
SARS-CoV-2 Exploits Non-Canonical Autophagic Processes to Replicate, Mature, and Egress the Infected Vero E6 Cells

 , and
, and {kind=link}
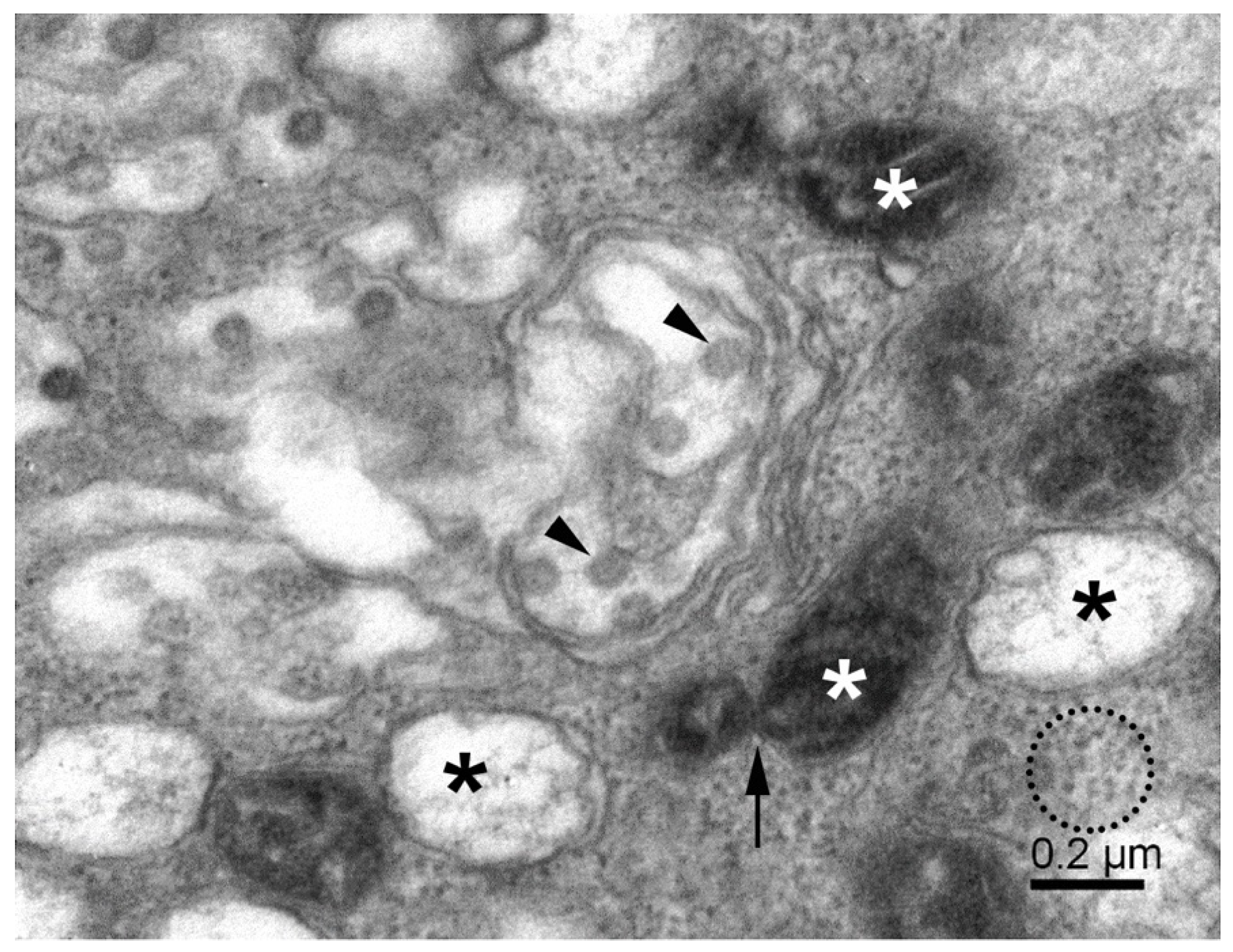{kind=link}
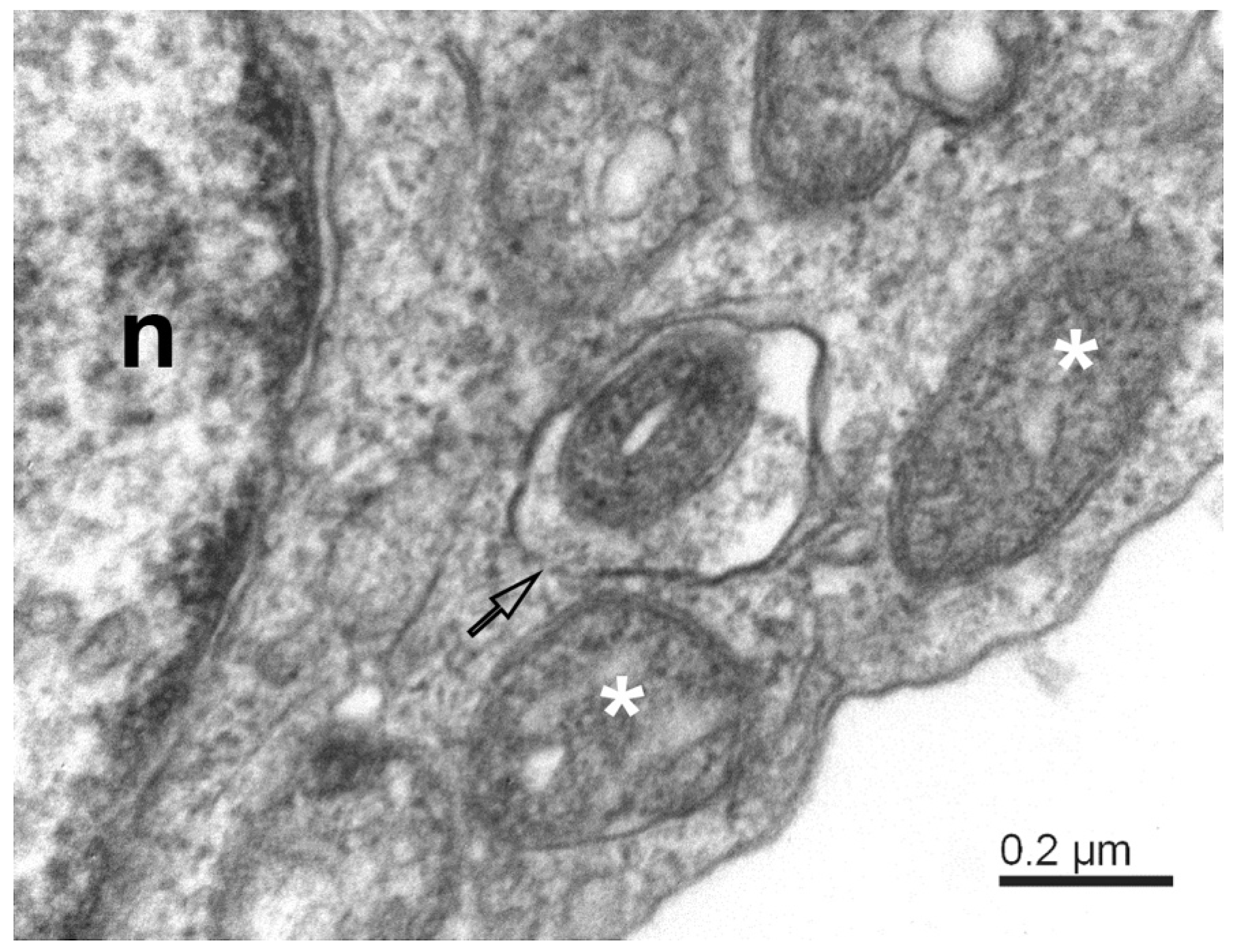{kind=link}
{kind=link}
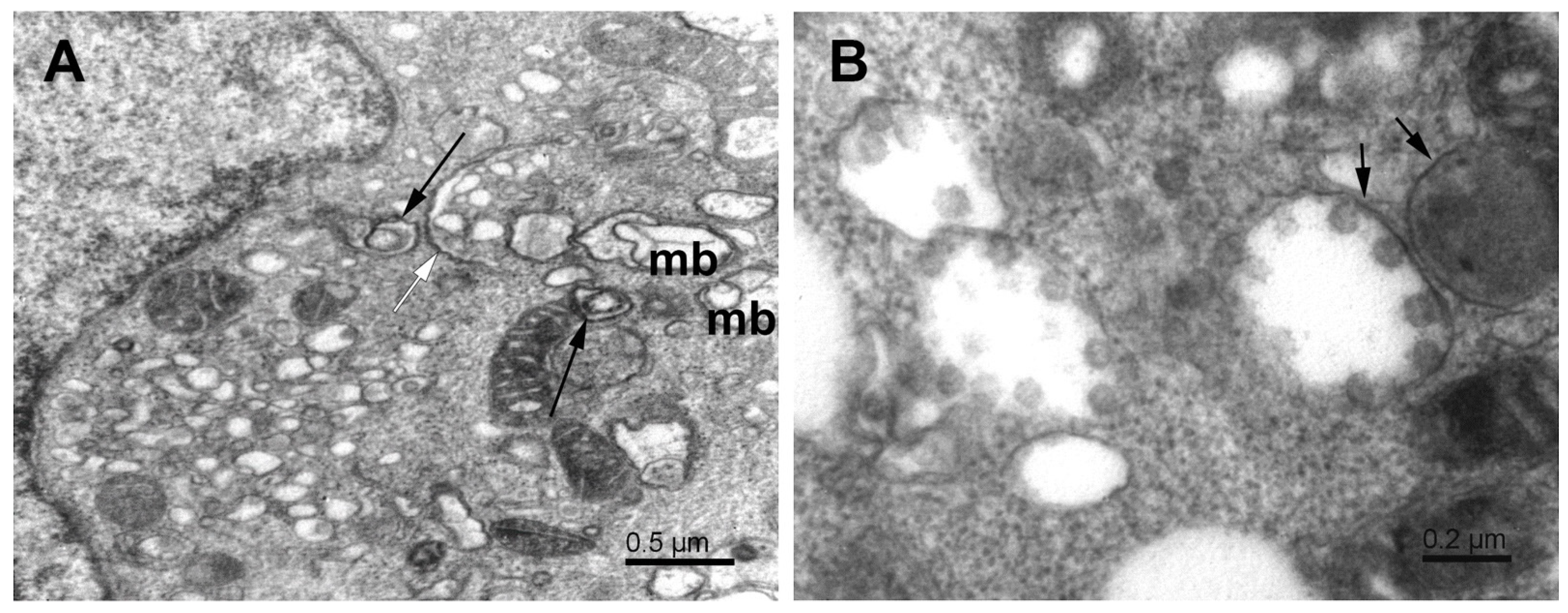{kind=link}
{kind=link}
{kind=link}
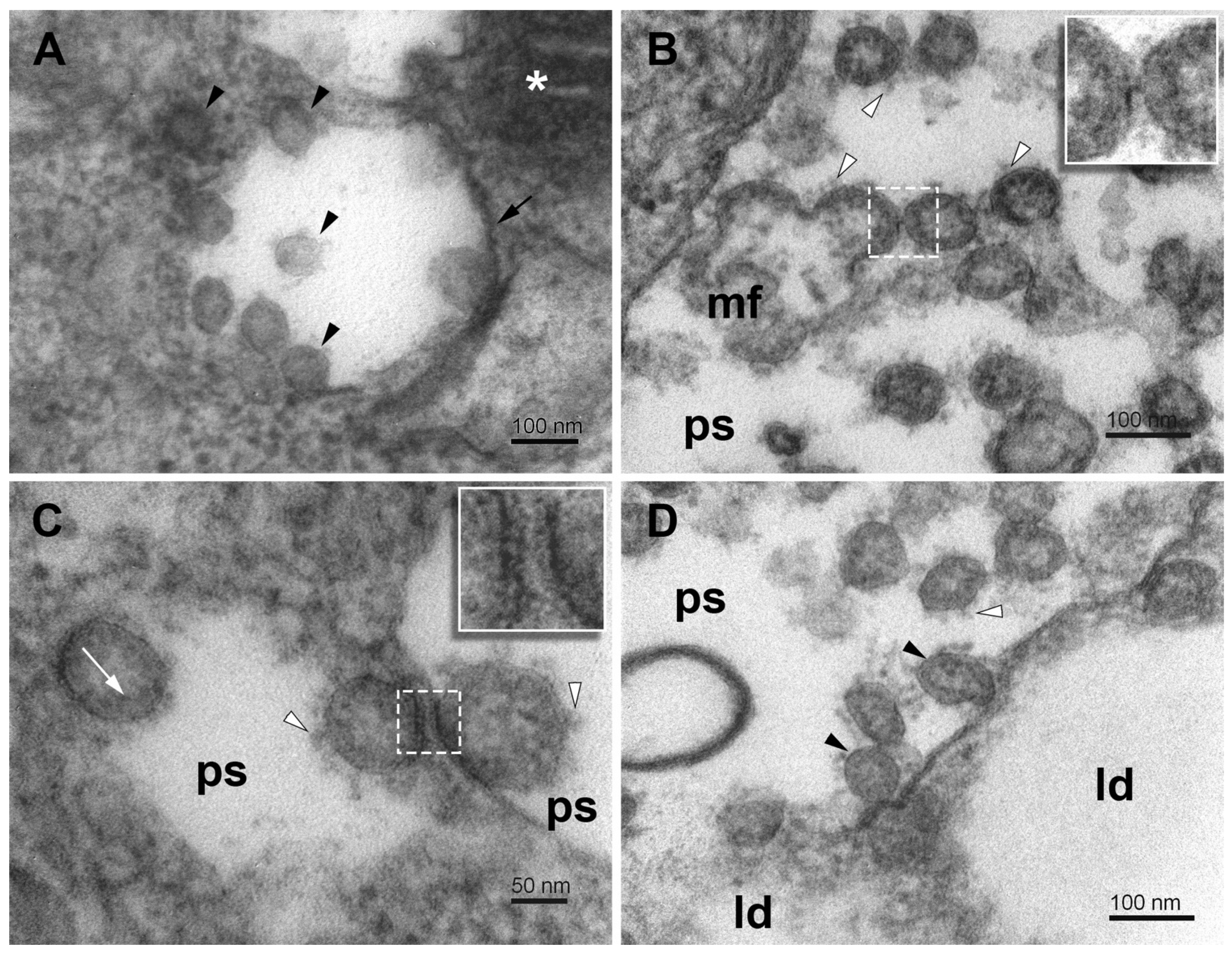{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
2.1. Cell Cultures and Infection with SARS-CoV-2
2.2. SARS-CoV-2 Replication In Vitro and TCID50 Assay
2.3. Transmission Electron Microscopy
2.4. RT2 Profiler Autophagy PCR Array in SARS-CoV-2-Infected Cells
3. Results and Discussion
3.1. Morphology of Infected Cells
3.2. Mitochondria, Fission, and Mitophagy
3.3. Phagophore, Phagosomes, and Autophagy
3.4. Lipid Droplets
3.5. Virus Assembly, Maturation, and Egress
3.6. SARS-CoV-2 Replication Dynamics and Autophagic Gene Expression
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Siddell, S.; Wege, H.; ter Meulen, V. The Structure and Replication of Coronaviruses. Curr. Top. Microbiol. Immunol. 1982, 99, 131–163. [Google Scholar] [PubMed] [Green Version]
- Klein, S.; Cortese, M.; Winter, S.L.; Wachsmuth-Melm, M.; Neufeldt, C.J.; Cerikan, B.; Stanifer, M.L.; Boulant, S.; Bartenschlager, R.; Chlanda, P. SARS-CoV-2 Structure and Replication Characterized by in Situ Cryo-Electron Tomography. Nat. Commun. 2020, 11, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Caldas, L.A.; Carneiro, F.A.; Monteiro, F.L.; Augusto, I.; Higa, L.M.; Miranda, K.; Tanuri, A.; de Souza, W. Intracellular Host Cell Membrane Remodelling Induced by SARS-CoV-2 Infection in Vitro. Biol. Cell 2021, 113, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Pöhlmann, S. A Multibasic Cleavage Site in the Spike Protein of SARS-CoV-2 Is Essential for Infection of Human Lung Cells. Mol. Cell 2020, 78, 779–784. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Krüger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- De Wilde, A.H.; Snijder, E.J.; Kikkert, M.; Van Hemert, M.J.; De Wilde, A.H.; Snijder, Á.E.J.; Kikkert, Á.M.; Van Hemert, Á.M.J. Host Factors in Coronavirus Replication. Curr. Top. Microbiol. Immunol. 2017, 419, 1–42. [Google Scholar] [CrossRef] [Green Version]
- de Breyne, S.; Vindry, C.; Guillin, O.; Condé, L.; Mure, F.; Gruffat, H.; Chavatte, L.; Ohlmann, T. Translational Control of Coronaviruses. Nucleic Acids Res. 2020, 48, 12502–12522. [Google Scholar] [CrossRef]
- Mihelc, E.M.; Baker, S.C.; Lanman, J.K. Coronavirus Infection Induces Progressive Restructuring of the Endoplasmic Reticulum Involving the Formation and Degradation of Double Membrane Vesicles. Virology 2021, 556, 9–22. [Google Scholar] [CrossRef]
- Lavi, E.; Wang, Q.; Weiss, S.R.; Gonatas, N.K. Syncytia Formation Induced by Coronavirus Infection Is Associated with Fragmentation and Rearrangement of the Golgi Apparatus. Virology 1996, 221, 325–334. [Google Scholar] [CrossRef] [Green Version]
- Knoops, K.; Kikkert, M.; Worm, S.H.E.V.D.; Zevenhoven-Dobbe, J.C.; Meer, Y.V.D.; Koster, A.J.; Mommaas, A.M.; Snijder, E.J. SARS-Coronavirus Replication Is Supported by a Reticulovesicular Network of Modified Endoplasmic Reticulum. PLoS Biol. 2008, 6, e226. [Google Scholar] [CrossRef] [Green Version]
- Wong, N.A.; Saier, M.H. The Sars-Coronavirus Infection Cycle: A Survey of Viral Membrane Proteins, Their Functional Interactions and Pathogenesis. Int. J. Mol. Sci. 2021, 22, 1308. [Google Scholar] [CrossRef]
- Ducatelle, R.; Hoorens, J. Significance of Lysosomes in the Morphogenesis of Coronaviruses. Arch. Virol. 1984, 79, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Dellibovi-Ragheb, T.A.; Kerviel, A.; Pak, E.; Qiu, Q.; Fisher, M.; Takvorian, P.M.; Bleck, C.; Hsu, V.W.; Fehr, A.R.; et al. β-Coronaviruses Use Lysosomes for Egress Instead of the Biosynthetic Secretory Pathway. Cell 2020, 183, 1520–1535. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, L.; Mostowy, S.; Sancho-Shimizu, V. Autophagy-Virus Interplay: From Cell Biology to Human Disease. Front. Cell Dev. Biol. 2018, 6, 155. [Google Scholar] [CrossRef] [Green Version]
