
Complete Genome Sequencing of Leptospira interrogans Isolates from Malaysia Reveals Massive Genome Rearrangement but High Conservation of Virulence-Associated Genes


 ,
,
Abstract
:1. Introduction
2. Results
2.1. Clinical Cases
2.2. Classification by Serotyping
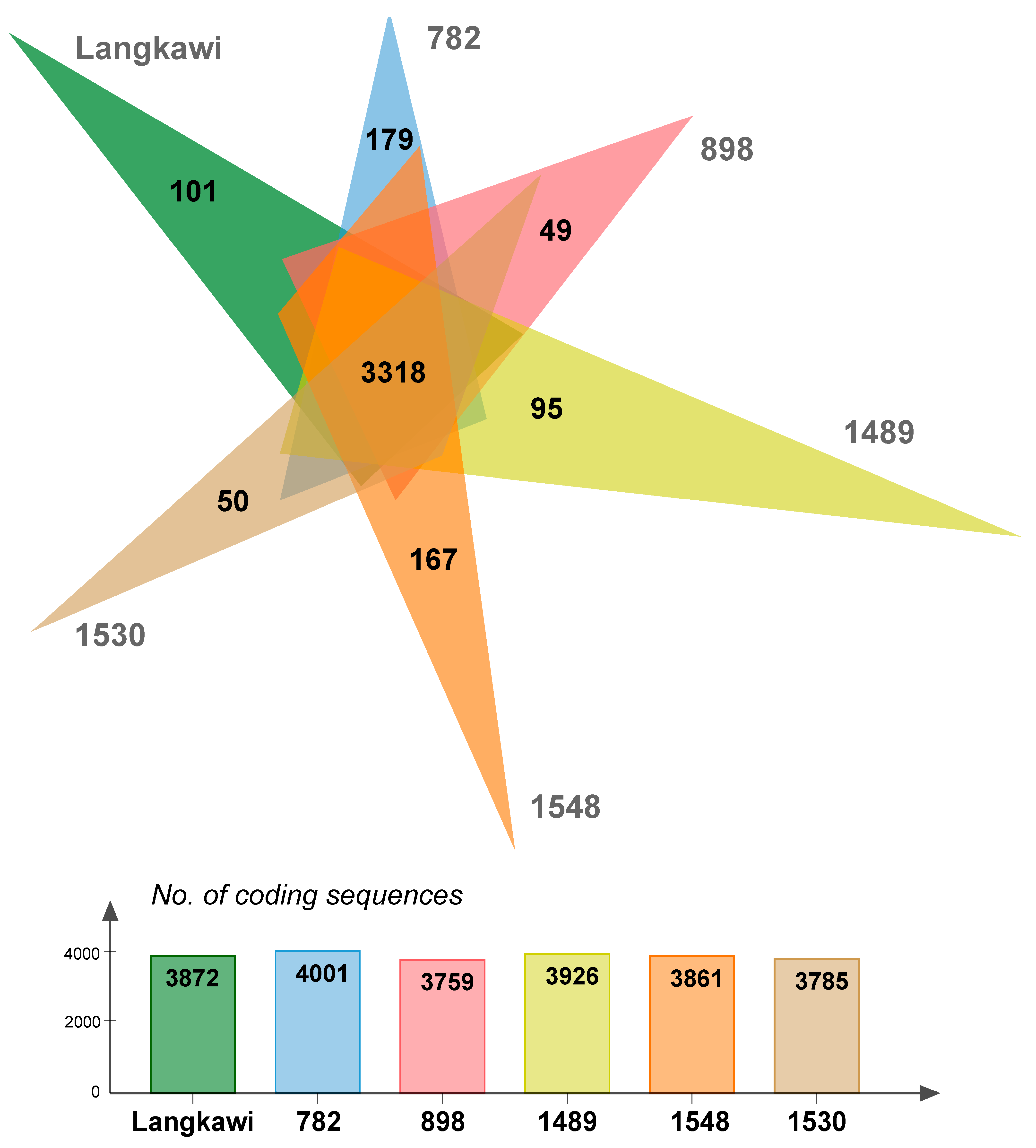
2.3. Complete Genome Sequences
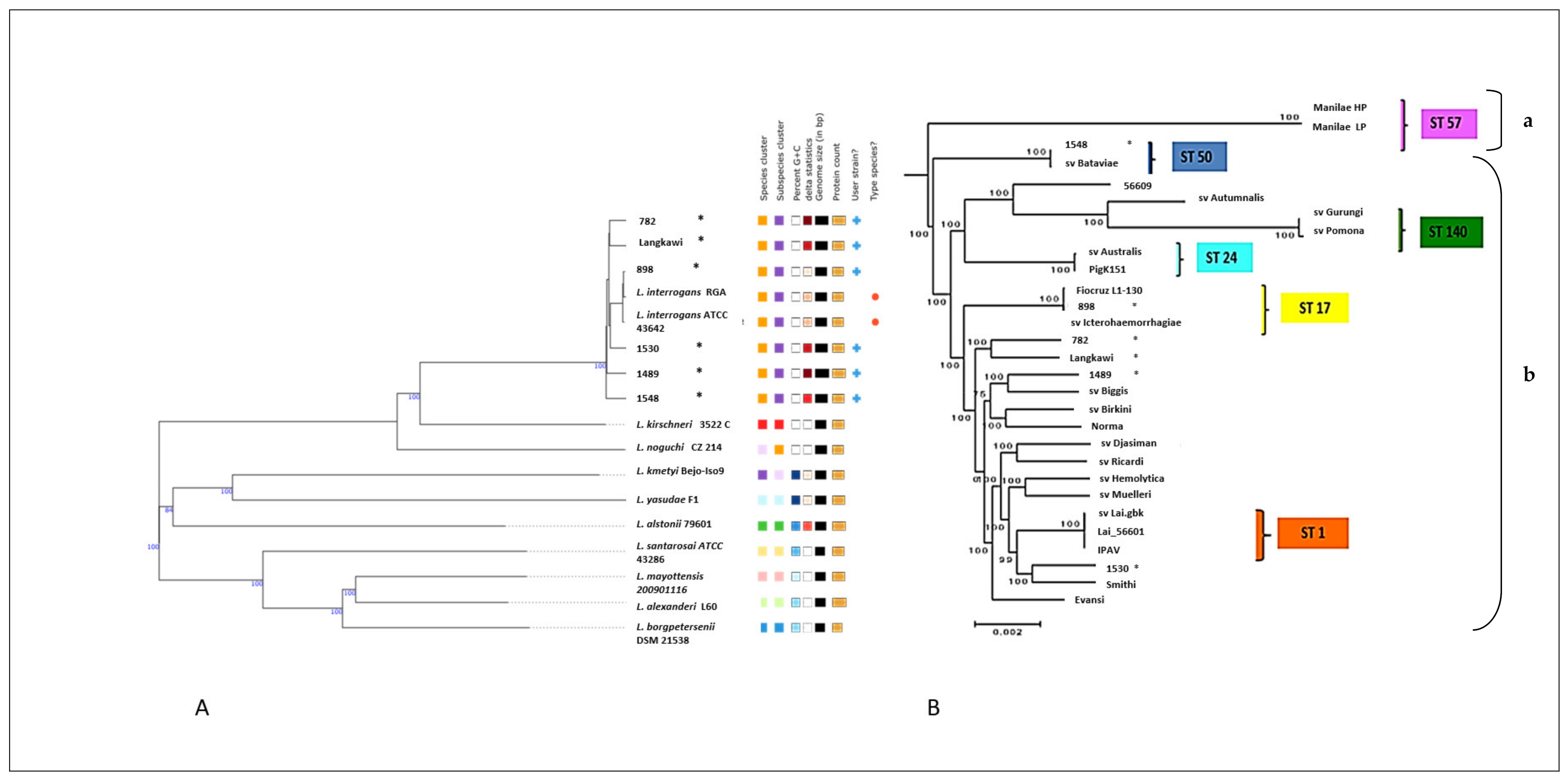
2.4. Whole-Genome-Based Typing and Phylogenetic Classification
2.5. Genome Organization and Rearrangements
2.6. Arrangement of the rfb Locus
2.7. Mobile Genetic Elements
2.8. Genes Associated with Virulence
2.9. Alginate Biosynthesis and Other Virulence Factors
2.10. Potential Virulence Factors by Homology Search against Other Prokaryotes
2.11. Antibiotic Resistance Genes
3. Discussion
4. Materials and Methods
4.1. Culture Collection
4.2. DNA Extraction
4.3. PacBio Library Preparation and Sequencing
4.4. Illumina Library Preparation and Sequencing
4.5. Genome Assembly and Annotation
4.6. Additional L. interrogans Genome Sequences
4.7. Serotyping
4.8. Cross Agglutination Absorption Test
4.9. In Silico DNA Hybridization
4.10. Genome BLAST Distance Phylogeny (GBDP) Species Typing
4.11. Multilocus Sequence Typing (MLST)
4.12. Single Nucleotide Polymorphism (SNP) Phylogeny Analysis
4.13. Core Genome Analysis
4.14. Protein Orthology
4.15. Genome Synteny
4.16. Plasmids
4.17. Prophages
4.18. Clustered Regularly Interspaced Short Palindromic Repeats (CRISPR)
4.19. Insertion Sequences (IS)
4.20. Antimicrobial Resistance Gene
4.21. O-Antigen Analysis
4.22. Bacterial Virulence Factors
4.23. Alginate Biosynthesis
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Mwachui, M.A.; Crump, L.; Hartskeerl, R.; Zinsstag, J.; Hattendorf, J. Environmental and behavioural determinants of leptospirosis transmission: A systematic review. PLoS Negl. Trop. Dis. 2015, 9, e0003843. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andre-Fontaine, G.; Aviat, F.; Thorin, C. Waterborne leptospirosis: Survival and preservation of the virulence of pathogenic Leptospira spp. in fresh water. Curr. Microbiol. 2015, 71, 136–142. [Google Scholar] [CrossRef] [PubMed]
