
Nasopharyngeal Carcinoma: The Role of the EGFR in Epstein–Barr Virus Infection


Abstract
:1. Epstein–Barr Virus (EBV)
1.1. Etiology of EBV
1.2. EBV Causes Disease by Infecting B Cells and Epithelial Cells
1.3. EBV Infection can Promote the Progression of Nasopharyngeal Carcinoma
1.4. LMP1 Protein Encoded by EBV Is Involved in the Progression of Nasopharyngeal Carcinoma
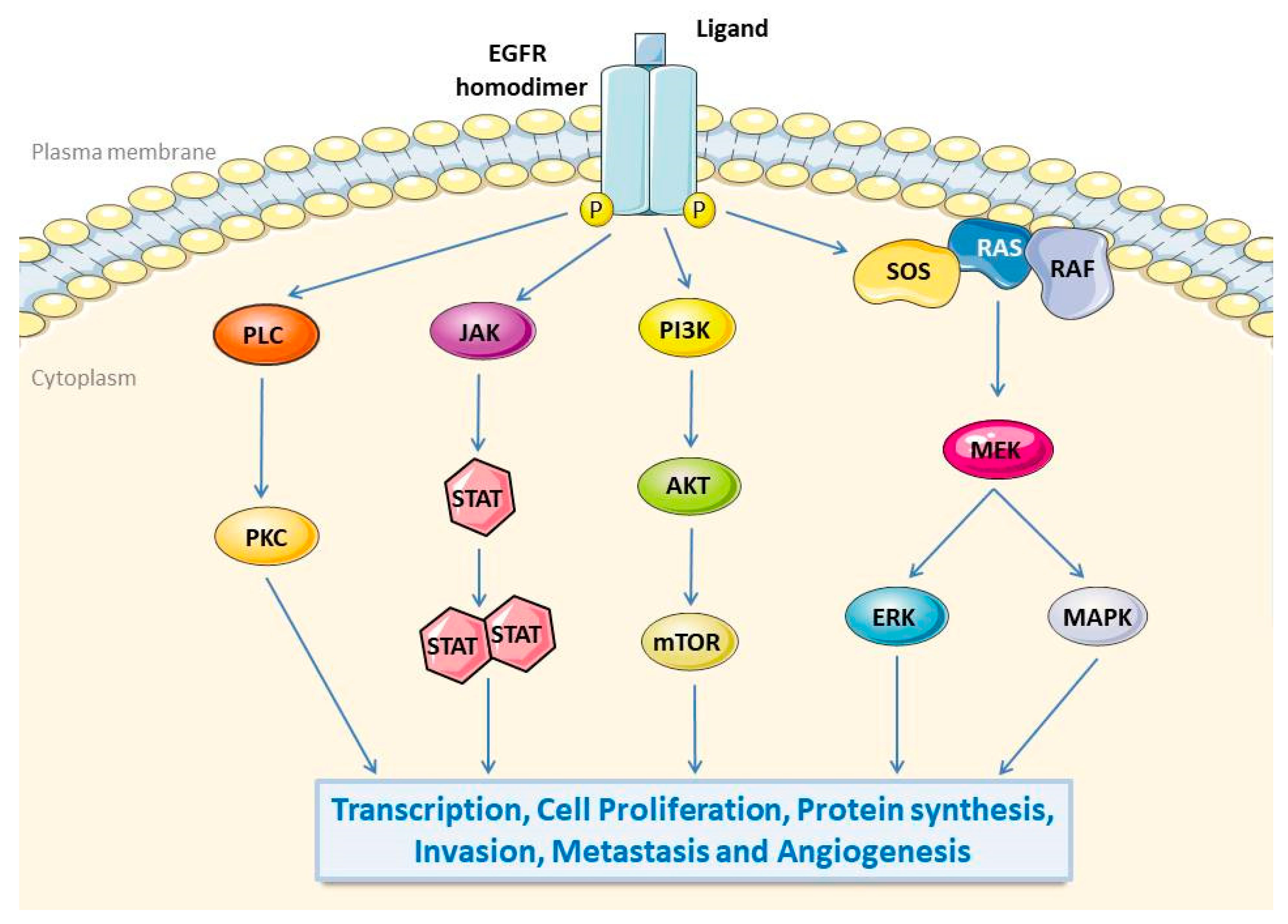
2. Epidermal Growth Factor Receptor (EGFR)
2.1. Biological Function of EGFR
2.2. EGFR Promotes Tumorigenesis and Progression
3. The Mechanism of LMP1-Mediated EGFR Expression and Nuclear Translocation
3.1. LMP1 Activates the EGFR through the NF-κB Pathway in NPC
3.2. LMP1 Activates EGFR through STAT3 in NPC
3.3. Others
4. The Role of EGFR Pathways in Nasopharyngeal Carcinoma
4.1. The Relationship between EGFR and Nasopharyngeal Carcinoma
4.2. EGFR Signalling Affects the Growth of Nasopharyngeal Carcinoma Cells
4.3. EGFR Promotes Invasion and Metastasis of Nasopharyngeal Carcinoma Cells
4.4. Other Roles of EGFR in Nasopharyngeal Carcinoma
5. EGFR Effects on EBV Infections
5.1. EGFR Is Overexpressed in EBV-Infected Cells
5.2. EGFR Enhances the Internalization and Fusion of EBV
6. The Role of EGFR Targeting in Nasopharyngeal Carcinoma
6.1. EGFR Monoclonal Antibody
6.1.1. Cetuximab for Nasopharyngeal Carcinoma
6.1.2. Nimotuzumab for Nasopharyngeal Carcinoma
6.2. Small Molecule EGFR Tyrosine Kinase Inhibitors (TKIs)
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fitzsimmons, L.; Kelly, G.L. EBV and Apoptosis: The Viral Master Regulator of Cell Fate? Viruses 2017, 9, 339. [Google Scholar] [CrossRef] [Green Version]
- Yin, H.; Qu, J.; Peng, Q.; Gan, R. Molecular mechanisms of EBV-driven cell cycle progression and oncogenesis. Med. Microbiol. Immunol. 2019, 208, 573–583. [Google Scholar] [CrossRef] [Green Version]
- Thorley-Lawson, D.A. EBV Persistence—Introducing the Virus. Curr. Top. Microbiol. Immunol. 2015, 390 Pt 1, 151–209. [Google Scholar]
- Münz, C. Latency and lytic replication in Epstein–Barr virus-associated oncogenesis. Nat. Rev. Genet. 2019, 17, 691–700. [Google Scholar] [CrossRef] [Green Version]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein–Barr virus infection and nasopharyngeal carcinoma. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160270. [Google Scholar] [CrossRef]
- Young, L.; Yap, L.-F.; Murray, P.G. Epstein–Barr virus: More than 50 years old and still providing surprises. Nat. Rev. Cancer 2016, 16, 789–802. [Google Scholar] [CrossRef]
- Kanda, T. EBV-Encoded Latent Genes. Adv. Exp. Med. Biol. 2018, 1045, 377–394. [Google Scholar]
- Edwards, R.H.; DeKroon, R.; Raab-Traub, N. Alterations in cellular expression in EBV infected epithelial cell lines and tumors. PLoS Pathog. 2019, 15, e1008071. [Google Scholar] [CrossRef]
- Zhang, H.; Li, Y.; Wang, H.-B.; Zhang, A.; Chen, M.-L.; Fang, Z.-X.; Dong, X.-D.; Li, S.-B.; Du, Y.; Xiong, D.; et al. Ephrin receptor A2 is an epithelial cell receptor for Epstein–Barr virus entry. Nat. Microbiol. 2018, 3, 164–171. [Google Scholar] [CrossRef]
- Wang, H.-B.; Zhang, H.; Zhang, J.-P.; Li, Y.; Zhao, B.; Feng, G.-K.; Du, Y.; Xiong, D.; Zhong, Q.; Liu, W.-L.; et al. Neuropilin 1 is an entry factor that promotes EBV infection of nasopharyngeal epithelial cells. Nat. Commun. 2015, 6, 6240. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Longnecker, R. Epithelial cell infection by Epstein–Barr virus. FEMS Microbiol. Rev. 2019, 43, 674–683. [Google Scholar] [CrossRef] [PubMed]
- Ferressini Gerpe, N.M.; Vistarop, A.G.; Moyano, A.; De Matteo, E.; Preciado, M.V.; Chabay, P.A. Distinctive EBV infection characteristics in children from a developing country. Int. J. Infect. Dis. 2020, 93, 139–145. [Google Scholar] [CrossRef]
- Dalton, T.; Doubrovina, E.; Pankov, D.; Reynolds, I.R.C.; Scholze, H.; Selvakumar, A.; Vizconde, T.; Savalia, B.; Dyomin, V.; Weigel, C.; et al. Epigenetic reprogramming sensitizes immunologically silent EBV+ lymphomas to virus-directed immunotherapy. Blood 2020, 135, 1870–1881. [Google Scholar] [CrossRef] [PubMed]
- Fu, W.; Verma, D.; Burton, A.; Swaminathan, S. Cellular RNA Helicase DHX9 Interacts with the Essential Epstein-Barr Virus (EBV) Protein SM and Restricts EBV Lytic Replication. J. Virol. 2019, 93, e01244-18. [Google Scholar] [CrossRef] [Green Version]