- Miller, K.; McGrath, M.E.; Hu, Z.; Ariannejad, S.; Weston, S.; Frieman, M.; Jackson, W.T. Coronavirus Interactions with the Cellular Autophagy Machinery. Autophagy 2020, 16, 2131–2139. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Z.; Lu, K.; Mao, B.; Liu, S.; Trilling, M.; Huang, A.; Lu, M.; Lin, Y. The Interplay between Emerging Human Coronavirus Infections and Autophagy. Emerg. Microbes Infect. 2021, 10, 196–205. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Qin, Y.; Chen, M. Viral Strategies for Triggering and Manipulating Mitophagy. Autophagy 2018, 14, 1665–1673. [Google Scholar] [CrossRef] [Green Version]
- Shi, C.-S.; Qi, H.-Y.; Boularan, C.; Huang, N.-N.; Abu-Asab, M.; Shelhamer, J.H.; Kehrl, J.H. SARS-Coronavirus Open Reading Frame-9b Suppresses Innate Immunity by Targeting Mitochondria and the MAVS/TRAF3/TRAF6 Signalosome. J. Immunol. 2014, 193, 3080–3089. [Google Scholar] [CrossRef] [Green Version]
- Carlsson, S.R.; Simonsen, A. Membrane Dynamics in Autophagosome Biogenesis. J. Cell Sci. 2015, 128, 193–205. [Google Scholar] [CrossRef] [Green Version]
- Delorme-Axford, E.; Klionsky, D.J. Highlights in the Fight against COVID-19: Does Autophagy Play a Role in SARS-CoV-2 Infection? Autophagy 2020, 16, 2123–2127. [Google Scholar] [CrossRef]
- Maier, H.J.; Britton, P. Involvement of Autophagy in Coronavirus Replication. Viruses 2012, 4, 3440–3451. [Google Scholar] [CrossRef] [PubMed]