- Costa, F.; Hagan, J.E.; Calcagno, J.; Kane, M.; Torgerson, P.; Martinez-Silveira, M.S.; Stein, C.; Abela-Ridder, B.; Ko, A.I. Global morbidity and mortality of leptospirosis: A systematic review. PLoS Negl. Trop. Dis. 2015, 9, e0003898. [Google Scholar] [CrossRef] [Green Version]
- Garba, B.; Bahaman, A.R.; Khairani-Bejo, S.; Zakaria, Z.; Mutalib, A.R. Retrospective study of leptospirosis in Malaysia. EcoHealth 2017, 14, 389–398. [Google Scholar] [CrossRef] [PubMed]
- Vincent, A.T.; Schiettekatte, O.; Goarant, C.; Neela, V.K.; Bernet, E.; Thibeaux, R.; Ismail, N.; Khalid, M.K.N.M.; Amran, F.; Masuzawa, T. Revisiting the taxonomy and evolution of pathogenicity of the genus Leptospira through the prism of genomics. PLoS Negl. Trop. Dis. 2019, 13, e0007270. [Google Scholar] [CrossRef] [Green Version]
- Cosson, J.-F.; Picardeau, M.; Mielcarek, M.; Tatard, C.; Chaval, Y.; Suputtamongkol, Y.; Buchy, P.; Jittapalapong, S.; Herbreteau, V.; Morand, S. Epidemiology of Leptospira transmitted by rodents in Southeast Asia. PLoS Negl. Trop. Dis. 2014, 8, e2902. [Google Scholar] [CrossRef]
- Hochedez, P.; Theodose, R.; Olive, C.; Bourhy, P.; Hurtrel, G.; Vignier, N.; Mehdaoui, H.; Valentino, R.; Martinez, R.; Delord, J.-M. Factors associated with severe leptospirosis, Martinique, 2010–2013. Emerg. Infect. Dis. 2015, 21, 2221. [Google Scholar] [CrossRef] [Green Version]
- Ren, S.-X.; Fu, G.; Jiang, X.-G.; Zeng, R.; Miao, Y.-G.; Xu, H.; Zhang, Y.-X.; Xiong, H.; Lu, G.; Lu, L.-F. Unique physiological and pathogenic features of Leptospira interrogans revealed by whole-genome sequencing. Nature 2003, 422, 888–893. [Google Scholar] [CrossRef]
- Picardeau, M. Virulence of the zoonotic agent of leptospirosis: Still terra incognita? Nat. Rev. Microbiol. 2017, 15, 297. [Google Scholar] [CrossRef] [PubMed]
- Nascimento, A.; Ko, A.I.; Martins, E.A.L.; Monteiro-Vitorello, C.B.; Ho, P.L.; Haake, D.; Verjovski-Almeida, S.; Hartskeerl, R.; Marques, M.d.V.; Oliveira, M.C.d. Comparative genomics of two Leptospira interrogans serovars reveals novel insights into physiology and pathogenesis. J. Bacteriol. 2004, 186, 2164–2172. [Google Scholar] [CrossRef] [Green Version]
