
Epstein–Barr Virus Infection in Lung Cancer: Insights and Perspectives

 , , and
, , and 
Abstract
:1. Introduction
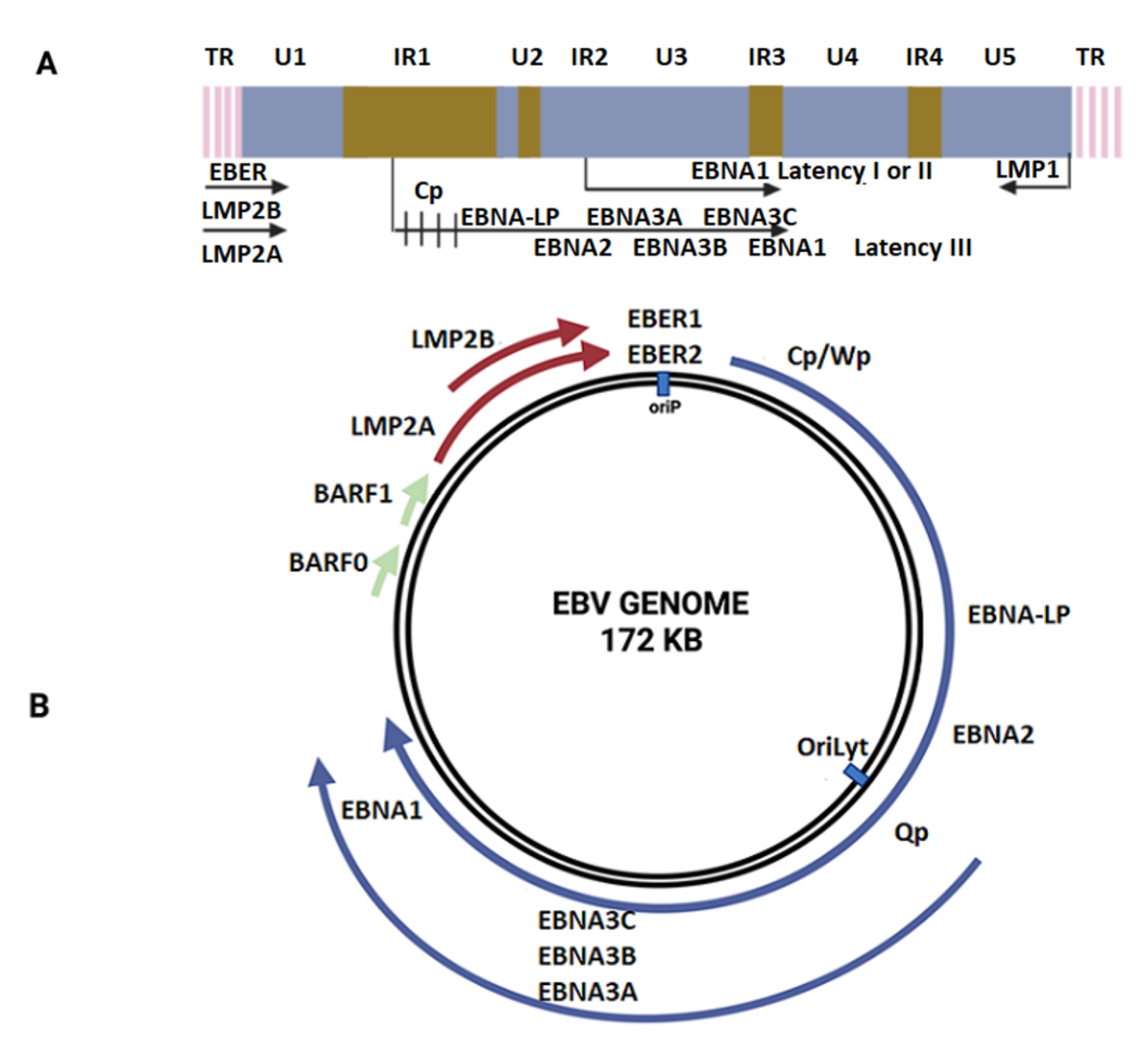
2. Epstein–Barr Virus: Structure, Replication, and Role in Cancer
2.1. Epstein–Barr Virus: Structure and Replication Cycle
2.2. Role of EBV in Epithelial Cancers
2.3. EBV Latency and Lytic Cycle in Cancer
3. EBV Infection in Lung Cancer
3.1. Pathogenesis of Lung Cancer
3.2. Epidemiology of EBV in Lung Cancer
3.3. Potential EBV-Mediated Lung Cancer Mechanisms
3.3.1. Interactions with Tobacco Components
Tobacco Smoke May Facilitate EBV Latency Establishment in Lung Epithelial Cells
Tobacco Smoke Impairs Immune Response Facilitating EBV Infection in Lung Epithelial Cells
Tobacco Smoke Can Activate the EBV Lytic Switch
3.3.2. Lung Cancer Cells May Facilitate EBV Entry through Increased CD21 Expression
3.3.3. EBV Interactions with other Environmental Contaminants in Lung Cancer
4. Conclusions and Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Sung, H.; Ferlay, J.; Siegel, R.L.; Laversanne, M.; Soerjomataram, I.; Jemal, A.; Bray, F. Global Cancer Statistics 2020: Globocan Estimates of Incidence and Mortality Worldwide for 36 Cancers in 185 Countries. CA Cancer J. Clin. 2021, 71, 209–249. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Fillmore, C.M.; Hammerman, P.S.; Kim, C.F.; Wong, K.-K. Non-small-cell lung cancers: A heterogeneous set of diseases. Nat. Cancer 2014, 14, 535–546. [Google Scholar] [CrossRef]
- Semenova, E.A.; Nagel, R.; Berns, A. Origins, genetic landscape, and emerging therapies of small cell lung cancer. Genes Dev. 2015, 29, 1447–1462. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Oser, M.G.; Niederst, M.J.; Sequist, L.V.; Engelman, J.A. Transformation from non-small-cell lung cancer to small-cell lung cancer: Molecular drivers and cells of origin. Lancet Oncol. 2015, 16, e165–e172. [Google Scholar] [CrossRef] [Green Version]
- Reis-Filho, J.S.; Simpson, P.T.; Martins, A.; Preto, A.; Gärtner, F.; Schmitt, F.C.; Archiv, V. Distribution of p63, cytokeratins 5/6 and cytokeratin 14 in 51 normal and 400 neoplastic human tissue samples using TARP-4 multi-tumor tissue microarray. Virchows Arch. 2003, 443, 122–132. [Google Scholar] [PubMed] [Green Version]
- Su, Y.-C.; Hsu, Y.-C.; Chai, C.-Y. Role of TTF-1, CK20, and CK7 Immunohistochemistry for Diagnosis of Primary and Secondary Lung Adenocarcinoma. Kaohsiung J. Med Sci. 2006, 22, 14–19. [Google Scholar] [CrossRef] [Green Version]
- Sholl, L.M. Large-cell carcinoma of the lung: A diagnostic category redefined by immunohistochemistry and genomics. Curr. Opin. Pulm. Med. 2014, 20, 324–331. [Google Scholar] [CrossRef]
- Hecht, S.S. Tobacco smoke carcinogens and lung cancer. J. Natl. Cancer Inst. 1999, 91, 1194–1210. [Google Scholar] [CrossRef] [Green Version]
- Rivlin, N.; Brosh, R.; Oren, M.; Rotter, V. Mutations in the p53 Tumor Suppressor Gene: Important Milestones at the Various Steps of Tumorigenesis. Genes Cancer 2011, 2, 466–474. [Google Scholar] [CrossRef] [Green Version]
- Larsen, J.E.; Minna, J.D. Molecular Biology of Lung Cancer: Clinical Implications. Clin. Chest Med. 2011, 32, 703–740. [Google Scholar] [CrossRef] [Green Version]
- Abbas, T.; Dutta, A. p21 in cancer: Intricate networks and multiple activities. Nat. Cancer 2009, 9, 400–414. [Google Scholar] [CrossRef]
- Hubaux, R.; Becker-Santos, D.D.; Enfield, K.S.; Lam, S.; Lam, W.L.; Martinez, V.D. Arsenic, asbestos and radon: Emerging players in lung tumorigenesis. Environ. Health 2012, 11, 89. [Google Scholar] [CrossRef]
- Czoli, C.D.; Hammond, D. TSNA Exposure: Levels of NNAL among Canadian Tobacco Users. Nicotine Tob. Res. 2015, 17, 825–830. [Google Scholar] [CrossRef] [Green Version]
- IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Some non-heterocyclic polycyclic aromatic hydrocarbons and some related exposures. IARC Monogr. Eval. Carcinog. Risks Hum. 2010, 92, 1–853. [Google Scholar]
- Manisalidis, I.; Stavropoulou, E.; Stavropoulos, A.; Bezirtzoglou, E. Environmental and Health Impacts of Air Pollution: A Review. Front. Public Health 2020, 8, 14. [Google Scholar] [CrossRef] [Green Version]
- Lakhani, A. Source Apportionment of Particle Bound Polycyclic Aromatic Hydrocarbons at an Industrial Location in Agra, India. Sci. World J. 2012, 2012, 781291. [Google Scholar] [CrossRef] [Green Version]
- Peng, N.; Li, Y.; Liu, Z.; Liu, T.; Gai, C. Emission, distribution and toxicity of polycyclic aromatic hydrocarbons (PAHs) during municipal solid waste (MSW) and coal co-combustion. Sci. Total Environ. 2016, 565, 1201–1207. [Google Scholar] [CrossRef]
- Syrjänen, K.J. HPV infections and lung cancer. J. Clin. Pathol. 2002, 55, 885–891. [Google Scholar] [CrossRef] [Green Version]
- Joh, J.; Jenson, A.B.; Moore, G.D.; Rezazedeh, A.; Slone, S.P.; Ghim, S.-J.; Kloecker, G.H. Human papillomavirus (HPV) and Merkel cell polyomavirus (MCPyV) in non small cell lung cancer. Exp. Mol. Pathol. 2010, 89, 222–226. [Google Scholar] [CrossRef]
- Palmarini, M.; Sharp, J.M.; de las Heras, M.; Fan, H. Jaagsiekte sheep retrovirus is necessary and sufficient to induce a contagious lung cancer in sheep. J. Virol. 1999, 73, 6964–6972. [Google Scholar] [CrossRef] [Green Version]
- Zheng, H.; Aziz, H.A.; Nakanishi, Y.; Masuda, S.; Saito, H.; Tsuneyama, K.; Takano, Y. Oncogenic role of JC virus in lung cancer. J. Pathol. 2007, 212, 306–315. [Google Scholar] [CrossRef] [PubMed]
- Becnel, D.; Abdelghani, R.; Nanbo, A.; Avilala, J.; Kahn, J.; Li, L.; Lin, Z. Pathogenic Role of Epstein—Barr Virus in Lung Cancers. Viruses 2021, 13, 877. [Google Scholar] [CrossRef] [PubMed]
- Hawkins, J.B.; Delgado-Eckert, E.; Thorley-Lawson, D.A.; Shapiro, M. The Cycle of EBV Infection Explains Persistence, the Sizes of the Infected Cell Populations and Which Come under CTL Regulation. PLoS Pathog. 2013, 9, e1003685. [Google Scholar] [CrossRef] [PubMed]
- Shannon-Lowe, C.; Adland, E.; Bell, A.I.; Delecluse, H.-J.; Rickinson, A.B.; Rowe, M. Features Distinguishing Epstein-Barr Virus Infections of Epithelial Cells and B Cells: Viral Genome Expression, Genome Maintenance, and Genome Amplification. J. Virol. 2009, 83, 7749–7760. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niedobitek, G.; Meru, N.; Delecluse, H.J. Epstein-Barr virus infection and human malignancies. Int. J. Exp. Pathol. 2001, 82, 149–170. [Google Scholar] [CrossRef]
- Aguayo, F.; Boccardo, E.; Corvalán, A.; Calaf, G.M.; Blanco, R. Interplay between Epstein-Barr virus infection and environmental xenobiotic exposure in cancer. Infect. Agents Cancer 2021, 16, 50. [Google Scholar] [CrossRef]
- Nanbo, A.; Noda, T.; Ohba, Y. Epstein–Barr Virus Acquires Its Final Envelope on Intracellular Compartments With Golgi Markers. Front. Microbiol. 2018, 9, 454. [Google Scholar] [CrossRef]
- Young, L.S.; Murray, P.G. Epstein–Barr virus and oncogenesis: From latent genes to tumours. Oncogene 2003, 22, 5108–5121. [Google Scholar] [CrossRef] [Green Version]
- Abbott, R.J.; Pachnio, A.; Pedroza-Pacheco, I.; Leese, A.M.; Begum, J.; Long, H.M.; Croom-Carter, D.; Stacey, A.; Moss, P.A.H.; Hislop, A.D.; et al. Asymptomatic Primary Infection with Epstein-Barr Virus: Observations on Young Adult Cases. J. Virol. 2017, 91, e00382-17. [Google Scholar] [CrossRef] [Green Version]
