
Non-Canonical Host Intracellular Niche Links to New Antimicrobial Resistance Mechanism


Abstract
:1. Introduction
2. P. aeruginosa as an Intracellular Pathogen
2.1. Overview
2.2. Intracellular Lifestyle of P. aeruginosa
2.2.1. P. aeruginosa Invasion of Host Cells
2.2.2. P. aeruginosa Intracellular Survival Strategies
2.3. Development of Intracellular P. aeruginosa Antibiotic Persister Cells
2.4. Summary
3. S. aureus as an Intracellular Pathogen
3.1. Overview
3.2. Intracellular Lifestyle of S. aureus
3.2.1. Internalization and Cell Entry
3.2.2. S. aureus Intracellular Survival Strategies
3.2.3. S. aureus Intracellular Escape and Replication
3.3. S. aureus Influence over Autophagy and Host Cell Death Pathways
3.3.1. Intracellular S. aureus Influence over Autophagy
3.3.2. Intracellular S. aureus Influence over Host Cell Death
3.4. Summary
4. Treating Intracellular P. aeruginosa and S. aureus by Harnessing the Host Immune Response in Combination with Using Antimicrobials
5. Surveillance and Prevention Are Instrumental in Fighting the AMR Crisis
5.1. Overview
5.2. Surveillance Is the Cornerstone of AMR Mitigation
5.3. The Use of Vaccines Is Critical for Limiting Antimicrobial Use, and Thereby AMR Development
6. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Dong, E.; Du, H.; Gardner, L. An interactive web-based dashboard to track COVID-19 in real time. Lancet Inf. Dis. 2020, 20, 533–534. [Google Scholar] [CrossRef]
- Adalja, A.A.; Watson, M.; Toner, E.S.; Cicero, A.; Inglesby, T.V. Characteristics of microbes most likely to cause pandemics and global catastrophes. Curr. Top. Microbiol. Immunol. 2019, 424, 1–20. [Google Scholar] [CrossRef] [PubMed]
- Global Antimicrobial Resistance and Use Surveillance System (GLASS) Report 2021; Licence: CC BY-NC-SA 3.0 IGO; World Health Organization: Geneva, Switzerland, 2021.
- Nadimpalli, M.L.; Chan, C.W.; Doron, S. Antibiotic resistance: A call to action to prevent the next epidemic of inequality. Nat. Med. 2021, 27, 187–188. [Google Scholar] [CrossRef] [PubMed]
- Pelfrene, E.; Botgros, R.; Cavaleri, M. Antimicrobial multidrug resistance in the era of COVID-19: A forgotten plight? Antimicrob. Resist. Infect. Control 2021, 10, 21. [Google Scholar] [CrossRef]
- Knight, G.M.; Glover, R.E.; McQuaid, C.F.; Olaru, I.D.; Gallandat, K.; Leclerc, Q.J.; Fuller, N.M.; Willcocks, S.J.; Hasan, R.; van Kleef, E.; et al. Antimicrobial resistance and COVID-19: Intersections and implications. eLife 2021, 10, e64139. [Google Scholar] [CrossRef]
- May, M. Tomorrow’s biggest microbial threats. Nat. Med. 2021, 27, 358–359. [Google Scholar] [CrossRef]
- Cars, O.; Chandy, S.J.; Mpundu, M.; Peralta, A.Q.; Zorzet, A.; So, A.D. Resetting the agenda for antibiotic resistance through a health systems perspective. Lancet Glob. Health 2021, 9, e1022–e1027. [Google Scholar] [CrossRef]
- World Health Organization. Fact Sheets—Antimicrobial resistance. Available online: https://www.who.int/news-room/fact-sheets/detail/antimicrobial-resistance (accessed on 17 November 2021).
