L | 98.2 | 96.8 | ||
| G17259T | Missense | S5665I | Helicase | 94.6 | 99.7 | |
| C21614T | Missense | L18F | S | Surface Glycoprotein | 96.4 | 99.9 |
| C21621A | Missense | T20N | 94.6 | 99.8 | ||
| C21638T | Missense | P26S | 96.4 | 99.1 | ||
| G21974T | Missense | D138Y | 96.4 | 100.0 | ||
| G22132T | Missense | R190S | 96.4 | 98.4 | ||
| A22812C | Missense | K417T | 96.4 | 83.4 | ||
| G23012A | Missense | E484K | 96.4 | 99.9 | ||
| A23063T | Missense | N501Y | 96.4 | 99.8 | ||
| A23403G | Missense | D614G | 100.0 | 97.7 | ||
| C23525T | Missense | H655Y | 96.4 | 100.0 | ||
| C24642T | Missense | T1027I | 96.4 | 99.9 | ||
| G25088T | Missense | V1176F | 100.0 | 99.9 | ||
| G25855T | Missense | D155Y | ORF3a | ORF3a Protein | 19.6 | 1.6 |
| T26149C | Missense | S253P | 94.6 | 98.7 | ||
| G28167A | Missense | E92K | ORF8 | ORF8 Protein | 94.6 | 99.8 |
| C28512G | Missense | P80R | N | Nucleocapsid Phosphoprotein | 96.4 | 98.3 |
| C28789T | Synonymous | Y172Y | 19.6 | 1.3 | ||
| AGTAGGG 28877–28883 TCTAAAC | Missense | RG203-204KR | 96.4 | 99.8 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Franceschi, V.B.; Caldana, G.D.; Perin, C.; Horn, A.; Peter, C.; Cybis, G.B.; Ferrareze, P.A.G.; Rotta, L.N.; Cadegiani, F.A.; Zimerman, R.A.; et al. Predominance of the SARS-CoV-2 Lineage P.1 and Its Sublineage P.1.2 in Patients from the Metropolitan Region of Porto Alegre, Southern Brazil in March 2021. Pathogens 2021, 10, 988. https://doi.org/10.3390/pathogens10080988
Franceschi VB, Caldana GD, Perin C, Horn A, Peter C, Cybis GB, Ferrareze PAG, Rotta LN, Cadegiani FA, Zimerman RA, et al. Predominance of the SARS-CoV-2 Lineage P.1 and Its Sublineage P.1.2 in Patients from the Metropolitan Region of Porto Alegre, Southern Brazil in March 2021. Pathogens. 2021; 10(8):988. https://doi.org/10.3390/pathogens10080988
Chicago/Turabian StyleFranceschi, Vinícius Bonetti, Gabriel Dickin Caldana, Christiano Perin, Alexandre Horn, Camila Peter, Gabriela Bettella Cybis, Patrícia Aline Gröhs Ferrareze, Liane Nanci Rotta, Flávio Adsuara Cadegiani, Ricardo Ariel Zimerman, and et al. 2021. "Predominance of the SARS-CoV-2 Lineage P.1 and Its Sublineage P.1.2 in Patients from the Metropolitan Region of Porto Alegre, Southern Brazil in March 2021" Pathogens 10, no. 8: 988. https://doi.org/10.3390/pathogens10080988
APA StyleFranceschi, V. B., Caldana, G. D., Perin, C., Horn, A., Peter, C., Cybis, G. B., Ferrareze, P. A. G., Rotta, L. N., Cadegiani, F. A., Zimerman, R. A., & Thompson, C. E. (2021). Predominance of the SARS-CoV-2 Lineage P.1 and Its Sublineage P.1.2 in Patients from the Metropolitan Region of Porto Alegre, Southern Brazil in March 2021. Pathogens, 10(8), 988. https://doi.org/10.3390/pathogens10080988