- Kang, M.S.; Kieff, E. Epstein-Barr virus latent genes. Exp. Mol. Med. 2015, 47, e131. [Google Scholar] [CrossRef] [Green Version]
- Thorley-Lawson, D.A. Epstein-Barr virus: Exploiting the immune system. Nat. Rev. Immunol. 2001, 1, 75–82. [Google Scholar] [CrossRef]
- Chesnokova, L.S.; Nishimura, S.L.; Hutt-Fletcher, L.M. Fusion of epithelial cells by Epstein-Barr virus proteins is triggered by binding of viral glycoproteins gHgL to integrins alphavbeta6 or alphavbeta8. Proc. Natl. Acad. Sci. USA 2009, 106, 20464–20469. [Google Scholar] [CrossRef] [Green Version]
- Shaw, P.; Kirschner, A.N.; Jardetzky, T.S.; Longnecker, R. Characteristics of Epstein–Barr virus envelope protein gp42. Virus Genes 2010, 40, 307–319. [Google Scholar] [CrossRef]
- Jiang, R.; Gu, X.; Nathan, C.-A.; Hutt-Fletcher, L. Laser-capture microdissection of oropharyngeal epithelium indicates restriction of Epstein-Barr virus receptor/CD21 mRNA to tonsil epithelial cells. J. Oral Pathol. Med. 2008, 37, 626–633. [Google Scholar] [CrossRef] [Green Version]
- Chen, J.; Sathiyamoorthy, K.; Zhang, X.; Schaller, S.; White, B.E.P.; Jardetzky, T.S.; Longnecker, R. Ephrin receptor A2 is a functional entry receptor for Epstein–Barr virus. Nat. Microbiol. 2018, 3, 172–180. [Google Scholar] [CrossRef]
- Hutt-Fletcher, L.M. Epstein-Barr Virus Entry. J. Virol. 2007, 81, 7825–7832. [Google Scholar] [CrossRef] [Green Version]
- Döhner, K.; Cornelius, A.; Serrero, M.C.; Sodeik, B. The journey of herpesvirus capsids and genomes to the host cell nucleus. Curr. Opin. Virol. 2021, 50, 147–158. [Google Scholar] [CrossRef]
- Lee, C.-P.; Chen, M.-R. Conquering the Nuclear Envelope Barriers by EBV Lytic Replication. Viruses 2021, 13, 702. [Google Scholar] [CrossRef]
- Aydin, I.; Schelhaas, M. Viral Genome Tethering to Host Cell Chromatin: Cause and Consequences. Traffic 2016, 17, 327–340. [Google Scholar] [CrossRef]
- Frappier, L. The Epstein-Barr Virus EBNA1 Protein. Scientifica 2012, 2012, 438204. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Liu, S.; Hu, J.; Luo, X.; Li, N.; Bode, A.M.; Cao, Y. Epstein-Barr virus lytic reactivation regulation and its pathogenic role in carcinogenesis. Int. J. Biol. Sci. 2016, 12, 1309–1318. [Google Scholar] [CrossRef] [Green Version]
- Feederle, R.; Shannon-Lowe, C.; Baldwin, G.; Delecluse, H.J. Defective Infectious Particles and Rare Packaged Genomes Produced by Cells Carrying Terminal-Repeat-Negative Epstein-Barr Virus. J. Virol. 2005, 79, 7641–7647. [Google Scholar] [CrossRef] [Green Version]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein—Barr virus infection and nasopharyngeal carcinoma. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20160270. [Google Scholar] [CrossRef]
- Tokunaga, M.; Land, C.E.; Uemura, Y.; Tokudome, T.; Tanaka, S.; Sato, E. Epstein-Barr virus in gastric carcinoma. Am. J. Pathol. 1993, 143, 1250–1254. [Google Scholar]
- Arbach, H.; Viglasky, V.; Lefeu, F.; Guinebretière, J.-M.; Ramirez, V.; Bride, N.; Boualaga, N.; Bauchet, T.; Peyrat, J.-P.; Mathieu, M.-C.; et al. Epstein-Barr Virus (EBV) Genome and Expression in Breast Cancer Tissue: Effect of EBV Infection of Breast Cancer Cells on Resistance to Paclitaxel (Taxol). J. Virol. 2006, 80, 845–853. [Google Scholar] [CrossRef] [Green Version]
- Bedri, S.; Sultan, A.A.; Alkhalaf, M.; Al Moustafa, A.-E.; Vranic, S. Epstein-Barr virus (EBV) status in colorectal cancer: A mini review. Hum. Vaccines Immunother. 2018, 15, 603–610. [Google Scholar] [CrossRef]
- Whitaker, N.J.; Glenn, W.K.; Sahrudin, A.; Orde, M.M.; Delprado, W.; Lawson, J.S. Human papillomavirus and Epstein Barr virus in prostate cancer: Koilocytes indicate potential oncogenic influences of human papillomavirus in prostate cancer. Prostate 2013, 73, 236–241. [Google Scholar] [CrossRef]
- Young, L.S.; Dawson, C.W. Epstein-Barr virus and nasopharyngeal carcinoma. Chin. J. Cancer 2014, 33, 581–590. [Google Scholar] [CrossRef]
- Pfeffer, S.; Zavolan, M.; Grässer, F.A.; Chien, M.; Russo, J.J.; Ju, J.; John, B.; Enright, A.J.; Marks, D.; Sander, C.; et al. Identification of Virus-Encoded MicroRNAs. Science 2004, 304, 734–736. [Google Scholar] [CrossRef]
- Iizasa, H.; Kim, H.; Kartika, A.V.; Kanehiro, Y.; Yoshiyama, H. Corrigendum: Role of Viral and Host microRNAs in Immune Regulation of Epstein-Barr Virus-Associated Diseases. Front. Immunol. 2020, 11, 498. [Google Scholar] [CrossRef] [Green Version]
- Richardo, T.; Prattapong, P.; Ngernsombat, C.; Wisetyaningsih, N.; Iizasa, H.; Yoshiyama, H.; Janvilisri, T. Epstein-Barr Virus Mediated Signaling in Nasopharyngeal Carcinoma Carcinogenesis. Cancers 2020, 12, 2441. [Google Scholar] [CrossRef] [PubMed]
- Murphy, G.; Pfeiffer, R.; Camargo, M.C.; Rabkin, C.S. Meta-analysis Shows That Prevalence of Epstein–Barr Virus-Positive Gastric Cancer Differs Based on Sex and Anatomic Location. Gastroenterology 2009, 137, 824–833. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Woellmer, A.; Hammerschmidt, W. Epstein–Barr virus and host cell methylation: Regulation of latency, replication and virus reactivation. Curr. Opin. Virol. 2013, 3, 260–265. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon-Lowe, C.; Rickinson, A. The Global Landscape of EBV-Associated Tumors. Front. Oncol. 2019, 9, 713. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peng, R.-J.; Han, B.-W.; Cai, Q.-Q.; Zuo, X.-Y.; Xia, T.; Chen, J.-R.; Feng, L.-N.; Lim, J.Q.; Chen, S.-W.; Zeng, M.; et al. Genomic and transcriptomic landscapes of Epstein-Barr virus in extranodal natural killer T-cell lymphoma. Leukemia 2019, 33, 1451–1462. [Google Scholar] [CrossRef]
- Alao, J.P. The regulation of cyclin D1 degradation: Roles in cancer development and the potential for therapeutic invention. Mol. Cancer 2007, 6, 24. [Google Scholar] [CrossRef] [Green Version]
- Tsang, C.M.; Yip, Y.L.; Lo, K.W.; Deng, W.; To, K.F.; Hau, P.M.; Lau, V.M.Y.; Takada, K.; Lui, V.W.Y.; Lung, M.L.; et al. Cyclin D1 overexpression supports stable EBV infection in nasopharyngeal epithelial cells. Proc. Natl. Acad. Sci. USA 2012, 109, E3473–E3482. [Google Scholar] [CrossRef] [Green Version]
- Kumar, P.; Murakami, M.; Kaul, R.; Saha, A.; Cai, Q.; Robertson, E.S. Deregulation of the cell cycle machinery by Epstein—Barr virus nuclear antigen 3C. Future Virol. 2009, 4, 79–91. [Google Scholar] [CrossRef] [Green Version]
- Frappier, L. Epstein-Barr virus: Current questions and challenges. Tumour Virus Res. 2021, 12, 200218. [Google Scholar] [CrossRef]
- Hirai, K.; Shirakata, M. Replication Licensing of the EBV oriP Minichromosome. Curr. Top. Microbiol. Immunol. 2001, 258, 13–33. [Google Scholar] [CrossRef]
- Tsao, S.W.; Tsang, C.M.; Lo, K.W. Epstein-Barr virus infection and persistence in nasopharyngeal epithelial cells. Chin. J. Cancer 2014, 33, 549–555. [Google Scholar]
- Klein, G. Viral latency and transformation: The strategy of Epstein-Barr virus. Cell 1989, 58, 5–8. [Google Scholar] [CrossRef]
- Zhao, Z.; Liu, W.; Liu, J.; Wang, J.; Luo, B. The effect of EBV on WIF1, NLK, and APC gene methylation and expression in gastric carcinoma and nasopharyngeal cancer. J. Med. Virol. 2017, 89, 1844–1851. [Google Scholar] [CrossRef]