- Gandra, S.; Barter, D.M.; Laxminarayan, R. Economic burden of antibiotic resistance: How much do we really know? Clin. Microbiol. 2014, 20, 973–980. [Google Scholar] [CrossRef] [Green Version]
- Phiri, A.; Abia, A.; Amoako, D.G.; Mkakosya, R.; Sundsfjord, A.; Essack, S.Y.; Simonsen, G.S. Burden, Antibiotic resistance, and clonality of Shigella spp. Implicated in community-acquired acute diarrhoea in Lilongwe, Malawi. Trop. Med. Infect. Dis. 2021, 6, 63. [Google Scholar] [CrossRef]
- Zhen, X.; Stålsby Lundborg, C.; Sun, X.; Zhu, N.; Gu, S.; Dong, H. Economic burden of antibiotic resistance in China: A national level estimate for inpatients. Antimicrob. Resist. Infect. Control 2021, 10, 5. [Google Scholar] [CrossRef]
- Woolhouse, M.; Waugh, C.; Perry, M.R.; Nair, H. Global disease burden due to antibiotic resistance—State of the evidence. J. Glob. Health 2016, 6, 010306. [Google Scholar] [CrossRef]
- de Kraker, M.E.; Davey, P.G.; Grundmann, H.; BURDEN Study Group. Mortality and hospital stay associated with resistant Staphylococcus aureus and Escherichia coli bacteremia: Estimating the burden of antibiotic resistance in Europe. PLoS Med. 2011, 8, e1001104. [Google Scholar] [CrossRef] [Green Version]
- Zhen, X.; Lundborg, C.S.; Sun, X.; Hu, X.; Dong, H. Economic burden of antibiotic resistance in ESKAPE organisms: A systematic review. Antimicrob. Resist. Infect. Control 2019, 8, 137. [Google Scholar] [CrossRef] [Green Version]
- Teillant, A.; Gandra, S.; Barter, D.; Morgan, D.J.; Laxminarayan, R. Potential burden of antibiotic resistance on surgery and cancer chemotherapy antibiotic prophylaxis in the USA: A literature review and modelling study. Lancet Infect. Dis. 2015, 15, 1429–1437. [Google Scholar] [CrossRef]
- O’Neill, J. Review on antimicrobial resistance: Tackling a crisis for the health and wealth of nations. London: The Review on Antimicrobial Resistance chaired by Jim O’Neill. 2014. Available online: https://wellcomecollection.org/works/rdpck35v/items (accessed on 17 November 2021).
- Murray, C.J.; Ikuta, K.S.; Sharara, F.; Swetschinski, L.; Aguilar, G.R.; Gray, A.; Han, C.; Bisignano, C.; Rao, P.; Wool, E.; et al. Global burden of bacterial antimicrobial resistance in 2019: A systematic analysis. Lancet, 2022; ahead of print. [Google Scholar] [CrossRef]
- Ehrlich, P.; Halta, S. Die Experimentelle Chemotherapie der Spirillosen; Springer: Berlin, Germany, 1910. [Google Scholar]
- Fleming, A. On the antibacterial action of cultures of a penicillium, with special reference to their use in the isolation of B. influenzæ. Br. J. Exp. Pathol. 1929, 10, 226–236. [Google Scholar] [CrossRef]
- Chain, E.; Florey, H.W.; Gardner, A.D.; Heatley, N.G.; Jennings, M.A.; Orr-Ewing, J.; Sanders, A.G. THE CLASSIC: Penicillin as a chemotherapeutic agent. 1940. Clin. Orth. Relat. Res. 2005, 439, 23–26. [Google Scholar] [CrossRef]
- Aminov, R.I. A brief history of the antibiotic era: Lessons learned and challenges for the future. Front Microbiol. 2010, 1, 134. [Google Scholar] [CrossRef] [Green Version]
- Reygaert, W.C. An overview of the antimicrobial resistance mechanisms of bacteria. AIMS Microbiol 2018, 4, 482–501. [Google Scholar] [CrossRef]
- Silva, M.T.; Pestana, N.T. The in vivo extracellular life of facultative intracellular bacterial parasites: Role in pathogenesis. Immunobiology 2013, 218, 325–337. [Google Scholar] [CrossRef]
- Kamaruzzaman, N.F.; Kendall, S.; Good, L. Targeting the hard to reach: Challenges and novel strategies in the treatment of intracellular bacterial infections. Br. J. Pharm. 2017, 174, 2225–2236. [Google Scholar] [CrossRef] [PubMed]
- Tulkens, P.M. Intracellular distribution and activity of antibiotics. Eur. J. Clin. Microbiol. Infect. Dis. 1991, 10, 100–106. [Google Scholar] [CrossRef] [PubMed]
- Nagai, J.; Takano, M. Molecular aspects of renal handling of aminoglycosides and strategies for preventing the nephrotoxicity. Drug Metab. Pharm. 2004, 19, 159–170. [Google Scholar] [CrossRef] [PubMed]