- Elgui de Oliveira, D.; Müller-Coan, B.G.; Pagano, J.S. Viral Carcinogenesis beyond Malignant Transformation: EBV in the Pro-gression of Human Cancers. Trends Microbiol. 2016, 24, 649–664. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.I. Primary Immunodeficiencies Associated with EBV Disease. Curr. Top. Microbiol. Immunol. 2015, 390, 241–265. [Google Scholar]
- Wu, H.-C.; Lin, Y.-J.; Lee, J.-J.; Liu, Y.-J.; Liang, S.-T.; Peng, Y.; Chiu, Y.-W.; Wu, C.-W.; Lin, C.-T. Functional Analysis of EBV in Nasopharyngeal Carcinoma Cells. Lab. Investig. 2003, 83, 797–812. [Google Scholar] [CrossRef] [Green Version]
- Nilsson, J.S.; Forslund, O.; Andersson, F.C.; Lindstedt, M.; Greiff, L. Intralesional EBV-DNA load as marker of prognosis for naso-pharyngeal cancer. Sci. Rep. 2019, 9, 15432. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dawson, C.W.; Port, R.J.; Young, L.S. The role of the EBV-encoded latent membrane proteins LMP1 and LMP2 in the pathogenesis of nasopharyngeal carcinoma (NPC). Semin. Cancer Biol. 2012, 22, 144–153. [Google Scholar] [CrossRef]
- Edwards, R.H.; Marquitz, A.R.; Raab-Traub, N. Changes in Expression Induced by Epstein-Barr Virus LMP1-CTAR1: Potential Role of bcl3. mBio 2015, 6, e00441-15. [Google Scholar] [CrossRef] [Green Version]
- Saridakis, V.; Sheng, Y.; Sarkari, F.; Holowaty, M.N.; Shire, K.; Nguyen, T.; Zhang, R.G.; Liao, J.; Lee, W.; Edwards, A.M.; et al. Structure of the p53 binding domain of HAUSP/USP7 bound to Epstein-Barr nuclear antigen 1 implications for EBV-mediated immortaliza-tion. Mol. Cell 2005, 18, 25–36. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Shi, Y.; Yuan, Q.; Liu, X.; Yan, B.; Chen, L.; Tao, Y.; Cao, Y. Epstein-Barr Virus encoded LMP1 regulates cyclin D1 promoter activity by nuclear EGFR and STAT3 in CNE1 cells. J. Exp. Clin. Cancer Res. 2013, 32, 90. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, F.; Zhou, M.; Shang, L.; Du, Q.; Li, Y.; Xie, L.; Liu, X.; Tang, M.; Luo, X.; Fan, J.; et al. EBV(LMP1)-induced metabolic reprogramming inhibits necroptosis through the hypermethylation of the RIP3 promoter. Theranostics 2019, 9, 2424–2438. [Google Scholar] [CrossRef] [PubMed]
- Schlessinger, J. Receptor Tyrosine Kinases: Legacy of the First Two Decades. Cold Spring Harb. Perspect. Biol. 2014, 6, a008912. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kumagai, S.; Koyama, S.; Nishikawa, H. Antitumour immunity regulated by aberrant ERBB family signalling. Nat. Rev. Cancer 2021, 21, 181–197. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Wang, P.; Zhang, C.; Ma, Z.-L. Epidermal growth factor receptor (EGFR): A rising star in the era of precision medicine of lung cancer. Oncotarget 2017, 8, 50209–50220. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Parker, M.I.; Nikonova, A.S.; Sun, D.; Golemis, E.A. Proliferative signaling by ERBB proteins and RAF/MEK/ERK effectors in pol-ycystic kidney disease. Cell. Signal. 2020, 67, 109497. [Google Scholar] [CrossRef]
- Eskilsson, E.; Røsland, G.V.; Solecki, G.; Wang, Q.; Harter, P.N.; Graziani, G.; Verhaak, R.G.W.; Winkler, F.; Bjerkvig, R.; Miletic, H. EGFR heterogeneity and implications for therapeutic intervention in glioblastoma. Neuro-Oncology 2018, 20, 743–752. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mirghani, H.; Amen, F.; Moreau, F.; Guigay, J.; Hartl, D.; Guily, J.L.S. Oropharyngeal cancers: Relationship between epidermal growth factor receptor alterations and human papillomavirus status. Eur. J. Cancer 2014, 50, 1100–1111. [Google Scholar] [CrossRef]
- Chen, J.; He, W.; Hu, X.; Shen, Y.; Cao, J.; Wei, Z.; Luan, Y.; He, L.; Jiang, F.; Tao, Y. A role for ErbB signaling in the induction of reactive astrogliosis. Cell Discov. 2017, 3, 17044. [Google Scholar] [CrossRef] [Green Version]
- Jotte, R.M.; Spigel, D.R. Advances in molecular-based personalized non-small-cell lung cancer therapy: Targeting epidermal growth factor receptor and mechanisms of resistance. Cancer Med. 2015, 4, 1621–1632. [Google Scholar] [CrossRef] [PubMed]
- Xu, M.J.; Johnson, D.E.; Grandis, J.R. EGFR-targeted therapies in the post-genomic era. Cancer Metast. Rev. 2017, 36, 463–473. [Google Scholar] [CrossRef] [PubMed]
- Ho, J.; Moyes, D.; Tavassoli, M.; Naglik, J.R. The Role of ErbB Receptors in Infection. Trends Microbiol. 2017, 25, 942–952. [Google Scholar] [CrossRef]