- Bulach, D.M.; Zuerner, R.L.; Wilson, P.; Seemann, T.; McGrath, A.; Cullen, P.A.; Davis, J.; Johnson, M.; Kuczek, E.; Alt, D.P. Genome reduction in Leptospira borgpetersenii reflects limited transmission potential. Proc. Natl. Acad. Sci. USA 2006, 103, 14560–14565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhong, Y.; Chang, X.; Cao, X.-J.; Zhang, Y.; Zheng, H.; Zhu, Y.; Cai, C.; Cui, Z.; Zhang, Y.; Li, Y.-Y. Comparative proteogenomic analysis of the Leptospira interrogans virulence-attenuated strain IPAV against the pathogenic strain 56601. Cell Res. 2011, 21, 1210–1229. [Google Scholar] [CrossRef] [Green Version]
- Palaniappan, R.U.; Chang, Y.-F.; Jusuf, S.; Artiushin, S.; Timoney, J.F.; McDonough, S.P.; Barr, S.C.; Divers, T.J.; Simpson, K.W.; McDonough, P.L. Cloning and molecular characterization of an immunogenic LigA protein of Leptospira interrogans. Infect. Immun. 2002, 70, 5924–5930. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xu, Y.; Zhu, Y.; Wang, Y.; Chang, Y.-F.; Zhang, Y.; Jiang, X.; Zhuang, X.; Zhu, Y.; Zhang, J.; Zeng, L. Whole genome sequencing revealed host adaptation-focused genomic plasticity of pathogenic Leptospira. Sci. Rep. 2016, 6, 20020. [Google Scholar] [CrossRef] [PubMed]
- Fouts, D.E.; Matthias, M.A.; Adhikarla, H.; Adler, B.; Amorim-Santos, L.; Berg, D.E.; Bulach, D.; Buschiazzo, A.; Chang, Y.-F.; Galloway, R.L. What makes a bacterial species pathogenic?: Comparative genomic analysis of the genus Leptospira. PLoS Negl. Trop. Dis. 2016, 10, e0004403. [Google Scholar] [CrossRef] [Green Version]
- Noda, A.A.; Grillová, L.; Mariet, J.-F.; Paiffer, N.B.; Ruiz, Y.C.; Rodríguez, I.; Echevarría, E.; Obregón, A.M.; Lienhard, R.; Picardeau, M. A first insight into the genomic diversity of Leptospira strains isolated from patients in Cuba. PLoS ONE 2020, 15, e0229673. [Google Scholar]
- Santos, L.A.; Adhikarla, H.; Yan, X.; Wang, Z.; Fouts, D.E.; Vinetz, J.M.; Alcantara, L.C.; Hartskeerl, R.A.; Goris, M.G.; Picardeau, M. Genomic Comparison Among Global Isolates of L. interrogans Serovars Copenhageni and Icterohaemorrhagiae Identified Natural Genetic Variation Caused by an Indel. Front. Cell Infect. Microbiol. 2018, 8, 193. [Google Scholar] [CrossRef] [Green Version]