- Grant, S.S.; Hung, D.T. Persistent bacterial infections, antibiotic tolerance, and the oxidative stress response. Virulence 2013, 4, 273–283. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, H.A.; Denis, O.; Vergison, A.; Theunis, A.; Tulkens, P.M.; Struelens, M.J.; Van Bambeke, F. Intracellular activity of antibiotics in a model of human THP-1 macrophages infected by a Staphylococcus aureus small-colony variant strain isolated from a cystic fibrosis patient: Pharmacodynamic evaluation and comparison with isogenic normal-phenotype and revertant strains. Antimicrob. Agents Chemother. 2009, 53, 1434–1442. [Google Scholar] [CrossRef] [Green Version]
- Rao, M.; Ippolito, G.; Mfinanga, S.; Ntoumi, F.; Yeboah-Manu, D.; Vilaplana, C.; Zumla, A.; Maeurer, M. Latent TB Infection (LTBI)—Mycobacterium tuberculosis pathogenesis and the dynamics of the granuloma battleground. Int. J. Infect. Dis. 2019, 80, S58–S61. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, K.; Salgame, P. Host innate immune response to Mycobacterium tuberculosis. J. Clin. Immunol. 2007, 27, 347–362. [Google Scholar] [CrossRef]
- Wolf, A.J.; Desvignes, L.; Linas, B.; Banaiee, N.; Tamura, T.; Takatsu, K.; Ernst, J.D. Initiation of the adaptive immune response to Mycobacterium tuberculosis depends on antigen production in the local lymph node, not the lungs. J. Exp. Med. 2008, 205, 105–115. [Google Scholar] [CrossRef]
- Wayne, L.G.; Sohaskey, C.D. Nonreplicating persistence of Mycobacterium tuberculosis. Annu. Rev. Microbiol. 2001, 55, 139–163. [Google Scholar] [CrossRef]
- Stover, C.; Phamm, X.; Erwin, A.; Mizoguchi, S.D.; Warrener, P.; Hickey, M.J.; Brinkman, F.S.; Hufnagle, W.O.; Kowalik, D.J.; Lagrou, M.; et al. Complete genome sequence of Pseudomonas aeruginosa PAO1, an opportunistic pathogen. Nature 2000, 406, 959–964. [Google Scholar] [CrossRef]
- Lyczak, J.B.; Cannon, C.L.; Pier, G.B. Establishment of Pseudomonas aeruginosa infection: Lessons from a versatile opportunist. Microbes Infect. 2000, 2, 1051–1060. [Google Scholar] [CrossRef]
- Alonso, B.; Fernández-Barat, L.; Di Domenico, E.G.; Marín, M.; Cercenado, E.; Merino, I.; de Pablos, M.; Muñoz, P.; Guembe, M. Correction to: Characterization of the virulence of Pseudomonas aeruginosa strains causing ventilator-associated pneumonia. BMC Infect. Dis. 2000, 20, 951. [Google Scholar] [CrossRef] [PubMed]
- Bleves, S.; Viarre, V.; Salacha, R.; Michel, G.P.F.; Filloux, A.; Voulhoux, R. Protein secretion systems in Pseudomonas aeruginosa: A wealth of pathogenic weapons. Int. J. Med. Microbiol. 2010, 300, 534–543. [Google Scholar] [CrossRef] [PubMed]
- Pang, Z.; Raudonis, R.; Glick, B.R.; Lin, T.; Cheng, Z. Antibiotic resistance in Pseudomonas aeruginosa: Mechanisms and alternative therapeutic strategies. Biotechnol. Adv. 2019, 37, 177–192. [Google Scholar] [CrossRef]
- Lambert, P.A. Mechanisms of antibiotic resistance in Pseudomonas aeruginosa. J. R. Soc. Med. 2002, 95, 22–26. [Google Scholar]
- Drenkard, E. Antimicrobial resistance of Pseudomonas aeruginosa biofilms. Microbes Infect. 2003, 5, 1213–1219. [Google Scholar] [CrossRef]
- Klockgether, J.; Cramer, N.; Wiehlmann, L.; Davenport, C.F.; Tümmler, B. Pseudomonas aeruginosa genomic structure and diversity. Front. Microbiol. 2011, 2, 150. [Google Scholar] [CrossRef] [Green Version]
- Poulsen, B.E.; Yang, R.; Clatworthy, A.E.; White, T.; Osmulski, S.J.; Li, L.; Penaranda, C.; Lander, E.S.; Shoresh, N.; Hung, D.T. Defining the core essential genome of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2019, 116, 10072–10080. [Google Scholar] [CrossRef] [Green Version]
- Subedi, D.; Vijay, A.K.; Kohli, G.S.; Rice, S.A.; Willcox, M. Comparative genomics of clinical strains of Pseudomonas aeruginosa strains isolated from different geographic sites. Sci. Rep. 2018, 8, 15668. [Google Scholar] [CrossRef] [Green Version]