- Hsu, M.; Wu, S.-Y.; Chang, S.-S.; Su, I.-J.; Tsai, C.-H.; Lai, S.-J.; Shiau, A.-L.; Takada, K.; Chang, Y. Epstein-Barr Virus Lytic Transactivator Zta Enhances Chemotactic Activity through Induction of Interleukin-8 in Nasopharyngeal Carcinoma Cells. J. Virol. 2008, 82, 3679–3688. [Google Scholar] [CrossRef] [Green Version]
- Germini, D.; Sall, F.B.; Shmakova, A.; Wiels, J.; Dokudovskaya, S.; Drouet, E.; Vassetzky, Y. Oncogenic Properties of the EBV ZEBRA Protein. Cancers 2020, 12, 1479. [Google Scholar] [CrossRef]
- Blanco, R.; Aguayo, F. Role of BamHI—A Rightward Frame 1 in Epstein-Barr Virus-Associated Epithelial Malignancies. Biology 2020, 9, 461. [Google Scholar] [CrossRef]
- Kenney, S.C.; Mertz, J.E. Regulation of the latent-lytic switch in Epstein–Barr virus. Semin. Cancer Biol. 2014, 26, 60–68. [Google Scholar] [CrossRef] [Green Version]
- Birdwell, C.E.; Prasai, K.; Dykes, S.; Jia, Y.; Munroe, T.G.; Bienkowska-Haba, M.; Scott, R.S. Epstein—Barr virus stably confers an invasive phenotype to epithelial cells through reprogramming of the WNT pathway. Oncotarget 2018, 9, 10417–10435. [Google Scholar] [CrossRef] [Green Version]
- Matsusaka, K.; Funata, S.; Fukuyo, M.; Seto, Y.; Aburatani, H.; Fukayama, M.; Kaneda, A. Epstein—Barr virus infection induces genome-wide de novo DNA methylation in non-neoplastic gastric epithelial cells. J. Pathol. 2017, 242, 391–399. [Google Scholar] [CrossRef] [Green Version]
- Uozaki, H.; Fukayama, M. Epstein-Barr Virus and Gastric Carcinoma—Viral Carcinogenesis through Epigenetic Mechanisms. Int. J. Clin. Exp. Pathol. 2008, 1, 198–216. [Google Scholar]
- Woellmer, A.; Arteaga-Salas, J.M.; Hammerschmidt, W. BZLF1 governs CpG-methylated chromatin of Epstein—Barr Virus reversing epigenetic repression. PLoS Pathog 2012, 8, e1002902. [Google Scholar] [CrossRef] [PubMed]
- Massion, P.P.; Carbone, D.P. The molecular basis of lung cancer: Molecular abnormalities and therapeutic implications. Respir. Res. 2003, 4, 12. [Google Scholar] [CrossRef] [Green Version]
- Cooper, W.A.; Lam, D.C.L.; O’Toole, S.A.; Minna, J.D. Molecular biology of lung cancer. J. Thorac. Dis. 2013, 5, S479–S490. [Google Scholar] [CrossRef] [PubMed]
- Sansregret, L.; Swanton, C. The Role of Aneuploidy in Cancer Evolution. Cold Spring Harb. Perspect. Med. 2017, 7, a028373. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zojer, N.; Dekan, G.; Ackermann, J.; Fiegl, M.; Kaufmann, H.; Drach, J.; Huber, H. Aneuploidy of chromosome 7 can be detected in invasive lung cancer and associated premalignant lesions of the lung by fluorescence in situ hybridisation. Lung Cancer 2000, 28, 225–235. [Google Scholar] [CrossRef]
- Jonsson, S.; Varella-Garcia, M.; Miller, Y.E.; Wolf, H.J.; Byers, T.; Braudrick, S.; Kiatsimkul, P.; Lewis, M.; Kennedy, T.C.; Keith, R.L.; et al. Chromosomal Aneusomy in Bronchial High-Grade Lesions Is Associated with Invasive Lung Cancer. Am. J. Respir. Crit. Care Med. 2008, 177, 342–347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Campbell, J.M.; Lockwood, W.W.; Buys, T.P.H.; Chari, R.; Coe, B.P.; Lam, S.; Lam, W.L. Integrative genomic and gene expression analysis of chromosome 7 identified novel oncogene loci in non-small cell lung cancer. Genome 2008, 51, 1032–1039. [Google Scholar] [CrossRef] [PubMed]
- Gawrychowski, J.; Lackowska, B.; Gabriel, A. Prognosis of the surgical treatment of patients with non-small cell lung cancer (NSCLC)-relation to DNA ploidy. Eur. J. Cardio-Thorac. Surg. 2003, 23, 870–877. [Google Scholar] [CrossRef] [Green Version]
- Oliani, C.; Barana, D.; Cazzadori, A.; Zanolin, E.; Santo, A.; Pasini, F.; Padovani, M.; Mazzini, G.; Cetto, G.L. Cytofluorimetric evaluation of DNA ploidy in lung cancer: A bronchoscopic study. Int. J. Biol. Markers. 2005, 20, 87–92. [Google Scholar] [CrossRef]
- Hofmann, H.S.; Knolle, J.; Bahn, H.; Klapperstück, T.; Lautenschläger, C.; Neef, H. Flow cytometric analysis of DNA content and proliferation and immunohistochemical staining of Ki-67 in non-small cell lung cancer. J. Cardiovasc. Surg. 2001, 42, 555–560. [Google Scholar]
- Choma, D.; Daurès, J.-P.; Quantin, X.; Pujol, J.L. Aneuploidy and prognosis of non-small-cell lung cancer: A meta-analysis of published data. Br. J. Cancer 2001, 85, 14–22. [Google Scholar] [CrossRef] [Green Version]
- Mao, L.; Lee, J.S.; Kurie, J.M.; Fan, Y.H.; Lippman, S.M.; Broxson, A.; Khuri, F.R.; Hong, W.K.; Yu, R.; Ro, J.Y.; et al. Clonal Genetic Alterations in the Lungs of Current and Former Smokers. J. Natl. Cancer Inst. 1997, 89, 857–862. [Google Scholar] [CrossRef] [Green Version]
- Sanz-Ortega, J.; Saez, M.C.; Sierra, E.; Torres, A.; Balibrea, J.L.; Hernando, F.; Sanz-Esponera, J.; Merino, M.J. 3p21, 5q21, and 9p21 allelic deletions are frequently found in normal bronchial cells adjacent to non-small-cell lung cancer, while they are unusual in patients with no evidence of malignancy. J. Pathol. 2001, 195, 429–434. [Google Scholar] [CrossRef]
- Wistuba, I.I.; Virmani, A.K.; Gazdar, A.F.; Lam, S.; Leriche, J.; Behrens, C.; Fong, K.; Samet, J.M.; Srivastava, S.; Minna, J.D. Molecular Damage in the Bronchial Epithelium of Current and Former Smokers. J. Natl. Cancer Inst. 1997, 89, 1366–1373. [Google Scholar] [CrossRef]
- Soda, M.; Choi, Y.L.; Enomoto, M.; Takada, S.; Yamashita, Y.; Ishikawa, S.; Fujiwara, S.-I.; Watanabe, H.; Kurashina, K.; Hatanaka, H.; et al. Identification of the transforming EML4–ALK fusion gene in non-small-cell lung cancer. Nature 2007, 448, 561–566. [Google Scholar] [CrossRef]
- Wang, Y.; Wang, S.; Xu, S.; Qu, J.; Liu, B. Clinicopathologic Features of Patients with Non-Small Cell Lung Cancer Harboring the EML4-ALK Fusion Gene: A Meta-Analysis. PLoS ONE 2014, 9, e110617. [Google Scholar] [CrossRef]
- Miyanaga, A.; Matsumoto, M.; Beck, J.A.; Horikawa, L.; Oike, T.; Okayama, H.; Tanaka, H.; Burkett, S.S.; Robles, A.I.; Khan, M.; et al. EML4-ALK induces cellular senescence in mortal normal human cells and promotes anchorage-independent growth in hTERT-transduced normal human cells. BMC Cancer 2021, 21, 310. [Google Scholar] [CrossRef]
- Halvorsen, A.R.; Silwal-Pandit, L.; Meza-Zepeda, L.A.; Vodák, D.; Vu, P.; Sagerup, C.; Hovig, E.; Myklebost, O.; Børresen-Dale, A.-L.; Brustugun, O.T.; et al. TP53 Mutation Spectrum in Smokers and Never Smoking Lung Cancer Patients. Front. Genet. 2016, 7, 85. [Google Scholar] [CrossRef] [Green Version]
- Demirhan, O.; Taştemir, D.; Hastürk, S.; Kuleci, S.; Hanta, I. Alterations in p16 and p53 genes and chromosomal findings in patients with lung cancer: Fluorescence in situ hybridization and cytogenetic studies. Cancer Epidemiol. 2010, 34, 472–477. [Google Scholar] [CrossRef]
- Deben, C.; Deschoolmeester, V.; Lardon, F.; Rolfo, C.; Pauwels, P. TP53 and MDM2 genetic alterations in non-small cell lung cancer: Evaluating their prognostic and predictive value. Crit. Rev. Oncol. 2016, 99, 63–73. [Google Scholar] [CrossRef]
- Tanaka, R.; Wang, D.; Morishita, Y.; Inadome, Y.; Minami, Y.; Iijima, T.; Fukai, S.; Goya, T.; Noguchi, M. Loss of function of p16 gene and prognosis of pulmonary adenocarcinoma. Cancer 2005, 103, 608–615. [Google Scholar] [CrossRef] [PubMed]
- Tominaga, M.; Sueoka, N.; Irie, K.; Iwanaga, K.; Tokunaga, O.; Hayashi, S.-I.; Nakachi, K.; Sueoka, E. Detection and discrimination of preneoplastic and early stages of lung adenocarcinoma using hnRNP B1 combined with the cell cycle-related markers p16, cyclin D1, and Ki-67. Lung Cancer 2003, 40, 45–53. [Google Scholar] [CrossRef]