- Wagenaar, J.F.; de Vries, P.J.; Hartskeerl, R.A. Leptospirosis with pulmonary hemorrhage, caused by a new strain of serovar lai: Langkawi. J. Travel Med. 2004, 11, 379–382. [Google Scholar] [CrossRef] [Green Version]
- Fukunaga, M.; Mifuchi, I. Unique organization of Leptospira interrogans rRNA genes. J. Bacteriol. 1989, 171, 5763–5767. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tindall, B. ATCC 43642 replaces ATCC 23581 as the type strain of Leptospira interrogans (Stimson 1907) Wenyon 1926. Opinion 91. Judicial Commission of the International Committee on Systematics of Prokaryotes. Int. J. Syst. Evol. Microbiol. 2014, 64, 3584–3585. [Google Scholar] [CrossRef]
- Boonsilp, S.; Thaipadungpanit, J.; Amornchai, P.; Wuthiekanun, V.; Bailey, M.S.; Holden, M.T.; Zhang, C.; Jiang, X.; Koizumi, N.; Taylor, K.; et al. A single multilocus sequence typing (MLST) scheme for seven pathogenic Leptospira species. PLoS Negl. Trop. Dis. 2013, 7, e1954. [Google Scholar] [CrossRef] [Green Version]
- Amran, F. In Leptospirosis in Malaysia. In Proceedings of the 12th Western Pasific Chemotherapy and Infectious Diseases Conference, Singapore, 2–5 December 2010. [Google Scholar]
- Levett, P.N. Systematics of Leptospiraceae. In Leptospira and Leptospirosis; Springer: Berlin/Heidelberg, Germany, 2015; pp. 11–20. [Google Scholar]
- Peñafiel, E.M.; Ramírez, F.F.; Kawabe, L.K. The use of bacteriophages in the development of new alternatives in therapy. Battle Against Microb. Pathog. Basic Sci. Technol. Adv. Educ. Programs 2015, 1, 346–353. [Google Scholar]
- Que-Gewirth, N.L.; Ribeiro, A.A.; Kalb, S.R.; Cotter, R.J.; Bulach, D.M.; Adler, B.; Saint Girons, I.; Werts, C.; Raetz, C.R. A methylated phosphate group and four amide-linked acyl chains in Leptospira interrogans lipid A The membrane anchor of an unusual lipopolysaccharide that activates TLR2. J. Biol. Chem. 2004, 279, 25420–25429. [Google Scholar] [CrossRef] [Green Version]