- Schneider, M.; Ackermann, K.; Stuart, M.; Wex, C.; Protzer, U.; Schätzl, H.M.; Gilch, S. Severe Acute Respiratory Syndrome Coronavirus Replication Is Severely Impaired by MG132 Due to Proteasome-Independent Inhibition of M-Calpain. J. Virol. 2012, 86, 10112–10122. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, N.; Shen, H.M. Targeting the Endocytic Pathway and Autophagy Process as a Novel Therapeutic Strategy in COVID-19. Int. J. Biol. Sci. 2020, 16, 1724. [Google Scholar] [CrossRef] [PubMed]
- Gassen, N.C.; Niemeyer, D.; Muth, D.; Corman, V.M.; Martinelli, S.; Gassen, A.; Hafner, K.; Papies, J.; Mösbauer, K.; Zellner, A.; et al. SKP2 Attenuates Autophagy through Beclin1-Ubiquitination and Its Inhibition Reduces MERS-Coronavirus Infection. Nat. Commun. 2019, 10, 1–16. [Google Scholar] [CrossRef] [Green Version]
- Ramirez, S.; Fernandez-Antunez, C.; Galli, A.; Underwood, A.; Pham, L.V.; Ryberg, L.A.; Feng, S.; Pedersen, M.S.; Mikkelsen, L.S.; Belouzard, S.; et al. Overcoming Culture Restriction for SARS-CoV-2 in Human Cells Facilitates the Screening of Compounds Inhibiting Viral Replication. Antimicrob. Agents Chemother. 2021, 65, e00097-21. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Landt, O.; Kaiser, M.; Molenkamp, R.; Meijer, A.; Chu, D.K.W.; Bleicker, T.; Brünink, S.; Schneider, J.; Schmidt, M.L.; et al. Detection of 2019 Novel Coronavirus (2019-NCoV) by Real-Time RT-PCR. Eurosurveillance 2020, 25, 2000045. [Google Scholar] [CrossRef] [Green Version]
- Ge, L.; Schekman, R. The ER-Golgi Intermediate Compartment Feeds the Phagophore Membrane. Autophagy 2014, 10, 170–172. [Google Scholar] [CrossRef] [Green Version]
- Hagemeijer, M.C.; Verheije, M.H.; Ulasli, M.; Shaltiël, I.A.; de Vries, L.A.; Reggiori, F.; Rottier, P.J.M.; de Haan, C.A.M. Dynamics of Coronavirus Replication-Transcription Complexes. J. Virol. 2010, 84, 2134–2149. [Google Scholar] [CrossRef] [Green Version]
- V’kovski, P.; Kratzel, A.; Steiner, S.; Stalder, H.; Thiel, V. Coronavirus Biology and Replication: Implications for SARS-CoV-2. Nat. Rev. Microbiol. 2021, 19, 155–170. [Google Scholar] [CrossRef]