- Licchesi, J.D.; Westra, W.H.; Hooker, C.M.; Herman, J.G. Promoter Hypermethylation of Hallmark Cancer Genes in Atypical Adenomatous Hyperplasia of the Lung. Clin. Cancer Res. 2008, 14, 2570–2578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Higashiyama, M.; Doi, O.; Kodama, K.; Yokouchi, H.; Tateishi, R. Retinoblastoma Protein Expression in Lung Cancer: An Immunohistochemical Analysis. Oncology 1994, 51, 544–551. [Google Scholar] [CrossRef]
- Zagorski, W.A.; Knudsen, E.S.; Reed, M.F. Retinoblastoma Deficiency Increases Chemosensitivity in Lung Cancer. Cancer Res. 2007, 67, 8264–8273. [Google Scholar] [CrossRef] [Green Version]
- Meuwissen, R.; Linn, S.C.; Linnoila, R.; Zevenhoven, J.; Mooi, W.J.; Berns, A. Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 2003, 4, 181–189. [Google Scholar] [CrossRef] [Green Version]
- Niederst, M.J.; Sequist, L.V.; Poirier, J.T.; Mermel, C.H.; Lockerman, E.L.; Garcia, A.R.; Katayama, R.; Costa, C.; Ross, K.N.; Moran, T.; et al. RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun. 2015, 6, 6377. [Google Scholar] [CrossRef]
- Rengifo, C.E.; Blanco, R.; Blanco, D.; Cedeño, M.; Frómeta, M.; Calzado, E.R. Immunohistochemical Characterization of Three Monoclonal Antibodies Raised against the Epidermal Growth Factor and Its Receptor in Non-Small-Cell Lung Cancer: Their Potential Use in the Selection of Patients for Immunotherapy. J. Biomarkers 2013, 2013, 627845. [Google Scholar] [CrossRef]
- Prabhakar, C.N. Epidermal growth factor receptor in non-small cell lung cancer. Transl. Lung Cancer Res. 2015, 4, 110–118. [Google Scholar]
- Cuneo, K.C.; Nyati, M.K.; Ray, D.; Lawrence, T.S. EGFR targeted therapies and radiation: Optimizing efficacy by appropriate drug scheduling and patient selection. Pharmacol. Ther. 2015, 154, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Zhang, S.M.; Zhu, Q.G.; Ding, X.X.; Lin, S.; Zhao, J.; Guan, L.; Li, T.; He, B.; Zhang, H.Q. Prognostic value of EGFR and KRAS in resected non-small cell lung cancer: A systematic review and meta-analysis. Cancer Manag. Res. 2018, 10, 3393–3404. [Google Scholar] [CrossRef] [Green Version]
- Liu, L.; Shao, X.; Gao, W.; Bai, J.; Wang, R.; Huang, P.; Yin, Y.; Liu, P.; Shu, Y. The Role of Human Epidermal Growth Factor Receptor 2 as a Prognostic Factor in Lung Cancer: A Meta-Analysis of Published Data. J. Thorac. Oncol. 2010, 5, 1922–1932. [Google Scholar] [CrossRef] [Green Version]
- Pillai, R.N.; Behera, M.; Berry, L.D.; Rossi, M.R.; Kris, M.G.; Johnson, B.E.; Bunn, P.A.; Ramalingam, S.S.; Khuri, F.R. HER2 mutations in lung adenocarcinomas: A report from the Lung Cancer Mutation Consortium. Cancer 2017, 123, 4099–4105. [Google Scholar] [CrossRef] [Green Version]
- Tomizawa, K.; Suda, K.; Onozato, R.; Kosaka, T.; Endoh, H.; Sekido, Y.; Shigematsu, H.; Kuwano, H.; Yatabe, Y.; Mitsudomi, T. Prognostic and predictive implications of HER2/ERBB2/neu gene mutations in lung cancers. Lung Cancer 2011, 74, 139–144. [Google Scholar] [CrossRef]
- Li, C.; Sun, Y.; Fang, R.; Han, X.; Luo, X.; Wang, R.; Pan, Y.; Hu, H.; Zhang, Y.; Pao, W.; et al. Lung adenocarcinomas with HER2-activating mutations are associated with distinct clinical features and HER2/EGFR copy number gains. J. Thorac. Oncol. 2012, 7, 85–89. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.E.; Narasanna, A.; Perez-Torres, M.; Xiang, B.; Wu, F.Y.; Yang, S.; Carpenter, G.; Gazdar, A.F.; Muthuswamy, S.; Arteaga, C.L. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell 2006, 10, 25–38. [Google Scholar] [CrossRef] [Green Version]
- Scoccianti, C.; Vesin, A.; Martel, G.; Olivier, M.; Brambilla, E.; Timsit, J.F.; Tavecchio, L.; Brambilla, C.; Field, J.K.; Hainaut, P.; et al. Prognostic value of TP53, KRAS and EGFR mutations in nonsmall cell lung cancer: The EUELC cohort. Eur. Respir. J. 2012, 40, 177–184. [Google Scholar] [CrossRef] [Green Version]
- Villaruz, L.C.; Socinski, M.A.; Cunningham, D.E.; Chiosea, S.I.; Burns, T.F.; Siegfried, J.M.; Dacic, S. The prognostic and predictive value of KRAS oncogene substitutions in lung adenocarcinoma. Cancer 2013, 119, 2268–2274. [Google Scholar] [CrossRef] [Green Version]
- Martorell, P.M.; Huerta, M.; Quilis, A.C.; Abellán, R.; Seda, E.; Blesa, S.; Chaves, F.J.; Beltrán, D.D.; Keränen, S.R.; Franco, J.; et al. Coexistence of EGFR, KRAS, BRAF, and PIK3CA Mutations and ALK Rearrangement in a Comprehensive Cohort of 326 Consecutive Spanish Nonsquamous NSCLC Patients. Clin. Lung Cancer 2017, 18, e395–e402. [Google Scholar] [CrossRef]
- Okudela, K.; Hayashi, H.; Ito, T.; Yazawa, T.; Suzuki, T.; Nakane, Y.; Sato, H.; Ishi, H.; KeQin, X.; Masuda, A.; et al. K-ras Gene Mutation Enhances Motility of Immortalized Airway Cells and Lung Adenocarcinoma Cells via Akt Activation: Possible Contribution to Non-Invasive Expansion of Lung Adenocarcinoma. Am. J. Pathol. 2004, 164, 91–100. [Google Scholar] [CrossRef]
- Jackson, E.L.; Willis, N.; Mercer, K.; Bronson, R.T.; Crowley, D.; Montoya, R.; Jacks, T.; Tuveson, D.A. Analysis of lung tumor initiation and progression using conditional expression of oncogenic K-ras. Genes Dev. 2001, 15, 3243–3248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoshizawa, A.; Sumiyoshi, S.; Sonobe, M.; Kobayashi, M.; Uehara, T.; Fujimoto, M.; Tsuruyama, T.; Date, H.; Haga, H. HER2 status in lung adenocarcinoma: A comparison of immunohistochemistry, fluorescence in situ hybridization (FISH), dual-ISH, and gene mutations. Lung Cancer 2014, 85, 373–378. [Google Scholar] [CrossRef] [PubMed]
- Patel, A.S.; Yoo, S.; Kong, R.; Sato, T.; Sinha, A.; Karam, S.; Bao, L.; Fridrikh, M.; Emoto, K.; Nudelman, G.; et al. Prototypical oncogene family Myc defines unappreciated distinct lineage states of small cell lung cancer. Sci. Adv. 2021, 7, eabc2578. [Google Scholar] [CrossRef] [PubMed]
- Rapp, U.R.; Korn, C.; Ceteci, F.; Karreman, C.; Luetkenhaus, K.; Serafin, V.; Zanucco, E.; Castro, I.; Potapenko, T. Myc Is a Metastasis Gene for Non-Small-Cell Lung Cancer. PLoS ONE 2009, 4, e6029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, E.Y.; Kim, A.; Kim, S.K.; Chang, Y.S. MYC expression correlates with PD-L1 expression in non-small cell lung cancer. Lung Cancer 2017, 110, 63–67. [Google Scholar] [CrossRef]
- Zhou, C.; Che, G.; Zheng, X.; Qiu, J.; Xie, Z.; Cong, Y.; Pei, X.; Zhang, H.; Sun, H.; Ma, H. Expression and clinical significance of PD-L1 and c-Myc in non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2019, 145, 2663–2674. [Google Scholar] [CrossRef]
- Dragoj, M.; Bankovic, J.; Podolski-Renic, A.; Stojkovic Buric, S.; Pesic, M.; Tanic, N.; Stankovic, T. Association of Overexpressed MYC Gene with Altered PHACTR3 and E2F4 Genes Contributes to Non-small Cell Lung Carcinoma Pathogenesis. J. Med. Biochem. 2019, 38, 188–195. [Google Scholar] [CrossRef]
- Nau, M.M.; Brooks, B.J.; Battey, J.F.; Sausville, E.A.; Gazdar, A.F.; Kirsch, I.R.; McBride, O.W.; Bertness, V.L.; Hollis, G.F.; Minna, J.D. L-myc, a new myc-related gene amplified and expressed in human small cell lung cancer. Nat. Cell Biol. 1985, 318, 69–73. [Google Scholar] [CrossRef]
- Qin, J.; Xie, F.; Li, C.; Han, N.; Lu, H. MYCL1 Amplification and Expression of L-Myc and c-Myc in Surgically Resected Small-Cell Lung Carcinoma. Pathol. Oncol. Res. 2021, 27, 1609775. [Google Scholar] [CrossRef]