- Ricaldi, J.; Matthias, M.A.; Vinetz, J.M.; Lewis, A.L. Expression of sialic acids and other nonulosonic acids in Leptospira. BMC Microbiol. 2012, 12, 161. [Google Scholar] [CrossRef] [Green Version]
- Ristow, P.; Bourhy, P.; Kerneis, S.; Schmitt, C.; Prevost, M.-C.; Lilenbaum, W.; Picardeau, M. Biofilm formation by saprophytic and pathogenic leptospires. Microbiology 2008, 154, 1309–1317. [Google Scholar] [CrossRef] [Green Version]
- Franklin, M.; Nivens, D.; Weadge, J.; Howell, P. Biosynthesis of the Pseudomonas aeruginosa Extracellular Polysaccharides, Alginate, Pel, and Psl. Front. Microbiol. 2011, 2, 167. [Google Scholar] [CrossRef] [Green Version]
- Murray, G.L. The molecular basis of Leptospiral pathogenesis. In Leptospira and Leptospirosis; Springer: Berlin/Heidelberg, Germany, 2015; pp. 139–185. [Google Scholar]
- Bryant, J.; Chewapreecha, C.; Bentley, S.D. Developing insights into the mechanisms of evolution of bacterial pathogens from whole-genome sequences. Future Microbiol. 2012, 7, 1283–1296. [Google Scholar] [CrossRef] [Green Version]
- Schiettekatte, O.; Vincent, A.T.; Malosse, C.; Lechat, P.; Chamot-Rooke, J.; Veyrier, F.J.; Picardeau, M.; Bourhy, P. Characterization of LE3 and LE4, the only lytic phages known to infect the spirochete Leptospira. Sci. Rep. 2018, 8, 11781. [Google Scholar]
- Thakker, M.; Park, J.S.; Carey, V.; Lee, J.C. Staphylococcus aureus serotype 5 capsular polysaccharide is antiphagocytic and enhances bacterial virulence in a murine bacteremia model. Infect. Immun. 1998, 66, 5183–5189. [Google Scholar] [CrossRef] [Green Version]
- Marques, M.B.; Kasper, D.L.; Pangburn, M.K.; Wessels, M.R. Prevention of C3 deposition by capsular polysaccharide is a virulence mechanism of type III group B streptococci. Infect. Immun. 1992, 60, 3986–3993. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jerse, A.E.; Sharma, N.D.; Simms, A.N.; Crow, E.T.; Snyder, L.A.; Shafer, W.M. A Gonococcal Efflux Pump System Enhances Bacterial Survival in a Female Mouse Model of Genital Tract Infection. Infect. Immun. 2003, 71, 5576–5582. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, F.; Yang, J.; Zhang, X.; Chen, L.; Jiang, Y.; Yan, Y.; Tang, X.; Wang, J.; Xiong, Z.; Dong, J.; et al. Genome dynamics and diversity of Shigella species, the etiologic agents of bacillary dysentery. Nucleic Acids Res. 2005, 33, 6445–6458. [Google Scholar] [CrossRef] [PubMed]