- Weiser, R.; Green, A.E.; Bull, M.J.; Cunningham-Oakes, E.; Jolley, K.A.; Maiden, M.C.J.; Hall, A.J.; Winstanley, C.; Weightman, A.J.; Donoghue, D.; et al. Not all Pseudomonas aeruginosa are equal: Strains from industrial sources possess uniquely large mutlireplicon genomes. Microb. Genom. 2019, 5, e000276. [Google Scholar] [CrossRef]
- Sana, T.G.; Baumann, C.; Merdes, A.; Soscia, C.; Rattei, T.; Hachani, A.; Jones, C.; Bennett, K.L.; Filloux, A.; Superti-Furga, G.; et al. Internalization of Pseudomonas aeruginosa strain PAO1 into epithelial cells is promoted by interaction of a T6SS effector with the microtubule network. MBio 2015, 6, e00712–e00715. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Penaranda, C.; Chumbler, N.M.; Hung, D.T. Dual transcriptional analysis reveals adaptation of host and pathogen to intracellular survival of Pseudomonas aeruginosa associated with urinary tract infection. PLoS Pathog. 2015, 17, e1009534. [Google Scholar] [CrossRef] [PubMed]
- Angus, A.A.; Lee, A.A.; Augustin, D.K.; Lee, E.J.; Evans, D.J.; Fleiszig, S.M. Pseudomonas aeruginosa induces membrane blebs in epithelial cells, which are utilized as a niche for intracellular replication and motility. Infect. Immun. 2008, 76, 1992–2001. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Garcia-Medina, R.; Dunne, W.M.; Singh, P.K.; Brody, S.L. Pseudomonas aeruginosa acquires biofilm-like properties within airway epithelial cells. Infect. Immun. 2005, 73, 8298–8305. [Google Scholar] [CrossRef] [Green Version]
- Sana, T.G.; Hachani, A.; Bucior, I.; Soscia, C.; Garvis, S.; Termine, E.; Engel, J.; Filloux, A.; Bleves, S. The second type VI secretion system of Pseudomonas aeruginosa strain PAO1 is regulated by quorum sensing and fur and modulates internalization in epithelial cells. J. Biol. Chem. 2012, 287, 27095–27105. [Google Scholar] [CrossRef] [Green Version]
- Angus, A.A.; Evans, D.J.; Barbieri, J.T.; Fleiszig, S.M. The ADP-ribosylation domain of Pseudomonas aeruginosa ExoS is required for membrane bleb niche formation and bacterial survival within epithelial cells. Infect. Immun. 2010, 78, 4500–4510. [Google Scholar] [CrossRef] [Green Version]
- Garai, P.; Berry, L.; Moussouni, M.; Bleves, S.; Blanc-Potard, A.-B. Killing from the Inside: Intracellular role of T3SS in the fate of Pseudomonas aeruginosa within macrophages revealed by MgtC and OprF mutants. PLoS Pathog. 2019, 15, e1007812. [Google Scholar] [CrossRef] [Green Version]
- Kierbel, A.; Gassama-Diagne, A.; Mostov, K.; Engel, J.N. The phosphoinositol-3-kinase–protein Kinase B/Akt pathway is Critical for Pseudomonas aeruginosa strain PAK internalization. Mol. Biol. Cell 2005, 16, 2577–2585. [Google Scholar] [CrossRef] [Green Version]
- Kierbel, A.; Gassama-Diagne, A.; Rocha, C.; Radoshevich, L.; Olson, J.; Mostov, K.; Engel, J. Pseudomonas aeruginosa exploits a PIP3-dependent pathway to transform apical into basolateral membrane. J. Cell Biol. 2007, 177, 21–27. [Google Scholar] [CrossRef]
- Durand, E.; Cambillau, C.; Cascales, E.; Journet, L. VgrG, Tae, Tle, and beyond: The versatile arsenal of type VI secretion effectors. Trends Microbiol. 2014, 22, 498–507. [Google Scholar] [CrossRef]
- Kollman, J.M.; Merdes, A.; Mourey, L.; Agard, D.A. Microtubule nucleation by γ-tubulin complexes. Nat. Rev. Mol. Cell Biol. 2011, 12, 709–721. [Google Scholar] [CrossRef] [PubMed]
- Wolfgang, M.C.; Kulasekara, B.R.; Liang, X.; Boyd, D.; Wu, K.; Yang, Q.; Miyada, C.G.; Lory, S. Conservation of genome content and virulence determinants among clinical and environmental isolates of Pseudomonas aeruginosa. Proc. Natl. Acad. Sci. USA 2003, 100, 8484–8489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yahr, T.L.; Vallis, A.J.; Hancock, M.K.; Barbieri, J.T.; Frank, D.W. ExoY, an adenylate cyclase secreted by the Pseudomonas aeruginosa type III system. Proc. Natl. Acad. Sci. USA 1998, 95, 13899–13904. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barbieri, J.T.; Sun, J. Pseudomonas aeruginosa ExoS and ExoT. In Reviews of Physiology, Biochemistry and Pharmacology; Aktories, K., Just, I., Eds.; Springer: Berlin, Germany, 2004; Volume 152, pp. 79–92. [Google Scholar]