- Fiorentino, F.P.; Tokgün, E.; Solé-Sánchez, S.; Giampaolo, S.; Tokgün, O.; Jauset, T.; Kohno, T.; Perucho, M.; Soucek, L.; Yokota, J. Growth suppression by MYC inhibition in small cell lung cancer cells with TP53 and RB1 inactivation. Oncotarget 2016, 7, 31014–31028. [Google Scholar] [CrossRef] [Green Version]
- Soucek, L.; Whitfield, J.R.; Sodir, N.M.; Massó-Vallés, D.; Serrano, E.; Karnezis, A.N.; Swigart, L.B.; Evan, G.I. Inhibition of Myc family proteins eradicates KRas-driven lung cancer in mice. Genes Dev. 2013, 27, 504–513. [Google Scholar] [CrossRef] [Green Version]
- Zhu, C.-Q.; Cutz, J.-C.; Liu, N.; Lau, D.; Shepherd, F.A.; Squire, J.A.; Tsao, M.-S. Amplification of telomerase (hTERT) gene is a poor prognostic marker in non-small-cell lung cancer. Br. J. Cancer 2006, 94, 1452–1459. [Google Scholar] [CrossRef]
- Zalewska-Ziob, M.; Dobija-Kubica, K.; Biernacki, K.; Adamek, B.; Kasperczyk, J.; Bruliński, K.; Ostrowska, Z. Clinical and prognostic value of hTERT mRNA expression in patients with non-small-cell lung cancer. Acta Biochim. Pol. 2017, 64, 641–646. [Google Scholar] [CrossRef]
- Kawai, T.; Hiroi, S.; Nakanishi, K.; Meeker, A.K. Telomere length and telomerase expression in atypical adenomatous hyperplasia and small bronchioloalveolar carcinoma of the lung. Am. J. Clin. Pathol. 2007, 127, 254–262. [Google Scholar] [CrossRef]
- Hara, H.; Yamashita, K.; Shinada, J.; Yoshimura, H.; Kameya, T. Clinicopathologic significance of telomerase activity and hTERT mRNA expression in non-small cell lung cancer. Lung Cancer 2001, 34, 219–226. [Google Scholar] [CrossRef]
- Wang, L.; Soria, J.-C.; Kemp, B.L.; Liu, D.D.; Mao, L.; Khuri, F.R. hTERT expression is a prognostic factor of survival in patients with stage I non-small cell lung cancer. Clin. Cancer Res. 2002, 8, 2883–2889. [Google Scholar]
- Wang, K.; Wang, R.L.; Liu, J.J.; Zhou, J.; Li, X.; Hu, W.W.; Jiang, W.J.; Hao, N.B. The prognostic significance of hTERT overexpression in cancers: A systematic review and meta-analysis. Medicine 2018, 97, e11794. [Google Scholar] [CrossRef]
- Zhai, G.; Li, J.; Zheng, J.; An, P.; Chen, X.; Wang, X.; Li, C. Expression of Concern: hTERT promoter methylation promotes small cell lung cancer progression and radiotherapy resistance. J. Radiat. Res. 2021, 62, 556. [Google Scholar]
- Ambinder, R.F.; Mann, R.B. Epstein-Barr-encoded RNA in situ hybridization: Diagnostic applications. Hum. Pathol. 1994, 25, 602–605. [Google Scholar] [CrossRef]
- Gulley, M.L. Molecular Diagnosis of Epstein-Barr Virus-Related Diseases. J. Mol. Diagn. 2001, 3, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Grässer, F.A.; Murray, P.G.; Kremmer, E.; Klein, K.; Remberger, K.; Feiden, W.; Reynolds, G.; Niedobitek, G.; Young, L.S.; Mueller-Lantzsch, N. Monoclonal antibodies directed against the Epstein-Barr virus-encoded nuclear antigen 1 (EBNA1): Immunohistologic detection of EBNA1 in the malignant cells of Hodgkin’s disease. Blood 1994, 84, 3792–3798. [Google Scholar] [CrossRef] [PubMed]
- Hayes, D.P.; Brink, A.A.; Vervoort, M.B.; Middeldorp, J.M.; Meijer, C.J.; Brule, A.J.V.D. Expression of Epstein-Barr virus (EBV) transcripts encoding homologues to important human proteins in diverse EBV associated diseases. Mol. Pathol. 1999, 52, 97–103. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, T.; Haba, R.; Tanizawa, J.; Katsuki, N.; Kadota, K.; Miyai, Y.; Bando, K.; Shibuya, S.; Nakano, M.; Kushida, Y. Cytopathologic features and differential diagnostic considerations of primary lymphoepithelioma-like carcinoma of the lung. Diagn. Cytopathol. 2012, 40, 820–825. [Google Scholar] [CrossRef] [PubMed]
- Chau, S.-L.; Tong, J.H.-M.; Chow, C.; Kwan, J.S.-H.; Lung, R.W.-M.; Chung, L.-Y.; Tin, E.K.-Y.; Wong, S.S.-Y.; Cheung, A.H.-K.; Lau, R.W.-H.; et al. Distinct Molecular Landscape of Epstein–Barr Virus Associated Pulmonary Lymphoepithelioma-Like Carcinoma Revealed by Genomic Sequencing. Cancers 2020, 12, 2065. [Google Scholar] [CrossRef]
- Tang, L.; Chen, N.; He, W.; Zhou, J.; Zhang, J.; Lin, Z.; Wang, Z.; Hao, J.; Lin, F. The clinicopathological features and prognosis of primary pulmonary lymphoepithelioma-like carcinoma: A systematic review and meta-analysis. PLoS ONE 2020, 15, e0240729. [Google Scholar] [CrossRef]
- Han, A.-J.; Xiong, M.; Zong, Y.-S. Association of Epstein-Barr Virus With Lymphoepithelioma-Like Carcinoma of the Lung in Southern China. Am. J. Clin. Pathol. 2000, 114, 220–226. [Google Scholar] [CrossRef]
- Chen, F.F.; Yan, J.J.; Lai, W.W.; Jin, Y.T.; Su, I.J. Epstein-Barr virus-associated nonsmall cell lung carcinoma: Undifferentiated “lymphoepithelioma-like” carcinoma as a distinct entity with better prognosis. Cancer 1998, 82, 2334–2342. [Google Scholar] [CrossRef]
- Hong, S.; Liu, D.; Luo, S.; Fang, W.; Zhan, J.; Fu, S.; Zhang, Y.; Wu, X.; Zhou, H.; Chen, X.; et al. The genomic landscape of Epstein-Barr virus-associated pulmonary lymphoepithelioma-like carcinoma. Nat. Commun. 2019, 10, 3108. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yeh, Y.C.; Ho, H.L.; Lin, C.I.; Chou, T.Y.; Wang, Y.C. Whole-exome Sequencing of Epstein-Barr Virus-associated Pulmonary Carcinoma With Low Lymphocytic Infiltration Shows Molecular Features Similar to Those of Classic Pulmonary Lymphoepithelioma-like Carcinoma: Evidence to Support Grouping Together as One Disease Entity. Am. J. Surg. Pathol. 2021, 45, 1476–1486. [Google Scholar] [CrossRef] [PubMed]
- Yeh, Y.C.; Kao, H.L.; Lee, K.L.; Wu, M.H.; Ho, H.L.; Chou, T.Y. Epstein-Barr Virus-Associated Pulmonary Carcinoma: Proposing an Alternative Term and Expanding the Histologic Spectrum of Lymphoepithelioma-like Carcinoma of the Lung. Am. J. Surg. Pathol. 2019, 43, 211–219. [Google Scholar] [CrossRef] [PubMed]
- Li, C.-M.; Han, G.-L.; Zhang, S.-J. Detection of Epstein-Barr virus in lung carcinoma tissue by in situ hybridization. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi Zhonghua Shiyan He Linchuang Bingduxue Zazhi Chin. J. Exp. Clin. Virol. 2007, 21, 288–290. [Google Scholar]
- Li, C.-M.; Zhang, S.-J.; Zhu, J.-H.; Han, G.-L. Study on EB virus infection, LMP1 and Bcl-2 expression in lung cancer patients. Zhonghua Shi Yan He Lin Chuang Bing Du Xue Za Zhi Zhonghua Shiyan He Linchuang Bingduxue Zazhi Chin. J. Exp. Clin. Virol. 2011, 25, 277–279. [Google Scholar]
- Zhang, L.; Liu, H.; Wang, Z. Detection of EB virus in pulmonary carcinoma by in situ hybridization. Zhonghua Bing Li Xue Za Zhi Chin. J. Pathol. 1996, 25, 1–3. [Google Scholar]
- Wong, M.P.; Chung, L.P.; Yuen, S.T.; Leung, S.Y.; Chan, S.Y.; Wang, E.; Fu, K.H. In situ detection of Epstein-Barr virus in non-small cell lung carcinomas. J. Pathol. 1995, 177, 233–240. [Google Scholar] [CrossRef]
- Kasai, K.; Sato, Y.; Kameya, T.; Inoue, H.; Yoshimura, H.; Kon, S.; Kikuchi, K. Incidence of latent infection of Epstein-Barr virus in lung cancers—An analysis of EBER1 expression in lung cancers byin situ hybridization. J. Pathol. 1994, 174, 257–265. [Google Scholar] [CrossRef]
- Gómez-Román, J.J.; Martínez, M.N.; Fernández, S.L.; Val-Bernal, J.F. Epstein—Barr virus-associated adenocarcinomas and squamous-cell lung carcinomas. Mod. Pathol. 2009, 22, 530–537. [Google Scholar] [CrossRef] [Green Version]