- Chapon-Herve, V.; Akrim, M.; Latifi, A.; Williams, P.; Lazdunski, A.; Bally, M. Regulation of the xcp secretion pathway by multiple quorum-sensing modulons in Pseudomonas aeruginosa. Mol. Microbiol. 1997, 24, 1169–1178. [Google Scholar] [CrossRef] [PubMed]
- Stone, C.B.; Johnson, D.L.; Bulir, D.C.; Gilchrist, J.D.; Mahony, J.B. Characterization of the putative type III secretion ATPase CdsN (Cpn0707) of Chlamydophila pneumoniae. J. Bacteriol. 2008, 190, 6580–6588. [Google Scholar] [CrossRef] [Green Version]
- Ressner, R.A.; Griffith, M.E.; Beckius, M.L.; Pimentel, G.; Miller, R.S.; Mende, K.; Fraser, S.L.; Galloway, R.L.; Hospenthal, D.R.; Murray, C.K. Antimicrobial Susceptibilities of Geographically Diverse Clinical Human Isolates of Leptospira. Antimicrob. Agents Chemother. 2008, 52, 2750–2754. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siti, K.B.; Fairuz, A.; Zunita, Z.; Aziah, D.; Mazrura, S.; Evie, K. Manual for Laboratory Diagnosis of Leptospirosis: One Health Approach; Malaysia One Health University Network (MyOHUN): Selangor, Malaysia, 2017; p. 56. [Google Scholar]
- Zerbino, D.R.; Birney, E. Velvet: Algorithms for de novo short read assembly using de Bruijn graphs. Genome Res. 2008, 18, 821–829. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Koboldt, D.C.; Zhang, Q.; Larson, D.E.; Shen, D.; McLellan, M.D.; Lin, L.; Miller, C.A.; Mardis, E.R.; Ding, L.; Wilson, R.K. VarScan 2: Somatic mutation and copy number alteration discovery in cancer by exome sequencing. Genome Res. 2012, 22, 568–576. [Google Scholar] [CrossRef] [Green Version]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef]
- Hartskeerl, R. Leptospirosis: Current status and future trends. Indian J. Med. Microbiol. 2006, 24, 309. [Google Scholar] [CrossRef]
- TSCL, Subcomittee on the Taxonomy of Leptospira. Int. J. Syst. Bacteriol. 1987, 37, 472–473.