- Lee, V.T.; Smith, R.S.; Tümmler, B.; Lory, S. Activities of Pseudomonas aeruginosa effectors secreted by the type III secretion system in vitro and during infection. Infect. Immun. 2005, 73, 1695–1705. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kroken, A.R.; Chen, C.K.; Evans, D.J.; Yahr, T.L.; Fleiszig, S.M. The impact of ExoS on Pseudomonas aeruginosa internalization by epithelial cells is independent of FleQ and correlates with bistability of type three secretion system gene expression. MBio 2018, 9, e00668–e00718. [Google Scholar] [CrossRef] [Green Version]
- Borkar, D.S.; Acharya, N.R.; Leong, C.; Lalitha, P.; Srinivasan, M.; Oldenburg, C.E.; Cevallos, V.; Lietman, T.M.; Evans, D.J.; Fleiszig, S.M. Cytotoxic clinical isolates of Pseudomonas aeruginosa identified during the steroids for corneal ulcers trial show elevated resistance to fluoroquinolones. BMC Ophthalmol. 2014, 14, 54. [Google Scholar] [CrossRef] [Green Version]
- Fleiszig, S.M.; Wiener-Kronish, J.P.; Miyazaki, H.; Vallas, V.; Mostov, K.E.; Kanada, D.; Sawa, T.; Yen, T.S.; Frank, D.W. Pseudomonas aeruginosa-mediated cytotoxicity and invasion correlate with distinct genotypes at the loci encoding exoenzymes. Infect. Immun. 1997, 65, 579–586. [Google Scholar] [CrossRef] [Green Version]
- Heimer, S.R.; Evans, D.J.; Stern, M.E.; Barbieri, J.T.; Yahr, T.; Fleiszig, S.M. Pseudomonas aeruginosa utilizes the type III secreted toxin ExoS to avoid acidified compartments within epithelial cells. PLoS ONE 2013, 8, e73111. [Google Scholar] [CrossRef] [Green Version]
- Belon, C.; Soscia, C.; Bernut, A.; Laubier, A.; Bleves, S.; Blanc-Potard, A.-B. A macrophage subversion factor is shared by intracellular and extracellular pathogens. PLoS Pathog. 2015, 11, e1004969. [Google Scholar] [CrossRef]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-ΚB signaling in inflammation. Sig. Transduct. Target. 2017, 2, 17023. [Google Scholar] [CrossRef] [Green Version]
- Williamson, K.S.; Richards, L.A.; Perez-Osorio, A.C.; Pitts, B.; McInnerney, K.; Stewart, P.S.; Franklin, M.J. Heterogeneity in Pseudomonas aeruginosa biofilms includes expression of hibernation factors in the antibiotic-tolerant subpopulation and Hypoxia-induced stress response in the metabolically active population. J. Bacteriol. 2012, 194, 2062–2073. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulcahy, L.R.; Burns, J.L.; Lory, S.; Lewis, K. Emergence of Pseudomonas aeruginosa strains producing high levels of persister cells in patients with cystic fibrosis. J. Bacteriol. 2010, 192, 6191–6199. [Google Scholar] [CrossRef] [Green Version]
- Maisonneuve, E.; Gerdes, K. Molecular mechanisms underlying bacterial persisters. Cell 2014, 157, 539–548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schulte, M.; Olschewski, K.; Hensel, M. The protected physiological status of intracellular Salmonella enterica persisters reduces host cell-imposed stress. Commun. Biol. 2020, 4, 520. [Google Scholar] [CrossRef] [PubMed]
- Bakkeren, E.; Huisman, J.S.; Fattinger, S.A.; Hausmann, A.; Furter, M.; Egli, A.; Slack, E.; Sellin, M.E.; Bonhoeffer, S.; Regoes, R.R.; et al. Salmonella persisters promote the spread of antibiotic resistance plasmids in the gut. Nature 2019, 573, 276–280. [Google Scholar] [CrossRef] [PubMed]
- Anyanwu, N.J.; John, W.C. Conventional and Rapid Methods for Identification of Staphylococcus Aureus from Clinical Specimens. Sci. J. Microbiol. 2013, 2, 174–178. [Google Scholar] [CrossRef] [Green Version]