- Jafarian, A.H.; Omidi-Ashrafi, A.; Mohamadian-Roshan, N.; Karimi-Shahri, M.; Ghazvini, K.; Boroumand-Noughabi, S. Association of Epstein Barr virus deoxyribonucleic acid with lung carcinoma. Indian J. Pathol. Microbiol. 2013, 56, 359–364. [Google Scholar] [CrossRef]
- Gupta, P.; Haldar, D.; Naru, J.; Dey, P.; Aggarwal, A.N.; Minz, R.; Aggarwal, R. Prevalence of human papillomavirus, Epstein-Barr virus, and cytomegalovirus in fine needle aspirates from lung carcinoma: A case-control study with review of literature. Diagn. Cytopathol. 2016, 44, 987–993. [Google Scholar] [CrossRef]
- Carpagnano, G.E.; Lacedonia, D.; Natalicchio, M.I.; Cotugno, G.; Zoppo, L.; Martinelli, D.; Antonetti, R.; Foschino-Barbaro, M.P. Viral colonization in exhaled breath condensate of lung cancer patients: Possible role of EBV and CMV. Clin. Respir. J. 2016, 12, 418–424. [Google Scholar] [CrossRef]
- Xia, H.; Wu, J.; Chen, C.; Mao, Y.; Zhu, J.; Chang, Q.; Mi, K.; Zhao, J.; Zhang, M. The relationship between Epstein-Barr-virus infection and expression of p53, Bcl-2 and C-myc gene in lung cancer. Zhongguo Fei Ai Za Zhi 2000, 3, 265–268. [Google Scholar] [CrossRef]
- Wang, S.; Xiong, H.; Yan, S.; Wu, N.; Lu, Z. Identification and Characterization of Epstein-Barr Virus Genomes in Lung Carcinoma Biopsy Samples by Next-Generation Sequencing Technology. Sci. Rep. 2016, 6, 26156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kheir, F.; Zhao, M.; Strong, M.J.; Yu, Y.; Nanbo, A.; Flemington, E.K.; Morris, G.F.; Reiss, K.; Li, L.; Lin, Z. Detection of Epstein-Barr Virus Infection in Non-Small Cell Lung Cancer. Cancers 2019, 11, 759. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rosemarie, Q.; Sugden, B. Epstein–Barr Virus: How Its Lytic Phase Contributes to Oncogenesis. Microorganisms 2020, 8, 1824. [Google Scholar] [CrossRef] [PubMed]
- Conway, E.J.; Hudnall, S.D.; Lazarides, A.; Bahler, A.; Fraire, A.E.; Cagle, P.T. Absence of evidence for an etiologic role for Epstein-Barr virus in neoplasms of the lung and pleura. Mod. Pathol. 1996, 9, 491–495. [Google Scholar]
- Chu, P.G.; Cerilli, L.A.; Chen, Y.-Y.; Mills, S.E.; Weiss, L.M. Epstein–Barr virus plays no role in the tumorigenesis of small-cell carcinoma of the lung. Mod. Pathol. 2004, 17, 158–164. [Google Scholar] [CrossRef] [Green Version]
- Lim, W.-T.; Chuah, K.L.; Leong, S.S.; Tan, E.H.; Toh, C.K. Assessment of human papillomavirus and Epstein-Barr virus in lung adenocarcinoma. Oncol. Rep. 2009, 21, 971–975. [Google Scholar] [CrossRef] [Green Version]
- Koshiol, J.; Gulley, M.L.; Zhao, Y.; Rubagotti, M.; Marincola, F.M.; Rotunno, M.; Tang, W.; Bergen, A.W.; Bertazzi, P.A.; Roy, D.; et al. Epstein–Barr virus microRNAs and lung cancer. Br. J. Cancer 2011, 105, 320–326. [Google Scholar] [CrossRef] [Green Version]
- Kim, M.-H.; Cho, J.S.; Kim, Y.; Lee, C.H.; Lee, M.K.; Shin, D.H. Discriminating between Terminal- and Non-Terminal Respiratory Unit-Type Lung Adenocarcinoma Based on MicroRNA Profiles. PLoS ONE 2016, 11, e0160996. [Google Scholar] [CrossRef]
- Ma, Y.; Pan, X.; Xu, P.; Mi, Y.; Wang, W.; Wu, X.; He, Q.; Liu, X.; Tang, W.; An, H.-X. Plasma microRNA alterations between EGFR-activating mutational NSCLC patients with and without primary resistance to TKI. Oncotarget 2017, 8, 88529–88536. [Google Scholar] [CrossRef] [Green Version]
- Brouchet, L.; Valmary, S.; Dahan, M.; Didier, A.; Galateau-Salle, F.; Brousset, P.; Degano, B. Detection of oncogenic virus genomes and gene products in lung carcinoma. Br. J. Cancer 2005, 92, 743–746. [Google Scholar] [CrossRef] [Green Version]
- Skalska, L.; White, R.E.; Parker, G.A.; Turro, E.; Sinclair, A.J.; Paschos, K.; Allday, M.J. Induction of p16(INK4a) is the major barrier to proliferation when Epstein-Barr virus (EBV) transforms primary B cells into lymphoblastoid cell lines. PLoS Pathog 2013, 9, e1003187. [Google Scholar] [CrossRef]
- Ratschiller, D.; Heighway, J.; Gugger, M.; Kappeler, A.; Pirnia, F.; Schmid, R.; Borner, M.; Betticher, D. Cyclin D1 Overexpression in Bronchial Epithelia of Patients With Lung Cancer Is Associated With Smoking and Predicts Survival. J. Clin. Oncol. 2003, 21, 2085–2093. [Google Scholar] [CrossRef]
- Yanagawa, N.; Tamura, G.; Oizumi, H.; Takahashi, N.; Shimazaki, Y.; Motoyama, T. Frequent epigenetic silencing of the p16 gene in non-small cell lung cancers of tobacco smokers. Jpn. J. Cancer Res. 2002, 93, 1107–1113. [Google Scholar] [CrossRef]
- Ramos-García, P.; Gil-Montoya, J.A.; Scully, C.; Ayén, A.; González-Ruiz, L.; Navarro-Triviño, F.J.; González-Moles, M.A. An update on the implications of cyclin D1 in oral carcinogenesis. Oral Dis. 2016, 23, 897–912. [Google Scholar] [CrossRef]
- Arellano-Orden, E.; Calero-Acuña, C.; Moreno-Mata, N.; Gómez-Izquierdo, L.; Sánchez-López, V.; López-Ramírez, C.; Tobar, D.; López-Villalobos, J.L.; Gutiérrez, C.; Blanco-Orozco, A.; et al. Cigarette Smoke Decreases the Maturation of Lung Myeloid Dendritic Cells. PLoS ONE 2016, 11, e0152737. [Google Scholar] [CrossRef] [Green Version]
- Strzelak, A.; Ratajczak, A.; Adamiec, A.; Feleszko, W. Tobacco Smoke Induces and Alters Immune Responses in the Lung Triggering Inflammation, Allergy, Asthma and Other Lung Diseases: A Mechanistic Review. Int. J. Environ. Res. Public Health 2018, 15, 1033. [Google Scholar] [CrossRef] [Green Version]
- Qiu, F.; Liang, C.-L.; Liu, H.; Zeng, Y.-Q.; Hou, S.; Huang, S.; Lai, X.; Dai, Z. Impacts of cigarette smoking on immune responsiveness: Up and down or upside down? Oncotarget 2017, 8, 268–284. [Google Scholar] [CrossRef] [Green Version]
- Pan, F.; Yang, T.-L.; Chen, X.-D.; Chen, Y.; Gao, G.; Liu, Y.-Z.; Pei, Y.-F.; Sha, B.-Y.; Jiang, Y.; Xu, C.; et al. Impact of female cigarette smoking on circulating B cells in vivo: The suppressed ICOSLG, TCF3, and VCAM1 gene functional network may inhibit normal cell function. Immunogenetics 2010, 62, 237–251. [Google Scholar] [CrossRef] [Green Version]
- Fine, C.I.; Han, C.D.; Sun, X.; Liu, Y.; McCutcheon, J.A. Tobacco reduces membrane HLA class I that is restored by transfection with transporter associated with antigen processing 1 cDNA. J. Immunol. 2002, 169, 6012–6019. [Google Scholar] [CrossRef] [Green Version]
- Korkolopoulou, P.; Kaklamanis, L.; Pezzella, F.; Harris, A.; Gatter, K.C. Loss of antigen-presenting molecules (MHC class I and TAP-1) in lung cancer. Br. J. Cancer 1996, 73, 148–153. [Google Scholar] [CrossRef] [Green Version]
- Jiang, C.; Chen, Q.; Xie, M. Smoking increases the risk of infectious diseases: A narrative review. Tob. Induc. Dis. 2020, 18, 60. [Google Scholar] [CrossRef] [PubMed]
- Cerimele, F.; Battle, T.; Lynch, R.; Frank, D.A.; Murad, E.; Cohen, C.; Macaron, N.; Sixbey, J.; Smith, K.; Watnick, R.S.; et al. Reactive oxygen signaling and MAPK activation distinguish Epstein—Barr Virus (EBV)-positive versus EBV-negative Burkitt’s lymphoma. Proc. Natl. Acad. Sci. USA 2005, 102, 175–179. [Google Scholar] [CrossRef] [Green Version]
- Pezzuto, F.; Buonaguro, L.; Caponigro, F.; Ionna, F.; Starita, N.; Annunziata, C.; Buonaguro, F.M.; Tornesello, M.L. Update on Head and Neck Cancer: Current Knowledge on Epidemiology, Risk Factors, Molecular Features and Novel Therapies. Oncology 2015, 89, 125–136. [Google Scholar] [CrossRef]