- Meier-Kolthoff, J.P.; Auch, A.F.; Klenk, H.-P.; Göker, M. Genome sequence-based species delimitation with confidence intervals and improved distance functions. BMC Bioinform. 2013, 14, 60. [Google Scholar] [CrossRef] [Green Version]
- Meier-Kolthoff, J.P.; Göker, M. TYGS is an automated high-throughput platform for state-of-the-art genome-based taxonomy. Nat. Commun. 2019, 10, 2182. [Google Scholar] [CrossRef] [PubMed]
- Treangen, T.J.; Ondov, B.D.; Koren, S.; Phillippy, A.M. The Harvest suite for rapid core-genome alignment and visualization of thousands of intraspecific microbial genomes. Genome Biol. 2014, 15, 524. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lechner, M.; Findeiss, S.; Steiner, L.; Marz, M.; Stadler, P.F.; Prohaska, S.J. Proteinortho: Detection of (co-)orthologs in large-scale analysis. BMC Bioinform. 2011, 12, 124. [Google Scholar] [CrossRef] [Green Version]
- Bardou, P.; Mariette, J.; Escudié, F.; Djemiel, C.; Klopp, C. jvenn: An interactive Venn diagram viewer. BMC Bioinform. 2014, 15, 293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Darling, A.C.; Mau, B.; Blattner, F.R.; Perna, N.T. Mauve: Multiple alignment of conserved genomic sequence with rearrangements. Genome Res. 2004, 14, 1394–1403. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huerta-Cepas, J.; Forslund, K.; Coelho, L.P.; Szklarczyk, D.; Jensen, L.J.; von Mering, C.; Bork, P. Fast Genome-Wide Functional Annotation through Orthology Assignment by eggNOG-Mapper. Mol. Biol. Evol. 2017, 34, 2115–2122. [Google Scholar] [CrossRef] [Green Version]
- Arndt, D.; Grant, J.R.; Marcu, A.; Sajed, T.; Pon, A.; Liang, Y.; Wishart, D.S. PHASTER: A better, faster version of the PHAST phage search tool. Nucleic Acids Res. 2016, 44, W16–W21. [Google Scholar] [CrossRef] [Green Version]
- Couvin, D.; Bernheim, A.; Toffano-Nioche, C.; Touchon, M.; Michalik, J.; Neron, B.; Rocha, E.P.C.; Vergnaud, G.; Gautheret, D.; Pourcel, C. CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res. 2018, 46, W246–W251. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Altschul, S.F.; Madden, T.L.; Schaffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped BLAST and PSI-BLAST: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Siguier, P.; Perochon, J.; Lestrade, L.; Mahillon, J.; Chandler, M. ISfinder: The reference centre for bacterial insertion sequences. Nucleic Acids Res. 2006, 34, D32–D36. [Google Scholar] [CrossRef] [Green Version]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Sullivan, M.J.; Petty, N.K.; Beatson, S.A. Easyfig: A genome comparison visualizer. Bioinformatics 2011, 27, 1009–1010. [Google Scholar] [CrossRef]