- Sakr, A.; Brégeon, F.; Mège, J.-L.; Rolain, J.-M.; Blin, O. Staphylococcus aureus nasal colonization: An update on mechanisms, epidemiology, risk factors, and subsequent infections. Front. Microbiol. 2018, 9, 2419. [Google Scholar] [CrossRef]
- Tong, S.Y.; Davis, J.S.; Eichenberger, E.; Holland, T.L.; Fowler, V.G. Staphylococcus aureus infections: Epidemiology, pathophysiology, clinical manifestations, and management. Clin. Microbiol. Rev. 2015, 28, 603–661. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Guo, R.; Xie, B.; Lai, Q.; Xu, J.; Hu, N.; Wan, L.; Dai, M.; Zhang, B. Drug resistance of pathogens causing nosocomial infection in orthopedics from 2012 to 2017: A 6-year retrospective study. J. Orthop. Surg. Res. 2021, 16, 100. [Google Scholar] [CrossRef]
- Casadevall, A.; Fang, F.C. The intracellular pathogen concept. Mol. Microbiol. 2020, 113, 541–545. [Google Scholar] [CrossRef] [Green Version]
- Horn, J.; Stelzner, K.; Rudel, T.; Fraunholz, M. Inside job: Staphylococcus aureus host-pathogen interactions. Int. J. Microbiol. 2018, 308, 607–624. [Google Scholar] [CrossRef] [PubMed]
- Lowy, F.D. Is Staphylococcus aureus an intracellular pathogen? Trends Microbiol. 2000, 8, 341–343. [Google Scholar] [CrossRef]
- Kwiecinski, J.M.; Horswill, A.R. Staphylococcus aureus bloodstream infections: Pathogenesis and regulatory mechanisms. Curr. Opin. Microbiol. 2020, 53, 51–60. [Google Scholar] [CrossRef] [PubMed]
- Alder, K.D.; Lee, I.; Munger, A.M.; Kwon, H.-K.; Morris, M.T.; Cahill, S.V.; Back, J.H.; Yu, K.E.; Lee, F.Y. Intracellular Staphylococcus aureus in bone and joint infections: A mechanism of disease recurrence, inflammation, and bone and cartilage destruction. Bone 2020, 141, 115568. [Google Scholar] [CrossRef]
- Loss, G.; Simões, P.M.; Valour, F.; Cortês, M.F.; Gonzaga, L.; Bergot, M.; Trouillet-Assant, S.; Josse, J.; Diot, A.; Ricci, E.; et al. Staphylococcus aureus small colony variants (SCVs): News from a chronic prosthetic joint infection. Front. Cell. Infect. Microbiol. 2019, 9, 363. [Google Scholar] [CrossRef]
- Oberbach, A.; Schlichting, N.; Feder, S.; Lehmann, S.; Kullnick, Y.; Buschmann, T.; Blumert, C.; Horn, F.; Neuhaus, J.; Neujahr, R.; et al. New insights into valve-related intramural and intracellular bacterial diversity in infective endocarditis. PLoS ONE 2017, 12, e0175569. [Google Scholar] [CrossRef]
- Strobel, M.; Pförtner, H.; Tuchscherr, L.; Völker, U.; Schmidt, F.; Kramko, N.; Schnittler, H.-J.; Fraunholz, M.J.; Löffler, B.; Peters, G.; et al. Post-invasion events after infection with Staphylococcus aureus are strongly dependent on both the host cell type and the infecting S. aureus strain. Clin. Microbiol. Infect. 2016, 22, 799–809. [Google Scholar] [CrossRef] [Green Version]
- Edwards, A.M.; Potter, U.; Meenan, N.A.; Potts, J.R.; Massey, R.C. Staphylococcus aureus keratinocyte invasion is dependent upon multiple high-affinity fibronectin-binding repeats within FnBPA. PLoS ONE 2011, 6, e18899. [Google Scholar] [CrossRef] [Green Version]
- Fraunholz, M.; Sinha, B. Intracellular Staphylococcus aureus: Live-in and let die. Front. Cell Infect. Microbiol. 2012, 2, 43. [Google Scholar] [CrossRef] [Green Version]
- Liang, X.; Garcia, B.L.; Visai, L.; Prabhakaran, S.; Meenan, N.A.; Potts, J.R.; Humphries, M.J.; Höök, M. Allosteric regulation of fibronectin/α5β1 interaction by fibronectin-binding mscramms. PLoS ONE 2016, 11, e0159118. [Google Scholar] [CrossRef] [Green Version]
- Bur, S.; Preissner, K.T.; Herrmann, M.; Bischoff, M. The Staphylococcus aureus extracellular adherence protein promotes bacterial internalization by keratinocytes independent of fibronectin-binding proteins. J. Investig. Dermatol. 2013, 133, 2004–2012. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirschhausen, N.; Schlesier, T.; Schmidt, M.A.; Götz, F.; Peters, G.; Heilmann, C. A novel staphylococcal internalization mechanism involves the major autolysin atl and heat shock cognate protein HSC70 as host cell receptor. Cell Microbiol. 2010, 12, 1746–1764. [Google Scholar] [CrossRef] [PubMed]