- Tan, X.; Lambert, P.F.; Rapraeger, A.C.; Anderson, R.A. Stress-Induced EGFR Trafficking: Mechanisms, Functions, and Therapeutic Implications. Trends Cell Biol. 2016, 26, 352–366. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sigismund, S.; Avanzato, D.; Lanzetti, L. Emerging functions of the EGFR in cancer. Mol. Oncol. 2018, 12, 3–20. [Google Scholar] [CrossRef]
- Chung, I.; Akita, R.W.; Vandlen, R.; Toomre, D.; Schlessinger, J.; Mellman, I. Spatial control of EGF receptor activation by reversible dimerization on living cells. Nature 2010, 464, 783–787. [Google Scholar] [CrossRef] [PubMed]
- Wang, Z. ErbB Receptors and Cancer. Methods Mol. Biol. 2017, 1652, 3–35. [Google Scholar] [PubMed]
- Ayati, A.; Moghimi, S.; Salarinejad, S.; Safavi, M.; Pouramiri, B.; Foroumadi, A. A review on progression of epidermal growth factor receptor (EGFR) inhibitors as an efficient approach in cancer targeted therapy. Bioorg. Chem. 2020, 99, 103811. [Google Scholar] [CrossRef] [PubMed]
- Normanno, N.; De Luca, A.; Bianco, C.; Strizzi, L.; Mancino, M.; Maiello, M.R.; Carotenuto, A.; De Feo, G.; Caponigro, F.; Salomon, D.S. Epidermal growth factor receptor (EGFR) signaling in cancer. Gene 2006, 366, 2–16. [Google Scholar] [CrossRef]
- Roskoski, R. The ErbB/HER family of protein-tyrosine kinases and cancer. Pharmacol. Res. 2014, 79, 34–74. [Google Scholar] [CrossRef]
- Roskoski, R. Small molecule inhibitors targeting the EGFR/ErbB family of protein-tyrosine kinases in human cancers. Pharmacol. Res. 2019, 139, 395–411. [Google Scholar] [CrossRef] [PubMed]
- Waterman, H.; Alroy, I.; Strano, S.; Seger, R.; Yarden, Y. The C-terminus of the kinase-defective neuregulin receptor ErbB-3 confers mitogenic superiority and dictates endocytic routing. EMBO J. 1999, 18, 3348–3358. [Google Scholar] [CrossRef] [Green Version]
- Miller, W.E.; Earp, H.S.; Raab-Traub, N. The Epstein-Barr virus latent membrane protein 1 induces expression of the epidermal growth factor receptor. J. Virol. 1995, 69, 4390–4398. [Google Scholar] [CrossRef] [Green Version]
- Meckes, D.G.; Shair, K.H.Y.; Marquitz, A.R.; Kung, C.-P.; Edwards, R.H.; Raab-Traub, N. Human tumor virus utilizes exosomes for intercellular communication. Proc. Natl. Acad. Sci. USA 2010, 107, 20370–20375. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stewart, S.; Dawson, C.W.; Takada, K.; Curnow, J.; Moody, C.A.; Sixbey, J.W.; Young, L.S. Epstein-Barr virus-encoded LMP2A regulates viral and cellular gene expression by modulation of the NF-kappaB transcription factor pathway. Proc. Natl. Acad. Sci. USA 2004, 101, 15730–15735. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kung, C.-P.; Raab-Traub, N. Epstein-Barr Virus Latent Membrane Protein 1 Induces Expression of the Epidermal Growth Factor Receptor through Effects on Bcl-3 and STAT. J. Virol. 2008, 82, 5486–5493. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tao, Y.; Song, X.; Deng, X.; Xie, D.; Lee, L.M.; Liu, Y.; Li, W.; Li, L.; Deng, L.; Wu, Q.; et al. Nuclear accumulation of epidermal growth factor receptor and acceleration of G1/S stage by Epstein–Barr-encoded oncoprotein latent membrane protein. Exp. Cell Res. 2005, 303, 240–251. [Google Scholar] [CrossRef]
- Tao, Y.; Song, X.; Tan, Y.; Lin, X.; Zhao, Y.; Zeng, L.; Tang, M.; Li, W.; Wu, Q.; Cao, Y. Nuclear translocation of EGF receptor regulated by Epstein-Barr virus encoded latent membrane protein. Sci. China Ser. C Life Sci. 2004, 47, 258–267. [Google Scholar] [CrossRef] [PubMed]
- Baud, V.; Karin, M. Is NF-kappaB a good target for cancer therapy? Hopes and pitfalls. Nat. Rev. Drug Discov. 2009, 8, 33–40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [Green Version]
- Thornburg, N.J.; Pathmanathan, R.; Raab-Traub, N. Activation of nuclear factor-kappaB p50 homodimer/Bcl-3 complexes in nasopharyngeal carcinoma. Cancer Res. 2003, 63, 8293–8301. [Google Scholar] [PubMed]
- Kung, C.P.; Raab-Traub, N. Epstein-Barr virus latent membrane protein 1 modulates distinctive NF-kappaB pathways through C-terminus-activating region 1 to regulate epidermal growth factor receptor expression. J. Virol. 2010, 84, 6605–6614. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Salmerón, A.; Janzen, J.; Soneji, Y.; Bump, N.; Kamens, J.; Allen, H.; Ley, S.C. Direct phosphorylation of NF-kappaB1 p105 by the IkappaB kinase complex on serine 927 is essential for signal-induced p105 proteolysis. J. Biol. Chem. 2001, 276, 22215–22222. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heissmeyer, V.; Krappmann, D.; Wulczyn, F.G.; Scheidereit, C. NF-kappaB p105 is a target of IkappaB kinases and controls signal induction of Bcl-3-p50 complexes. EMBO J. 1999, 18, 4766–4778. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Luftig, M.; Yasui, T.; Soni, V.; Kang, M.S.; Jacobson, N.; Cahir-McFarland, E.; Seed, B.; Kieff, E. Epstein-Barr virus latent infection membrane protein 1 TRAF-binding site induces NIK/IKK alpha-dependent noncanonical NF-kappaB activation. Proc. Natl. Acad. Sci. USA 2004, 101, 141–146. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paine, E.; Scheinman, R.I.; Baldwin, A.S., Jr.; Raab-Traub, N. Expression of LMP1 in epithelial cells leads to the activation of a select subset of NF-kappa B/Rel family proteins. J. Virol. 1995, 69, 4572–4576. [Google Scholar] [CrossRef] [Green Version]
- Westerheide, S.D.; Mayo, M.W.; Anest, V.; Hanson, J.L.; Baldwin, A.S. The Putative Oncoprotein Bcl-3 Induces Cyclin D1 To Stimulate G1 Transition. Mol. Cell. Biol. 2001, 21, 8428–8436. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Thornburg, N.J.; Raab-Traub, N. Induction of epidermal growth factor receptor expression by Epstein-Barr virus latent mem-brane protein 1 C-terminal-activating region 1 is mediated by NF-kappaB p50 homodimer/Bcl-3 complexes. J. Virol. 2007, 81, 12954–12961. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baldassarre, F.; Mallardo, M.; Mezza, E.; Scala, G.; Quinto, I. Regulation of NF-kappa B through the nuclear processing of p105 (NF-kappa B1) in Epstein-Barr virus-immortalized B cell lines. J. Biol. Chem. 1995, 270, 31244–31248. [Google Scholar] [CrossRef] [Green Version]
- Chung, G.T.; Lou, W.P.; Chow, C.; To, K.F.; Choy, K.W.; Leung, A.W.; Tong, C.Y.; Yuen, J.W.; Ko, C.W.; Yip, T.T.; et al. Constitutive activation of distinct NF-κB signals in EBV-associated nasopharyngeal carcinoma. J. Pathol. 2013, 231, 311–322. [Google Scholar] [CrossRef]
- Siersbæk, R.D.; Scabia, V.; Nagarajan, S.; Chernukhin, I.; Papachristou, E.K.; Broome, R.; Johnston, S.J.; Joosten, S.E.; Green, A.R.; Kumar, S.; et al. IL6/STAT3 Signaling Hijacks Estrogen Receptor α Enhancers to Drive Breast Cancer Metastasis. Cancer Cell 2020, 38, 412–423.e9. [Google Scholar] [CrossRef]
- Teoh, P.J.; Chung, T.-H.; Chng, P.Y.; Toh, S.H.M.; Chng, W.J. IL6R-STAT3-ADAR1 (P150) interplay promotes oncogenicity in multiple myeloma with 1q21 amplification. Haematology 2019, 105, 1391–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsiao, J.R.; Jin, Y.T.; Tsai, S.T.; Shiau, A.L.; Wu, C.L.; Su, W.C. Constitutive activation of STAT3 and STAT5 is present in the majority of nasopharyngeal carcinoma and correlates with better prognosis. Br. J. Cancer 2003, 89, 344–349. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kung, C.P.; Meckes, D.G., Jr.; Raab-Traub, N. Epstein-Barr virus LMP1 activates EGFR, STAT3, and ERK through effects on PKCdelta. J. Virol. 2011, 85, 4399–4408. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Lee, J.M.; Zong, Y.; Borowitz, M.; Ng, M.H.; Ambinder, R.F.; Hayward, S.D. Linkage between STAT Regulation and Epstein-Barr Virus Gene Expression in Tumors. J. Virol. 2001, 75, 2929–2937. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Hutt-Fletcher, L.; Cao, L.; Hayward, S.D. A Positive Autoregulatory Loop of LMP1 Expression and STAT Activation in Epithelial Cells Latently Infected with Epstein-Barr Virus. J. Virol. 2003, 77, 4139–4148. [Google Scholar] [CrossRef] [Green Version]
- Park, O.K.; Schaefer, T.S.; Nathans, D. In vitro activation of Stat3 by epidermal growth factor receptor kinase. Proc. Natl. Acad. Sci. USA 1996, 93, 13704–13708. [Google Scholar] [CrossRef] [Green Version]
- Tu, C.; Zeng, Z.; Qi, P.; Li, X.; Guo, C.; Xiong, F.; Xiang, B.; Zhou, M.; Liao, Q.; Yu, J.; et al. Identification of genomic alterations in naso-pharyngeal carcinoma and nasopharyngeal carcinoma-derived Epstein-Barr virus by whole-genome sequencing. Carcinogenesis 2018, 39, 1517–1528. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Wu, S.K.; Wang, Y.; Fan, Z.X.; Li, C.R.; Feng, M.; Xu, P.; Wang, W.D.; Lang, J.Y. p53, MDM2, eIF4E and EGFR expression in na-sopharyngeal carcinoma and their correlation with clinicopathological characteristics and prognosis: A retrospective study. Oncol. Lett. 2015, 9, 113–118. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsu, C.-H.; Gao, M.; Chen, C.-L.; Yeh, P.-Y.; Cheng, A.-L. Inhibitors of Epidermoid Growth Factor Receptor Suppress Cell Growth and Enhance Chemosensitivity of Nasopharyngeal Cancer Cells in vitro. Oncology 2005, 68, 538–547. [Google Scholar] [CrossRef]