- Farkash, E.A.; Wilson, A.M.; Jentzen, J.M. Ultrastructural Evidence for Direct Renal Infection with SARS-CoV-2. J. Am. Soc. Nephrol. 2020, 31, 1683–1687. [Google Scholar] [CrossRef]
- Ogando, N.S.; Dalebout, T.J.; Zevenhoven-Dobbe, J.C.; Limpens, R.W.A.L.; van der Meer, Y.; Caly, L.; Druce, J.; de Vries, J.J.C.; Kikkert, M.; Bárcena, M.; et al. SARS-Coronavirus-2 Replication in Vero E6 Cells: Replication Kinetics, Rapid Adaptation and Cytopathology. J. Gen. Virol. 2020, 101, 925–940. [Google Scholar] [CrossRef] [PubMed]
- Gatti, P.; Ilamathi, H.S.; Todkar, K.; Germain, M. Mitochondria Targeted Viral Replication and Survival Strategies—Prospective on SARS-CoV-2. Front. Pharmacol. 2020, 11, 578599. [Google Scholar] [CrossRef] [PubMed]
- Yamashita, S.I.; Jin, X.; Furukawa, K.; Hamasaki, M.; Nezu, A.; Otera, H.; Saigusa, T.; Yoshimori, T.; Sakai, Y.; Mihara, K.; et al. Mitochondrial Division Occurs Concurrently with Autophagosome Formation but Independently of Drp1 during Mitophagy. J. Cell Biol. 2016, 215, 649–665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hailey, D.W.; Rambold, A.S.; Satpute-Krishnan, P.; Mitra, K.; Sougrat, R.; Kim, P.K.; Lippincott-Schwartz, J. Mitochondria Supply Membranes for Autophagosome Biogenesis during Starvation. Cell 2010, 141, 656–667. [Google Scholar] [CrossRef] [Green Version]
- Biazik, J.; Ylä-Anttila, P.; Vihinen, H.; Jokitalo, E.; Eskelinen, E.L. Ultrastructural Relationship of the Phagophore with Surrounding Organelles. Autophagy 2015, 11, 439–451. [Google Scholar] [CrossRef] [Green Version]
- Cook, K.L.; Soto-Pantoja, D.R.; Abu-Asab, M.; Clarke, P.A.G.; Roberts, D.D.; Clarke, R. Mitochondria Directly Donate Their Membrane to Form Autophagosomes during a Novel Mechanism of Parkin-Associated Mitophagy. Cell Biosci. 2014, 4, 1–9. [Google Scholar] [CrossRef] [Green Version]