- Schlesier, T.; Siegmund, A.; Rescher, U.; Heilmann, C. Characterization of the ATL-mediated staphylococcal internalization mechanism. Int. J. Med. Microbiol. Suppl. 2020, 310, 151463. [Google Scholar] [CrossRef] [PubMed]
- Herman-Bausier, P.; Labate, C.; Towell, A.M.; Derclaye, S.; Geoghegan, J.A.; Dufrêne, Y.F. Staphylococcus aureus clumping factor A is a force-sensitive molecular switch that activates bacterial adhesion. Proc. Natl. Acad Sci. USA 2018, 115, 5564–5569. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDonnell, C.J.; Garciarena, C.D.; Watkin, R.L.; McHale, T.M.; McLoughlin, A.; Claes, J.; Verhamme, P.; Cummins, P.M.; Kerrigan, S.W. Inhibition of major integrin αvβ3 reduces Staphylococcus aureus attachment to sheared human endothelial cells. J. Thromb. Haemost. 2016, 14, 2536–2547. [Google Scholar] [CrossRef]
- Ashraf, S.; Cheng, J.; Zhao, X. Clumping factor A of staphylococcus aureus interacts with annexin A2 on mammary epithelial cells. Sci. Rep. 2017, 7, 40608. [Google Scholar] [CrossRef] [Green Version]
- Claes, J.; Liesenborghs, L.; Peetermans, M.; Veloso, T.R.; Missiakas, D.; Schneewind, O.; Mancini, S.; Entenza, J.M.; Hoylaerts, M.F.; Heying, R.; et al. Clumping factor A, von Willebrand factor-binding protein and von willebrand factor anchor Staphylococcus aureus to the vessel wall. J. Thromb. Haemost. 2017, 15, 1009–1019. [Google Scholar] [CrossRef] [Green Version]
- Na, M.; Hu, Z.; Mohammad, M.; do Stroparo, M.; Ali, A.; Fei, Y.; Jarneborn, A.; Verhamme, P.; Schneewind, O.; Missiakas, D.; et al. The expression of von Willebrand factor-binding protein determines joint-invading capacity of Staphylococcus aureus, a core mechanism of septic arthritis. Mbio 2020, 11, e02472-20. [Google Scholar] [CrossRef]
- Viljoen, A.; Viela, F.; Mathelié-Guinlet, M.; Missiakas, D.; Pietrocola, G.; Speziale, P.; Dufrêne, Y.F. Staphylococcus aureus vWF-binding protein triggers a strong interaction between clumping factor A and host vWF. Commun. Biol. 2021, 4, 453. [Google Scholar] [CrossRef]
- Walsh, E.J.; Miajlovic, H.; Gorkun, O.V.; Foster, T.J. Identification of the Staphylococcus aureus MSCRAMM clumping factor B (clfb) binding site in the αC-domain of human fibrinogen. Microbiology 2008, 154, 550–558. [Google Scholar] [CrossRef]
- Walsh, E.J.; O’Brien, L.M.; Liang, X.; Hook, M.; Foster, T.J. Clumping factor B, a fibrinogen-binding MSCRAMM (microbial surface components recognizing adhesive matrix molecules) adhesin of Staphylococcus aureus, also binds to the tail region of type I cytokeratin 10. J. Biol. Chem. 2004, 279, 50691–50699. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulcahy, M.E.; Geoghegan, J.A.; Monk, I.R.; O’Keeffe, K.M.; Walsh, E.J.; Foster, T.J.; McLoughlin, R.M. Nasal colonisation by Staphylococcus aureus depends upon clumping factor B binding to the squamous epithelial cell envelope protein loricrin. PLoS Pathog. 2012, 8, e1003092. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vitry, P.; Valotteau, C.; Feuillie, C.; Bernard, S.; Alsteens, D.; Geoghegan, J.A.; Dufrêne, Y.F. Force-induced strengthening of the interaction between staphylococcus aureus clumping factor B and loricrin. Mbio 2017, 8, e01748-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zapotoczna, M.; Jevnikar, Z.; Miajlovic, H.; Kos, J.; Foster, T.J. Iron-regulated surface determinant B (ISDB) Promotes staphylococcus aureus adherence to and internalization by non-phagocytic human cells. Cell Microbiol. 2013, 15, 1026–1041. [Google Scholar] [CrossRef] [Green Version]
- Pietrocola, G.; Pellegrini, A.; Alfeo, M.J.; Marchese, L.; Foster, T.J.; Speziale, P. The iron-regulated surface determinant B (ISDB) protein from Staphylococcus aureus acts as a receptor for the host protein vitronectin. J. Biol. Chem. 2020, 295, 10008–10022. [Google Scholar] [CrossRef]