- Sheen, T.-S.; Huang, Y.-T.; Chang, Y.-L.; Ko, J.-Y.; Wu, C.-S.; Yu, Y.-C.; Tsai, C.-H.; Hsu, M.-M. Epstein-Barr Virus-encoded Latent Membrane Protein 1 Co-expresses with Epidermal Growth Factor Receptor in Nasopharyngeal Carcinoma. Jpn. J. Cancer Res. 1999, 90, 1285–1292. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.; Youhong, T.; Tan, Y.; He, Y.; Ban, Y.; Cai, J.; Li, X.; Xiong, W.; Zeng, Z.; Li, G.; et al. EGFR-PKM2 signaling promotes the metastatic potential of nasopharyngeal carcinoma through induction of FOSL1 and ANTXR. Carcinogenesis 2020, 41, 723–733. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chua, D.T.; Nicholls, J.M.; Sham, J.S.; Au, G.K. Prognostic value of epidermal growth factor receptor expression in patients with advanced stage nasopharyngeal carcinoma treated with induction chemotherapy and radiotherapy. Int. J. Radiat. Oncol. 2004, 59, 11–20. [Google Scholar] [CrossRef] [PubMed]
- Bourouba, M.; Benyelles-Boufennara, A.; Terki, N.; Baraka-Kerboua, E.; Bouzid, K.; Touil-Boukoffa, C. Epidermal growth factor receptor (EGFR) abundance correlates with p53 and Bcl-2 accumulation and patient age in a small cohort of North African nasopharyngeal carcinoma patients. Eur. Cytokine Netw. 2011, 22, 38–44. [Google Scholar] [CrossRef]
- Liang, Z.; Liu, Z.; Cheng, C.; Wang, H.; Deng, X.; Liu, J.; Liu, C.; Li, Y.; Fang, W. VPS33B interacts with NESG1 to modulate EGFR/PI3K/AKT/c-Myc/P53/miR-133a-3p signaling and induce 5-fluorouracil sensitivity in nasopharyngeal carcinoma. Cell Death Dis. 2019, 10, 1–16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, W.; Lu, Z. Nuclear PKM2 regulates the Warburg effect. Cell Cycle 2013, 12, 3343–3347. [Google Scholar] [CrossRef] [Green Version]
- Luo, Y.; Wang, J.; Wang, F.; Liu, X.; Lu, J.; Yu, X.; Ma, X.; Peng, X.; Li, X. Foxq1 promotes metastasis of nasopharyngeal carcinoma by inducing vasculogenic mimicry via the EGFR signaling pathway. Cell Death Dis. 2021, 12, 1–16. [Google Scholar] [CrossRef]
- A Elian, F.; Are, U.; Ghosh, S.; Nuin, P.; Footz, T.; McMullen, T.P.; Brindley, D.N.; A Walter, M. FOXQ1 is Differentially Expressed Across Breast Cancer Subtypes with Low Expression Associated with Poor Overall Survival. Breast Cancer Targets Ther. 2021, 13, 171–188. [Google Scholar] [CrossRef] [PubMed]
- Peng, L.-X.; Wang, M.-D.; Xie, P.; Yang, J.-P.; Sun, R.; Zheng, L.-S.; Mei, Y.; Meng, D.-F.; Peng, X.-S.; Lang, Y.-H.; et al. LACTB promotes metastasis of nasopharyngeal carcinoma via activation of ERBB3/EGFR-ERK signaling resulting in unfavorable patient survival. Cancer Lett. 2021, 498, 165–177. [Google Scholar] [CrossRef]
- Lin, Q.; Wang, H.; Lin, X.; Zhang, W.; Huang, S.; Zheng, Y. PTPN12 Affects Nasopharyngeal Carcinoma Cell Proliferation and Mi-gration Through Regulating EGFR. Cancer Biother. Radiopharm. 2018, 33, 60–64. [Google Scholar] [CrossRef] [PubMed]
- Li, F.; Zhao, X.; Sun, R.; Ou, J.; Huang, J.; Yang, N.; Xu, T.; Li, J.; He, X.; Li, C.; et al. EGFR-rich extracellular vesicles derived from highly metastatic nasopharyngeal carcinoma cells accelerate tumour metastasis through PI3K/AKT pathway-suppressed ROS. J. Extracell. Vesicles 2020, 10, e12003. [Google Scholar] [CrossRef] [PubMed]
- Frawley, T.; Piskareva, O. Extracellular Vesicle Dissemination of Epidermal Growth Factor Receptor and Ligands and Its Role in Cancer Progression. Cancers 2020, 12, 3200. [Google Scholar] [CrossRef]
- Zanetti-Domingues, L.C.; Bonner, S.E.; Martin-Fernandez, M.L.; Huber, V. Mechanisms of Action of EGFR Tyrosine Kinase Receptor Incorporated in Extracellular Vesicles. Cells 2020, 9, 2505. [Google Scholar] [CrossRef]
- Zanetti-Domingues, L.C.; Bonner, S.E.; Iyer, R.S.; Martin-Fernandez, M.L.; Huber, V. Cooperation and Interplay between EGFR Signalling and Extracellular Vesicle Biogenesis in Cancer. Cells 2020, 9, 2639. [Google Scholar] [CrossRef]
- Huang, W.; Liu, J.; Feng, X.; Chen, H.; Zeng, L.; Huang, G.; Liu, W.; Wang, L.; Jia, W.; Chen, J.; et al. DLC-1 induces mitochondrial apoptosis and epithelial mesenchymal transition arrest in nasopharyngeal carcinoma by targeting EGFR/Akt/NF-κB pathway. Med. Oncol. 2015, 32, 115. [Google Scholar] [CrossRef] [PubMed]
- Feng, X.; Li, C.; Liu, W.; Chen, H.; Zhou, W.; Wang, L.; Zhu, B.; Yao, K.; Jiang, X.; Ren, C. DLC-1, a candidate tumor suppressor gene, inhibits the proliferation, migration and tumorigenicity of human nasopharyngeal carcinoma cells. Int. J. Oncol. 2013, 42, 1973–1984. [Google Scholar] [CrossRef]