- Saka, H.A.; Valdivia, R. Emerging Roles for Lipid Droplets in Immunity and Host-Pathogen Interactions. Annu. Rev. Cell Dev. Biol. 2012, 28, 411–437. [Google Scholar] [CrossRef]
- Viktorova, E.G.; Nchoutmboube, J.A.; Ford-Siltz, L.A.; Iverson, E.; Belov, G.A. Phospholipid Synthesis Fueled by Lipid Droplets Drives the Structural Development of Poliovirus Replication Organelles. PLoS Pathog. 2018, 14, e1007280. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Z.; He, G.; Filipowicz, N.A.; Randall, G.; Belov, G.A.; Kopek, B.G.; Wang, X. Host Lipids in Positive-Strand RNA Virus Genome Replication. Front. Microbiol. 2019, 10, 286. [Google Scholar] [CrossRef] [Green Version]
- Pagliari, F.; Marafioti, M.G.; Genard, G.; Candeloro, P.; Viglietto, G.; Seco, J.; Tirinato, L. SsRNA Virus and Host Lipid Rearrangements: Is There a Role for Lipid Droplets in SARS-CoV-2 Infection? Front. Mol. Biosci. 2020, 7, 578964. [Google Scholar] [CrossRef]
- Stertz, S.; Reichelt, M.; Spiegel, M.; Kuri, T.; Martínez-Sobrido, L.; García-Sastre, A.; Weber, F.; Kochs, G. The Intracellular Sites of Early Replication and Budding of SARS-Coronavirus. Virology 2007, 361, 304–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- di Malta, C.; Cinque, L.; Settembre, C. Transcriptional Regulation of Autophagy: Mechanisms and Diseases. Front. Cell Dev. Biol. 2019, 7, 114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Robinson, P.C.; Liew, D.F.L.; Liew, J.W.; Monaco, C.; Richards, D.; Shivakumar, S.; Tanner, H.L.; Feldmann, M. The Potential for Repurposing Anti-TNF as a Therapy for the Treatment of COVID-19. Med 2020, 1, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Sato, A.; Linehan, M.M.; Iwasaki, A. Dual Recognition of Herpes Simplex Viruses by TLR2 and TLR9 in Dendritic Cells. Proc. Natl. Acad. Sci. USA 2006, 103, 17343–17348. [Google Scholar] [CrossRef] [Green Version]