- Takada, H.; Imadome, K.-I.; Shibayama, H.; Yoshimori, M.; Wang, L.; Saitoh, Y.; Uota, S.; Yamaoka, S.; Koyama, T.; Shimizu, N.; et al. EBV induces persistent NF-κB activation and contributes to survival of EBV-positive neoplastic T- or NK-cells. PLoS ONE 2017, 12, e0174136. [Google Scholar] [CrossRef] [Green Version]
- Chen, Y.-R.; Liu, M.-T.; Chang, Y.-T.; Wu, C.-C.; Hu, C.-Y.; Chen, J.-Y. Epstein-Barr Virus Latent Membrane Protein 1 Represses DNA Repair through the PI3K/Akt/FOXO3a Pathway in Human Epithelial Cells. J. Virol. 2008, 82, 8124–8137. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yi, M.; Cai, J.; Li, J.; Chen, S.; Zeng, Z.; Peng, Q.; Ban, Y.; Zhou, Y.; Li, Z.; Xiong, W.; et al. Rediscovery of NF-κB signaling in nasopharyngeal carcinoma: How genetic defects of NF-κB pathway interplay with EBV in driving oncogenesis? J. Cell Physiol. 2018, 233, 5537–5549. [Google Scholar] [CrossRef] [PubMed]
- Tsao, S.; Tsang, C.M.; To, K.; Lo, K. The role of Epstein–Barr virus in epithelial malignancies. J. Pathol. 2015, 235, 323–333. [Google Scholar] [CrossRef] [PubMed]
- Racero, I.T.; Gómez, N.C.; Leal, C.R.; Tinoco, E.L. Infections by Epstein-Barr virus and cytomegalovirus. Medicine 2014, 11, 2954–2964. [Google Scholar] [CrossRef]
- Feng, W.-H.; Cohen, J.I.; Fischer, S.; Li, L.; Sneller, M.; Goldbach-Mansky, R.; Raab-Traub, N.; Delecluse, H.-J.; Kenney, S.C. Reactivation of Latent Epstein-Barr Virus by Methotrexate: A Potential Contributor to Methotrexate-Associated Lymphomas. JNCI J. Natl. Cancer Inst. 2004, 96, 1691–1702. [Google Scholar] [CrossRef] [Green Version]
- Mrozek-Gorska, P.; Buschle, A.; Pich, D.; Schwarzmayr, T.; Fechtner, R.; Scialdone, A.; Hammerschmidt, W. Epstein–Barr virus reprograms human B lymphocytes immediately in the prelatent phase of infection. Proc. Natl. Acad. Sci. USA 2019, 116, 16046–16055. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J.; Jia, L.; Liu, T.; Yip, Y.L.; Tang, W.C.; Lin, W.; Deng, W.; Lo, K.W.; You, C.; Lung, M.L.; et al. mTORC2-mediated PDHE1α nuclear translocation links EBV-LMP1 reprogrammed glucose metabolism to cancer metastasis in nasopharyngeal carcinoma. Oncogene 2019, 38, 4669–4684. [Google Scholar] [CrossRef] [Green Version]
- Usui, G.; Matsusaka, K.; Mano, Y.; Urabe, M.; Funata, S.; Fukayama, M.; Ushiku, T.; Kaneda, A. DNA Methylation and Genetic Aberrations in Gastric Cancer. Digestion 2021, 102, 25–32. [Google Scholar] [CrossRef]
- Christensen, B.C.; Houseman, E.A.; Marsit, C.; Zheng, S.; Wrensch, M.R.; Wiemels, J.L.; Nelson, H.; Karagas, M.R.; Padbury, J.F.; Bueno, R.; et al. Aging and Environmental Exposures Alter Tissue-Specific DNA Methylation Dependent upon CpG Island Context. PLoS Genet. 2009, 5, e1000602. [Google Scholar] [CrossRef] [Green Version]
- Tagliamento, M.; Agostinetto, E.; Borea, R.; Brandão, M.; Poggio, F.; Addeo, A.; Lambertini, M. VISTA: A Promising Target for Cancer Immunotherapy? ImmunoTargets Ther. 2021, 10, 185–200. [Google Scholar] [CrossRef]
- Pertovaara, M.; Heliövaara, M.; Raitala, A.; Oja, S.S.; Knekt, P.; Hurme, M. The activity of the immunoregulatory enzyme indoleamine 2,3-dioxygenase is decreased in smokers. Clin. Exp. Immunol. 2006, 145, 469–473. [Google Scholar] [CrossRef]
- Liu, W.L.; Lin, Y.H.; Xiao, H.; Xing, S.; Chen, H.; Chi, P.D.; Zhang, G. Epstein—Barr virus infection induces indoleamine 2,3-dioxygenase expression in human monocyte-derived macrophages through p38/mitogen-activated protein kinase and NF-κB pathways: Impairment in T cell functions. J. Virol. 2014, 88, 6660–6671. [Google Scholar] [CrossRef] [Green Version]
- Zhu, Y.; Jiang, C.; Liu, Y.; Li, Y.; Wu, H.; Feng, J.; Xu, Y. Association between IDO activity and prognosis in patients with non-small cell lung cancer after radiotherapy. Ann. Transl. Med. 2020, 8, 1169. [Google Scholar] [CrossRef]
- Skok, M.; Grailhe, R.; Changeux, J.-P. Nicotinic receptors regulate B lymphocyte activation and immune response. Eur. J. Pharmacol. 2005, 517, 246–251. [Google Scholar] [CrossRef]
- He, Y.-Q.; Xue, W.-Q.; Xu, F.-H.; Xu, Y.-F.; Zhang, J.-B.; Yu, H.-L.; Feng, Q.-S.; Chen, L.-Z.; Cao, S.-M.; Liu, Q.; et al. The Relationship between Environmental Factors and the Profile of Epstein-Barr Virus Antibodies in the Lytic and Latent Infection Periods in Healthy Populations from Endemic and Non-Endemic Nasopharyngeal Carcinoma Areas in China. EBioMedicine 2018, 30, 184–191. [Google Scholar] [CrossRef] [Green Version]
- Yang, Q.-Y.; He, Y.-Q.; Xue, W.-Q.; Zhou, T.; Liao, Y.; Zheng, M.-Q.; Jia, Y.-J.; Yuan, L.-L.; Jia, W.-H. Association Between Serum Cotinine Level and Serological Markers of Epstein–Barr Virus in Healthy Subjects in South China Where Nasopharyngeal Carcinoma Is Endemic. Front. Oncol. 2019, 9, 865. [Google Scholar] [CrossRef] [Green Version]
- Xu, F.-H.; Xiong, D.; Xu, Y.-F.; Cao, S.-M.; Xue, W.-Q.; Qin, H.-D.; Liu, W.-S.; Cao, J.-Y.; Zhang, Y.; Feng, Q.-S.; et al. An Epidemiological and Molecular Study of the Relationship between Smoking, Risk of Nasopharyngeal Carcinoma, and Epstein–Barr Virus Activation. JNCI J. Natl. Cancer Inst. 2012, 104, 1396–1410. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, J.; Li, H.; Luo, X.; Li, Y.; Bode, A.; Cao, Y. The role of oxidative stress in EBV lytic reactivation, radioresistance and the potential preventive and therapeutic implications. Int. J. Cancer 2017, 141, 1722–1729. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.-Y.; Fang, C.-Y.; Wu, C.-C.; Tsai, C.-H.; Lin, S.-F.; Chen, J.-Y. Reactive Oxygen Species Mediate Epstein-Barr Virus Reactivation by N-Methyl-N’-Nitro-N-Nitrosoguanidine. PLoS ONE 2013, 8, e84919. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Camargo, M.C.; Koriyama, C.; Matsuo, K.; Kim, W.-H.; Herrera-Goepfert, R.; Liao, L.M.; Yu, J.; Carrasquilla, G.; Sung, J.J.; Alvarado-Cabrero, I.; et al. Case-case comparison of smoking and alcohol risk associations with Epstein-Barr virus-positive gastric cancer. Int. J. Cancer 2013, 134, 948–953. [Google Scholar] [CrossRef] [Green Version]
- Wang, G.-Z.; Zhang, L.; Zhao, X.-C.; Gao, S.-H.; Qu, L.-W.; Yu, H.; Fang, W.-F.; Zhou, Y.-C.; Liang, F.; Zhang, C.; et al. The Aryl hydrocarbon receptor mediates tobacco-induced PD-L1 expression and is associated with response to immunotherapy. Nat. Commun. 2019, 10, 1125. [Google Scholar] [CrossRef] [Green Version]
- El-Fattah, E.E.A.; Abdelhamid, A.M. Benzo[a]pyrene immunogenetics and immune archetype reprogramming of lung. Toxicology 2021, 463, 152994. [Google Scholar] [CrossRef]
- Ma, S.D.; Xu, X.; Jones, R.; Delecluse, H.-J.; Zumwalde, N.A.; Sharma, A.; Gumperz, J.E.; Kenney, S.C. PD-1/CTLA-4 Blockade Inhibits Epstein-Barr Virus-Induced Lymphoma Growth in a Cord Blood Humanized-Mouse Model. PLoS Pathog 2016, 12, e1005642. [Google Scholar] [CrossRef] [Green Version]
- Li, Q.X.; Young, L.; Niedobitek, G.; Dawson, C.W.; Birkenbach, M.; Wang, F.; Rickinson, A.B. Epstein–Barr virus infection and replication in a human epithelial cell system. Nat. Cell Biol. 1992, 356, 347–350. [Google Scholar] [CrossRef]
- Fingeroth, J.D.; Diamond, M.E.; Sage, D.R.; Hayman, J.; Yates, Y.L. CD21-Dependent infection of an epithelial cell line, 293, by Epstein-Barr virus. J. Virol. 1999, 73, 2115–2125. [Google Scholar] [CrossRef] [Green Version]
- Hutter, C.; Zenklusen, J.C. The Cancer Genome Atlas: Creating Lasting Value beyond Its Data. Cell 2018, 173, 283–285. [Google Scholar] [CrossRef]