- Yang, J.; Zhu, D.; Liu, S.; Shao, M.; Liu, Y.; Li, A.; Lv, Y.; Huang, M.; Lou, D.; Fan, Q. Curcumin enhances radiosensitization of nasopha-ryngeal carcinoma by regulating circRNA network. Mol. Carcinog. 2020, 59, 202–214. [Google Scholar] [CrossRef]
- Ma, N.; Kawanishi, M.; Hiraku, Y.; Murata, M.; Huang, G.-W.; Huang, Y.; Luo, D.-Z.; Mo, W.-G.; Fukui, Y.; Kawanishi, S. Reactive nitrogen species-dependent DNA damage in EBV-associated nasopharyngeal carcinoma: The relation to STAT3 activation and EGFR expression. Int. J. Cancer 2008, 122, 2517–2525. [Google Scholar] [CrossRef] [PubMed]
- Zachary, I.C. How neuropilin-1 regulates receptor tyrosine kinase signalling: The knowns and known unknowns. Biochem. Soc. Trans. 2011, 39, 1583–1591. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huang, J.; Huang, W.; Zhang, Z.; Lin, X.; Lin, H.; Peng, L.; Chen, T. Highly Uniform Synthesis of Selenium Nanoparticles with EGFR Targeting and Tumor Microenvironment-Responsive Ability for Simultaneous Diagnosis and Therapy of Nasopharyngeal Carcinoma. ACS Appl. Mater. Interfaces 2019, 11, 11177–11193. [Google Scholar] [CrossRef] [PubMed]
- Chong, C.R.; Jänne, P.A. The quest to overcome resistance to EGFR-targeted therapies in cancer. Nat. Med. 2013, 19, 1389–1400. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, C.; Zhou, Y.; Zhang, X.; Fu, S.; Lin, Z.; Fang, W.; Yang, Y.; Huang, Y.; Zhao, H.; Hong, S.; et al. Anti-epidermal growth factor receptor monoclonal antibody plus palliative chemotherapy as a first-line treatment for recurrent or metastatic nasopharyngeal car-cinoma. Cancer Med. 2020, 9, 1721–1732. [Google Scholar] [CrossRef] [Green Version]
- Campoli, M.; Ferris, R.L.; Ferrone, S.; Wang, X. Immunotherapy of Malignant Disease with Tumor Antigen-Specific Monoclonal Antibodies. Clin. Cancer Res. 2009, 16, 11–20. [Google Scholar] [CrossRef] [Green Version]
- Ali, S.M.; Yao, M.; Yao, J.; Wang, J.; Cheng, Y.; Schrock, A.B.; Chirn, G.-W.; Chen, H.; Mu, S.; Gay, L.; et al. Comprehensive genomic profiling of different subtypes of nasopharyngeal carcinoma reveals similarities and differences to guide targeted therapy. Cancer 2017, 123, 3628–3637. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, L.S.Y.; Wong, B.; Gangodu, N.R.; Lee, A.Z.E.; Kian Fong Liou, A.; Loh, K.S.; Li, H.; Yann Lim, M.; Salazar, A.M.; Lim, C.M. Enhancing the immune stimulatory effects of cetuximab therapy through TLR3 signalling in Epstein-Barr virus (EBV) positive nasopha-ryngeal carcinoma. Oncoimmunology 2018, 7, 1500109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, L.R.; Zhu, H.F.; Xu, J.; Jiang, X.S.; Yin, L.; Jiang, N.; Zong, D.; Wang, F.J.; Huang, S.F.; Bian, X.H.; et al. Effectiveness of Cetuximab in Combination with Concurrent Chemoradiotherapy in Locoregionally Advanced Nasopharyngeal Carcinoma: A 1:2 Pro-pensity Score-matched Analysis. J. Cancer 2018, 9, 1642–1651. [Google Scholar] [CrossRef] [Green Version]
- Peng, H.; Tang, L.L.; Liu, X.; Chen, L.; Li, W.F.; Mao, Y.P.; Zhang, Y.; Liu, L.Z.; Tian, L.; Guo, Y.; et al. Anti-EGFR targeted therapy delivered before versus during radiotherapy in locoregionally advanced nasopharyngeal carcinoma: A big-data, intelligence plat-form-based analysis. BMC Cancer 2018, 18, 323. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.-S.; Liang, Y.-J.; Li, X.-Y.; Liu, S.-L.; Chen, Q.-Y.; Tang, L.-Q.; Mai, H.-Q. Palliative chemotherapy with or without anti-EGFR therapy for de novo metastatic nasopharyngeal carcinoma: A propensity score-matching study. Drug Des. Dev. Ther. 2019, 13, 3207–3216. [Google Scholar] [CrossRef] [Green Version]
- Ueda, Y.; Enokida, T.; Okano, S.; Fujisawa, T.; Ito, K.; Tahara, M. Combination Treatment with Paclitaxel, Carboplatin, and Ce-tuximab (PCE) as First-Line Treatment in Patients with Recurrent and/or Metastatic Nasopharyngeal Carcinoma. Front. Oncol. 2020, 10, 571304. [Google Scholar] [CrossRef]
- Wang, Z.-Q.; Mei, Q.; Li, J.-B.; You, R.; Liu, Y.-P.; Sun, R.; Hu, G.-Y.; Chen, M.-Y.; Hua, Y.-J. The long-term survival of patients with III-IVb stage nasopharyngeal carcinoma treated with IMRT with or without Nimotuzumab: A propensity score-matched analysis. BMC Cancer 2019, 19, 1122. [Google Scholar]