- Sainz, B.; Mossel, E.C.; Peters, C.J.; Garry, R.F. Interferon-Beta and Interferon-Gamma Synergistically Inhibit the Replication of Severe Acute Respiratory Syndrome-Associated Coronavirus (SARS-CoV). Virology 2004, 329, 11–17. [Google Scholar] [CrossRef] [Green Version]
- Józefiak, A.; Larska, M.; Pomorska-Mól, M.; Ruszkowski, J.J. The IGF-1 Signaling Pathway in Viral Infections. Viruses 2021, 13, 1488. [Google Scholar] [CrossRef]
- Shin, G.C.; Kang, H.S.; Lee, A.R.; Kim, K.H. Hepatitis B Virus–Triggered Autophagy Targets TNFRSF10B/Death Receptor 5 for Degradation to Limit TNFSF10/TRAIL Response. Autophagy 2016, 12, 2451–2466. [Google Scholar] [CrossRef] [Green Version]
- Espert, L.; Denizot, M.; Grimaldi, M.; Robert-Hebmann, V.; Gay, B.; Varbanov, M.; Codogno, P.; Biard-Piechaczyk, M. Autophagy Is Involved in T Cell Death after Binding of HIV-1 Envelope Proteins to CXCR4. J. Clin. Investig. 2006, 116, 2161–2172. [Google Scholar] [CrossRef]
- Das, B.; Dobrowolski, C.; Luttge, B.; Valadkhan, S.; Chomont, N.; Johnston, R.; Bacchetti, P.; Hoh, R.; Gandhi, M.; Deeks, S.G.; et al. Estrogen Receptor-1 Is a Key Regulator of HIV-1 Latency That Imparts Gender-Specific Restrictions on the Latent Reservoir. Proc. Natl. Acad. Sci. USA 2018, 115, E7795–E7804. [Google Scholar] [CrossRef] [Green Version]
- Wang, L.; Tian, Y.; Ou, J.-H.J. HCV Induces the Expression of Rubicon and UVRAG to Temporally Regulate the Maturation of Autophagosomes and Viral Replication. PLoS Pathog. 2015, 11, e1004764. [Google Scholar] [CrossRef]
- Skytte Rasmussen, M.; Mouilleron, S.; Kumar Shrestha, B.; Wirth, M.; Lee, R.; Bowitz Larsen, K.; Abudu Princely, Y.; O’Reilly, N.; Sjøttem, E.; Tooze, S.A.; et al. ATG4B Contains a C-Terminal LIR Motif Important for Binding and Efficient Cleavage of Mammalian Orthologs of Yeast Atg8. Autophagy 2017, 13, 834–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beatman, E.L.; Massey, A.; Shives, K.D.; Burrack, K.S.; Chamanian, M.; Morrison, T.E.; Beckham, J.D. Alpha-Synuclein Expression Restricts RNA Viral Infections in the Brain. J. Virol. 