- Chen, L.; Zheng, D.; Liu, B.; Yang, J.; Jin, Q. VFDB 2016: Hierarchical and refined dataset for big data analysis—10 years on. Nucleic Acids Res. 2016, 44, D694–D697. [Google Scholar] [CrossRef] [PubMed]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Strain | Serogroup | Serovar |
|---|---|---|
| Langkawi * | Icterohaemorrhagiae | Lai-like |
| 898 | Icterohaemorrhagiae | Copenhageni/Icterohaemorrhagiae |
| 782 | Canicola | Bindjei |
| 1489 | Bataviae | Paidjan |
| 1548 | Bataviae | Bataviae |
| 1530 | Icterohaemorrhagiae | Yeoncheon |
| Feature | Strain | |||||
|---|---|---|---|---|---|---|
| Langkawi | 782 | 898 | 1489 | 1548 | 1530 | |
| Size (Mb) | 4.76 | 4.91 | 4.63 | 4.63 | 4.74 | 4.67 |
| Chromosome I (Mb) | 4.37 | 4.56 | 4.28 | 4.28 | 4.35 | 4.32 |
| Chromosome II (Mb) | 0.39 | 0.35 | 0.35 | 0.35 | 0.39 | 0.35 |
| No. of plasmids | 2 | 5 | 0 | 7 | 2 | ≥5 * |
| No. of contigs | 4 | 7 | 2 | 9 | 4 | 10 |
| CDS | 3872 | 4001 | 3759 | 3926 | 3861 | 3785 |
| tmRNA | 1 | 1 | 1 | 1 | 1 | 1 |
| Repeat region ^ | 1 | 2 | 1 | 2 | 3 | 2 |
| Gene | 4048 | 4476 | 3716 | 4516 | 4000 | 4087 |
| tRNA | 37 | 37 | 37 | 37 | 37 | 37 |
| 23S | 2 | 2 | 2 | 2 | 2 | 2 |
| 16S | 2 | 2 | 2 | 2 | 2 | 2 |
| 5S | 1 | 1 | 1 | 1 | 1 | 1 |
| Signal peptide | 132 | 129 | 124 | 133 | 131 | 125 |
| Miscellaneous RNA | 5 | 11 | 5 | 10 | 5 | 0 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramli, S.R.; Bunk, B.; Spröer, C.; Geffers, R.; Jarek, M.; Bhuju, S.; Goris, M.; Mustakim, S.; Pessler, F. Complete Genome Sequencing of Leptospira interrogans Isolates from Malaysia Reveals Massive Genome Rearrangement but High Conservation of Virulence-Associated Genes. Pathogens 2021, 10, 1198. https://doi.org/10.3390/pathogens10091198
Ramli SR, Bunk B, Spröer C, Geffers R, Jarek M, Bhuju S, Goris M, Mustakim S, Pessler F. Complete Genome Sequencing of Leptospira interrogans Isolates from Malaysia Reveals Massive Genome Rearrangement but High Conservation of Virulence-Associated Genes. Pathogens. 2021; 10(9):1198. https://doi.org/10.3390/pathogens10091198
Chicago/Turabian StyleRamli, Siti Roszilawati, Boyke Bunk, Cathrin Spröer, Robert Geffers, Michael Jarek, Sabin Bhuju, Marga Goris, Sahlawati Mustakim, and Frank Pessler. 2021. "Complete Genome Sequencing of Leptospira interrogans Isolates from Malaysia Reveals Massive Genome Rearrangement but High Conservation of Virulence-Associated Genes" Pathogens 10, no. 9: 1198. https://doi.org/10.3390/pathogens10091198
APA StyleRamli, S. R., Bunk, B., Spröer, C., Geffers, R., Jarek, M., Bhuju, S., Goris, M., Mustakim, S., & Pessler, F. (2021). Complete Genome Sequencing of Leptospira interrogans Isolates from Malaysia Reveals Massive Genome Rearrangement but High Conservation of Virulence-Associated Genes. Pathogens, 10(9), 1198. https://doi.org/10.3390/pathogens10091198