- Zhu, Y.; Yang, S.; Zhou, S.; Yang, J.; Qin, Y.; Gui, L.; Shi, Y.; He, X. Nimotuzumab plus platinum-based chemotherapy versus plati-num-based chemotherapy alone in patients with recurrent or metastatic nasopharyngeal carcinoma. Ther. Adv. Med. Oncol. 2020, 12, 1758835920953738. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhang, G.; Miao, X.-B.; Deng, X.-B.; Wu, Y.; Liu, Y.; Jin, Z.-R.; Li, X.-Q.; Liu, Q.-Z.; Sun, D.-X.; et al. Cancer stem-like cell properties are regulated by EGFR/AKT/β-catenin signaling and preferentially inhibited by gefitinib in nasopharyngeal carcinoma. FEBS J. 2013, 280, 2027–2041. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Li, Z.; Wu, L.; Wang, Z.; Wang, X.; Yü, Y.; Zhao, Q.; Luo, F. MiRNA-125a-5p: A regulator and predictor of gefitinib’s effect on nasopharyngeal carcinoma. Cancer Cell Int. 2014, 14, 24. [Google Scholar] [CrossRef] [Green Version]
- Yugui, F.; Wang, H.; Sun, D.; Zhang, X. Nasopharyngeal cancer combination chemoradiation therapy based on folic acid modified, gefitinib and yttrium 90 co-loaded, core-shell structured lipid-polymer hybrid nanoparticles. Biomed. Pharmacother. 2019, 114, 108820. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhou, F.; Zhang, J.; Zou, Q.; Fan, Q.; Zhang, F. Erlotinib enhanced chemoradiotherapy sensitivity via inhibiting DNA damage repair in nasopharyngeal carcinoma CNE2 cells. Ann. Palliat. Med. 2020, 9, 2559–2567. [Google Scholar] [CrossRef]
- Lan, M.-Y.; Hsu, Y.-B.; Chen, J.-P.; Lu, Y.-J. Polyethylene Glycol-Coated Graphene Oxide Loaded with Erlotinib as an Effective Therapeutic Agent for Treating Nasopharyngeal Cancer Cells. Int. J. Nanomed. 2020, 15, 7569–7582. [Google Scholar] [CrossRef]
- You, B.; Le Tourneau, C.; Chen, E.X.; Wang, L.; Jarvi, A.; Bharadwaj, R.R.; Kamel-Reid, S.; Perez-Ordonez, B.; Mann, V.; Siu, L.L. A Phase II Trial of Erlotinib as Maintenance Treatment After Gemcitabine Plus Platinum-based Chemotherapy in Patients with Recurrent and/or Metastatic Nasopharyngeal Carcinoma. Am. J. Clin. Oncol. 2012, 35, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Huang, F.; Liang, X.; Min, X.; Zhang, Y.; Wang, G.; Peng, Z.; Peng, F.; Li, M.; Chen, L.; Chen, Y. Simultaneous Inhibition of EGFR and HER2 via Afatinib Augments the Radiosensitivity of Nasopharyngeal Carcinoma Cells. J. Cancer 2019, 10, 2063–2073. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Xue, C.; Tian, Y.; Zhang, J.; Zhao, Y.; Zhan, J.; Fang, W. In vitro and in vivo efficacy of afatinib as a single agent or in combination with gemcitabine for the treatment of nasopharyngeal carcinoma. Drug Des. Dev. Ther. 2016, 10, 1299–1306. [Google Scholar] [CrossRef] [Green Version]
- Lin, M.; You, R.; Liu, Y.-P.; Zhang, Y.-N.; Zhang, H.-J.; Zou, X.; Yang, Q.; Li, C.-F.; Hua, Y.-J.; Yu, T.; et al. Beneficial effects of anti-EGFR agents, Cetuximab or Nimotuzumab, in combination with concurrent chemoradiotherapy in advanced nasopharyngeal carcinoma. Oral Oncol. 2018, 80, 1–8. [Google Scholar] [CrossRef]
- Talavera, A.; Friemann, R.; Gómez-Puerta, S.; Martinez-Fleites, C.; Garrido, G.; Rabasa, A.; Requena, A.L.; Pupo, A.; Johansen, R.F.; Sanchez, O.; et al. Nimotuzumab, an Antitumor Antibody that Targets the Epidermal Growth Factor Receptor, Blocks Ligand Binding while Permitting the Active Receptor Conformation. Cancer Res. 2009, 69, 5851–5859. [Google Scholar] [CrossRef] [Green Version]
- Si, X.; Wu, S.; Wang, H.; Zhang, X.; Wang, M.; Zeng, X.; Zhang, L. Nimotuzumab combined with chemotherapy as first-line treatment for advanced lung squamous cell carcinoma. Thorac. Cancer 2018, 9, 1056–1061. [Google Scholar] [CrossRef] [PubMed]
- Fei, Z.; Xu, T.; Li, M.; Chen, T.; Li, L.; Qiu, X.; Chen, C. Effectiveness and cost-effectiveness analysis of nimotuzumab for the radio-therapy of locoregionally advanced nasopharyngeal carcinoma. Radiat. Oncol. 2020, 15, 230. [Google Scholar] [CrossRef]
- Yao, J.J.; Zhang, L.L.; Gao, T.S.; Peng, Y.L.; Lawrence, W.R.; Zhang, W.J.; Zhang, F.; Zhou, G.Q.; Wang, S.Y.; Sun, Y. Comparing treatment outcomes of concurrent chemoradiotherapy with or without nimotuzumab in patients with locoregionally advanced naso-pharyngeal carcinoma. Cancer Biol. Ther. 2018, 19, 1102–1107. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- You-Ping, L.; Hua, Y.-J.; Liu, Y.-P.; Yang, Q.; Zhang, Y.-N.; Li, J.-B.; Li, C.-F.; Zou, X.; Jing-Yu, C.; Cao, J.-Y.; et al. Concurrent Chemoradiotherapy with or without Anti-EGFR-Targeted Treatment for Stage II-IVb Nasopharyngeal Carcinoma: Retrospective Analysis with a Large Cohort and Long Follow-up. Theranostics 2017, 7, 2314–2324. [Google Scholar]