- Li, R.; Zhou, R.; Zhang, J. Function of PM2.5 in the pathogenesis of lung cancer and chronic airway inflammatory diseases (Review). Oncol. Lett. 2018, 15, 7506–7514. [Google Scholar] [CrossRef] [Green Version]
- Tomczak, A.; Miller, A.B.; Weichenthal, S.A.; To, T.; Wall, C.; Van Donkelaar, A.; Martin, R.V.; Crouse, D.; Villeneuve, P.J. Long-term exposure to fine particulate matter air pollution and the risk of lung cancer among participants of the Canadian National Breast Screening Study. Int. J. Cancer 2016, 139, 1958–1966. [Google Scholar] [CrossRef] [Green Version]
- Lee, J.; Stone, J.; Desai, P.; Kosowicz, J.; Liu, J.; Ambinder, R. Arsenicals, the Integrated Stress Response, and Epstein–Barr Virus Lytic Gene Expression. Viruses 2021, 13, 812. [Google Scholar] [CrossRef]
- Ren, C.; Zhou, Y.; Liu, W.; Wang, Q. Paradoxical effects of arsenic in the lungs. Environ. Health Prev. Med. 2021, 26, 1–13. [Google Scholar] [CrossRef]
- Li, C.; Wang, C.; Yu, J.; Fan, Y.; Liu, D.; Zhou, W.; Shi, T. Residential Radon and Histological Types of Lung Cancer: A Meta-Analysis of Case–Control Studies. Int. J. Environ. Res. Public Health 2020, 17, 1457. [Google Scholar] [CrossRef] [Green Version]
- Alavanja, M.C.R. Biologic damage resulting from exposure to tobacco smoke and from radon: Implication for preventive interventions. Oncogene 2002, 21, 7365–7375. [Google Scholar] [CrossRef] [Green Version]
- Leng, S.; Thomas, C.L.; Snider, A.M.; Picchi, M.A.; Chen, W.; Willis, D.G.; Carr, T.G.; Krzeminski, J.; Desai, D.; Shantu, A.; et al. Radon Exposure, IL-6 Promoter Variants, and Lung Squamous Cell Carcinoma in Former Uranium Miners. Environ. Heal Perspect. 2016, 124, 445–451. [Google Scholar] [CrossRef] [Green Version]
- Moyano, P.; García, J.; García, J.M.; Pelayo, A.; Muñoz-Calero, P.; Frejo, M.T.; Anadon, M.J.; Lobo, M.; Del Pino, J. Chlorpyrifos-induced cell proliferation in human breast cancer cell lines differentially mediated by estrogen and aryl hydrocarbon receptors and KIAA1363 enzyme after 24 h and 14 days exposure. Chemosphere 2020, 251, 126426. [Google Scholar] [CrossRef]
- Kramer, S.; Hikel, S.M.; Adams, K.; Hinds, D.; Moon, K. Current Status of the Epidemiologic Evidence Linking Polychlorinated Biphenyls and Non-Hodgkin Lymphoma, and the Role of Immune Dysregulation. Environ. Health Perspect. 2012, 120, 1067–1075. [Google Scholar] [CrossRef] [Green Version]
- Park, E.Y.; Park, E.; Kim, J.; Oh, J.-K.; Kim, B.; Hong, Y.-C.; Lim, M.K. Impact of environmental exposure to persistent organic pollutants on lung cancer risk. Environ. Int. 2020, 143, 105925. [Google Scholar] [CrossRef]
- Fu, J.; Mai, B.; Sheng, G.; Zhang, G.; Wang, X.; Peng, P.; Xiao, X.; Ran, R.; Cheng, F.; Peng, X.; et al. Persistent organic pollutants in environment of the Pearl River Delta, China: An overview. Chemosphere 2003, 52, 1411–1422. [Google Scholar] [CrossRef]
- Bonner, M.R.; Williams, B.A.; Rusiecki, J.A.; Blair, A.; Freeman, L.E.B.; Hoppin, J.; Dosemeci, M.; Lubin, J.; Sandler, D.P.; Alavanja, M.C.R. Occupational exposure to terbufos and the incidence of cancer in the Agricultural Health Study. Cancer Causes Control 2010, 21, 871–877. [Google Scholar] [CrossRef]
- Kashuba, E.V.; Gradin, K.; Isaguliants, M.; Szekely, L.; Poellinger, L.; Klein, G.; Kazlauskas, A. Regulation of Transactivation Function of the Aryl Hydrocarbon Receptor by the Epstein-Barr Virus-encoded EBNA-3 Protein. J. Biol. Chem. 2006, 281, 1215–1223. [Google Scholar] [CrossRef] [Green Version]
- Inoue, H.; Mishima, K.; Yamamoto-Yoshida, S.; Ushikoshi, R.; Nakagawa, Y.; Yamamoto, K.; Ryo, K.; Ide, F.; Saito, I. Aryl Hydrocarbon Receptor-Mediated Induction of EBV Reactivation as a Risk Factor for Sjögren’s Syndrome. J. Immunol. 2012, 188, 4654–4662. [Google Scholar] [CrossRef] [Green Version]
- Attafi, I.M.; Bakheet, S.A.; Korashy, H.M. The role of NF-κB and AhR transcription factors in lead-induced lung toxicity in human lung cancer A549 cells. Toxicol. Mech. Methods 2020, 30, 197–207. [Google Scholar] [CrossRef]
- Zou, X.N.; Lu, S.H.; Liu, B. Volatile N-nitrosamines and their precursors in chinese salted fish—A possible etiological factor for NPC in China. Int. J. Cancer 1994, 59, 155–158. [Google Scholar] [CrossRef]
- Wang, S.Y.; Hu, Y.L.; Wu, Y.L.; Li, X.; Chi, G.B.; Chen, Y.; Dai, W.S. A comparative study of the risk factors for lung cancer in Guangdong, China. Lung Cancer 1996, 14 (Suppl. S1), S99–S105. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Tumor Types | Total | EBV (+) | (%) | EBV Gene | Methods | Country | Ref |
|---|---|---|---|---|---|---|---|
| AdC, SQC, SCLC, LCC | 80 | 5 | 6.3 | EBNA | ISH, IHC, PCR | Japan | Kasai [145] |
| NSCLC | 167 | 9 | 5.4 | EBNA | ISH, Southern Blot, IHC | China | Wong [144] |
| AC, mesothelioma | 130 | 0 | 0 | Absent | ISH | United States | Conway [154] |
| SQC, AdC, LCC, LELC | 127 | 11 | 8.7 | EBNA | ISH, IH | China | Chen [137] |
| LELC, SQC, AdC, LCC, SCC | 51 | 30 | 58.8 | EBER, LMP1, VCA | ISH, IHC | China | Han [136] |
| SCC | 23 | 1 | 4.3 | LMP-1 | ISH, IHC, PCR | United States | Chu [155] |
| SQC, AC, AdC-SCC, LCLC, SCLC | 122 | 0 | 0 | Absent | ISH, IHC, PCR | France | Brouchet [160] |
| NSCLC | 108 | 36 | 33.3 | EBER | ISH | China | Li [141] |
| SQC, AdC | 19 | 12 | 63.2 | EBER | ISH, PCR, IHC | Spain | Gomez-Roman [146] |
| AdC | 110 | 0 | 0 | Absent | ISH | Singapore | Lim [156] |
| SQC, AdC | 48 | 7 | 14.6 | BART1, BART2, and BHRF1–3 | qPCR, Microarray | United States | Koshiol [157] |
| SQC, AdC, SCC | 48 | 5 | 10.4 | LMP-1 | PCR | Iran | Jafarian [147] |
| NSCLC | 66 | 4 | 6.1 | LMP1 and EBNA1 | NGS | China | Wang [151] |
| NSCLC | 1017 | 3 | 0.3 | EBNA-1, LMP-1 and LMP-2 | NGS, ISH | United States | Kheir [152] |
| SQC, AdC, SCLC | 73 | 2 | 2.7 | Absent | PCR | Unites States | Gupta [148] |
| LELC | 91 | 91 | 100.0 | EBER | ISH-WES | China | Hong [138] |
| LC | 108 | 36 | 33.3 | EBER, LMP1, BCL-2 | IHC | China | Li [142] |
| SQC | 87 | 33 | 37.9 | Absent | ISH | China | Zhang [143] |
| NSCLC, SQC | 70 | 18 | 31.4 | Absent | qPCR | Italy | Carpagnano [149] |
| LC | 48 | 25 | 52 | Absent | PCR, in situ PCR | China | Xia [150] |
| AdC | 4 | - | - | EBER | Microarray, qPCR, ISH | Korea | Kim [158] |
| NSCLC | Sample mix | - | - | Absent | Microarray, qPCR | Korea | Ma [159] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Osorio, J.C.; Blanco, R.; Corvalán, A.H.; Muñoz, J.P.; Calaf, G.M.; Aguayo, F. Epstein–Barr Virus Infection in Lung Cancer: Insights and Perspectives. Pathogens 2022, 11, 132. https://doi.org/10.3390/pathogens11020132
Osorio JC, Blanco R, Corvalán AH, Muñoz JP, Calaf GM, Aguayo F. Epstein–Barr Virus Infection in Lung Cancer: Insights and Perspectives. Pathogens. 2022; 11(2):132. https://doi.org/10.3390/pathogens11020132
Chicago/Turabian StyleOsorio, Julio C., Rancés Blanco, Alejandro H. Corvalán, Juan P. Muñoz, Gloria M. Calaf, and Francisco Aguayo. 2022. "Epstein–Barr Virus Infection in Lung Cancer: Insights and Perspectives" Pathogens 11, no. 2: 132. https://doi.org/10.3390/pathogens11020132
APA StyleOsorio, J. C., Blanco, R., Corvalán, A. H., Muñoz, J. P., Calaf, G. M., & Aguayo, F. (2022). Epstein–Barr Virus Infection in Lung Cancer: Insights and Perspectives. Pathogens, 11(2), 132. https://doi.org/10.3390/pathogens11020132