2016, 90, 2767–2782. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mohamud, Y.; Xue, Y.C.; Liu, H.; Ng, C.S.; Bahreyni, A.; Jan, E.; Luo, H. The Papain-like Protease of Coronaviruses Cleaves ULK1 to Disrupt Host Autophagy. Biochem. Biophys. Res. Commun. 2021, 540, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Koren, I.; Reem, E.; Kimchi, A. DAP1, a Novel Substrate of MTOR, Negatively Regulates Autophagy. Curr. Biol. 2010, 20, 1093–1098. [Google Scholar] [CrossRef] [Green Version]
- Mehrzadi, S.; Karimi, M.Y.; Fatemi, A.; Reiter, R.J.; Hosseinzadeh, A. SARS-CoV-2 and Other Coronaviruses Negatively Influence Mitochondrial Quality Control: Beneficial Effects of Melatonin. Pharmacol. Ther. 2021, 224, 107825. [Google Scholar] [CrossRef]











Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Koči, J.; Novotová, M.; Sláviková, M.; Klempa, B.; Zahradník, I. SARS-CoV-2 Exploits Non-Canonical Autophagic Processes to Replicate, Mature, and Egress the Infected Vero E6 Cells. Pathogens 2022, 11, 1535. https://doi.org/10.3390/pathogens11121535
Koči J, Novotová M, Sláviková M, Klempa B, Zahradník I. SARS-CoV-2 Exploits Non-Canonical Autophagic Processes to Replicate, Mature, and Egress the Infected Vero E6 Cells. Pathogens. 2022; 11(12):1535. https://doi.org/10.3390/pathogens11121535
Chicago/Turabian StyleKoči, Juraj, Marta Novotová, Monika Sláviková, Boris Klempa, and Ivan Zahradník. 2022. "SARS-CoV-2 Exploits Non-Canonical Autophagic Processes to Replicate, Mature, and Egress the Infected Vero E6 Cells" Pathogens 11, no. 12: 1535. https://doi.org/10.3390/pathogens11121535
APA StyleKoči, J., Novotová, M., Sláviková, M., Klempa, B., & Zahradník, I. (2022). SARS-CoV-2 Exploits Non-Canonical Autophagic Processes to Replicate, Mature, and Egress the Infected Vero E6 Cells. Pathogens, 11(12), 1535. https://doi.org/10.3390/pathogens11121535