- Mao, L.; Tan, J.; Wang, F.; Luo, Y.; Liu, W.; Zeng, F.; Yu, B.; Huang, H.; Lu, J.; Peng, X.; et al. Retrospective study comparing anti-EGFR monoclonal antibody plus cisplatin-based chemoradiotherapy versus chemoradiotherapy alone for stage II-IVb nasopha-ryngeal carcinoma and prognostic value of EGFR and VEGF expression. Clin. Otolaryngol. 2019, 44, 572–580. [Google Scholar] [CrossRef]
- Lui, V.W.Y.; Lau, C.P.Y.; Ho, K.; Ng, M.H.L.; Cheng, S.H.; Tsao, S.-W.; Tsang, C.M.; Lei, K.I.K.; Chan, A.T.; Mok, T.S.K. Anti-invasion, anti-proliferation and anoikis-sensitization activities of lapatinib in nasopharyngeal carcinoma cells. Investig. New Drugs 2010, 29, 1241–1252. [Google Scholar] [CrossRef] [PubMed]
- Chua, D.T.; Wei, W.I.; Wong, M.P.; Sham, J.S.; Nicholls, J.; Au, G.K. Phase II study of gefitinib for the treatment of recurrent and met-astatic nasopharyngeal carcinoma. Head Neck 2008, 30, 863–867. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| The Category of Drugs | Target | Therapeutic/Experimental Regimen | The Type of the Test | Results | References |
|---|---|---|---|---|---|
| Anti-EGFR antibody | The extracellular domain of the receptor | Palliative chemotherapy plus an anti-EGFR agent (NTZ or CTX) in R/M NPC | Clinical trial | Prolong R/M patients’ OS, PFS | [92] |
| CTX plus Poly-ICLC treatment | In vitro experiment | Poly-ICLC could enhance both CTX-mediated innate and adaptive anti-tumor immunity against NPC | [95] | ||
| CCRT plus CTX in LA NPC | Clinical trial | Prolong LA NPC patients’ OS, PFS | [96] | ||
| IC+CTX/NTZ or CCRT + CTX/NTZ in LA NPC patients | Clinical trial | Prolong LA NPC patients’ PFS | [97] | ||
| PCT+CTX/NTZ in de novo metastatic NPC patients. | The clinical trial | Cannot improve de novo metastatic NPC patients‘ prognosis | [98] | ||
| paclitaxel + carboplatin + CTX (PCE) therapy for R/M NPC | Clinical trail | Potentially effective for R/M NPC | [99] | ||
| IMRT+cisplatin+NTZ therapy for LA NPC | Clinical trial | Prolong LA NPC patients’ OS | [100] | ||
| NTZ+ chemotherapy for R/M NPC | Clinical trail | Prolong R/M NPC patients’ OS | [101] | ||
| Small molecule EGFR tyrosine kinase inhibitors (TKI) | Tyrosine kinase domain | Gefitinib | In vitro and in vivo experiment | Suppress cancer stem-like cells of NPC xenografts. | [102] |
| Gefitinib | In vitro and in vivo experiment | Inhibit two NPC cell lines proliferation | [103] | ||
| FA-GEF-Y90-LPNP | In vitro and in vivo experiment | Exhibit the best in vivo tumor inhibition ability without more systemic toxicity | [104] | ||
| Erlotinib plus radiotherapy/chemotherapy | In vitro experiment | Enhance the sensitivity of tumor cells to radiotherapy/chemotherapy, and weaken radiotherapy/chemotherapy resistance of tumor cells | [105] | ||
| GO-PEG-Erlotinib | In vitro experiment | Suppress NPC cell proliferation, migration, and invasion | [106] | ||
| Erlotinib plus Cisplatin in patients with R/M NPC. | Clinical trial | Maintenance or second-line therapy with Erlotinib after chemotherapy was not effective in RM NPC | [107] | ||
| Afatinib | In vitro experiment | Increase NPC cell radiosensitivity | [108] | ||
| Afatinib or Afatinib combination with gemcitabine | In vitro and in vivo experiment | Single Afatinib induces cell cycle arrest and inhibits the proliferation of NPC cell lines. Afatinib + gemcitabine have an anti-tumor effect in an NPC xenograft model | [109] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Peng, X.; Zhou, Y.; Tao, Y.; Liu, S. Nasopharyngeal Carcinoma: The Role of the EGFR in Epstein–Barr Virus Infection. Pathogens 2021, 10, 1113. https://doi.org/10.3390/pathogens10091113
Peng X, Zhou Y, Tao Y, Liu S. Nasopharyngeal Carcinoma: The Role of the EGFR in Epstein–Barr Virus Infection. Pathogens. 2021; 10(9):1113. https://doi.org/10.3390/pathogens10091113
Chicago/Turabian StylePeng, Xintong, Yanling Zhou, Yongguang Tao, and Shuang Liu. 2021. "Nasopharyngeal Carcinoma: The Role of the EGFR in Epstein–Barr Virus Infection" Pathogens 10, no. 9: 1113. https://doi.org/10.3390/pathogens10091113
APA StylePeng, X., Zhou, Y., Tao, Y., & Liu, S. (2021). Nasopharyngeal Carcinoma: The Role of the EGFR in Epstein–Barr Virus Infection. Pathogens, 10(9), 1113. https://doi.org/10.3390/pathogens10091113