- Nguyen, M.-T.; Luqman, A.; Bitschar, K.; Hertlein, T.; Dick, J.; Ohlsen, K.; Bröker, B.; Schittek, B.; Götz, F. Staphylococcal (phospho)lipases promote biofilm formation and host cell invasion. Int. J. Med. Microbiol. Suppl. 2018, 308, 653–663. [Google Scholar] [CrossRef]
- Tribelli, P.M.; Luqman, A.; Nguyen, M.T.; Madlung, J.; Fan, S.H.; Macek, B.; Sass, P.; Bitschar, K.; Schittek, B.; Kretschmer, D.; et al. Staphylococcus aureus LPL protein triggers human host cell invasion via activation of hsp90 receptor. Cell Microbiol. 2020, 22, e13111. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.-H.; Jiang, Y.-L.; Zhang, J.; Wang, L.; Bai, X.-H.; Zhang, S.-J.; Ren, Y.-M.; Li, N.; Zhang, Y.-H.; Zhang, Z.; et al. Structural insights into SraP-mediated Staphylococcus aureus adhesion to host cells. PLoS Pathogens. 2014, 10, e1004169. [Google Scholar] [CrossRef]
- Corrigan, R.M.; Miajlovic, H.; Foster, T.J. Surface proteins that promote adherence of Staphylococcus aureus to human desquamated nasal epithelial cells. BMC Microbiol. 2009, 9, 22. [Google Scholar] [CrossRef] [Green Version]
- Askarian, F.; Uchiyama, S.; Valderrama, J.A.; Ajayi, C.; Sollid, J.U.; van Sorge, N.M.; Nizet, V.; van Strijp, J.A.; Johannessen, M. Serine-aspartate repeat protein D increases Staphylococcus aureus virulence and survival in blood. Infect. Immun. 2017, 85, e00559–e00616. [Google Scholar] [CrossRef] [Green Version]
- Claro, T.; Widaa, A.; O’Seaghdha, M.; Miajlovic, H.; Foster, T.J.; O’Brien, F.J.; Kerrigan, S.W. Staphylococcus aureus protein a binds to osteoblasts and triggers signals that weaken bone in osteomyelitis. PLoS ONE 2011, 6, e18748. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Edwards, A.M.; Bowden, M.G.; Brown, E.L.; Laabei, M.; Massey, R.C. Staphylococcus aureus extracellular adherence protein triggers TNFA release, promoting attachment to endothelial cells via protein A. PLoS ONE 2012, 7, e43046. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viela, F.; Prystopiuk, V.; Leprince, A.; Mahillon, J.; Speziale, P.; Pietrocola, G.; Dufrêne, Y.F. Binding of staphylococcus aureus protein A to von willebrand factor is regulated by Mechanical Force. mBio 2019, 10, e00555-19. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soong, G.; Martin, F.J.; Chun, J.; Cohen, T.S.; Ahn, D.S.; Prince, A. Staphylococcus aureus protein a mediates invasion across airway epithelial cells through activation of rhoa GTPase signaling and proteolytic activity. J. Biol. Chem. 2011, 286, 35891–35898. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ji, Z.; Su, J.; Hou, Y.; Yao, Z.; Yu, B.; Zhang, X. EGFR/Fak and c-src signalling pathways mediate the internalisation of staphylococcus aureus by osteoblasts. Cell Microbiol. 2020, 22, e13240. [Google Scholar] [CrossRef]
- Abel, J.; Goldmann, O.; Ziegler, C.; Höltje, C.; Smeltzer, M.S.; Cheung, A.L.; Bruhn, D.; Rohde, M.; Medina, E. Staphylococcus aureus evades the extracellular antimicrobial activity of mast cells by promoting its own uptake. J. Innate Immun. 2011, 3, 495–507. [Google Scholar] [CrossRef]
- Goldmann, O.; Tuchscherr, L.; Rohde, M.; Medina, E. A-hemolysin enhances staphylococcus aureus internalization and survival within mast cells by modulating the expression of β1 integrin. Cell Microbiol. 2016, 18, 807–819. [Google Scholar] [CrossRef]
- Hermann, I.; Räth, S.; Ziesemer, S.; Volksdorf, T.; Dress, R.J.; Gutjahr, M.; Müller, C.; Beule, A.G.; Hildebrandt, J.-P. Staphylococcus aureus hemolysin a disrupts cell–matrix adhesions in human airway epithelial cells. Am. J. Respir. Cell Mol. Biol. 2015, 52, 14–24. [Google Scholar] [CrossRef]
- Eiffler, I.; Behnke, J.; Ziesemer, S.; Müller, C.; Hildebrandt, J.-P. Staphylococcus aureus α-toxin-mediated cation entry depolarizes membrane potential and activates p38 MAP kinase in airway epithelial cells. Am. J. Physiol. Lung Cell Mol. Physiol. 2016, 311, L676–L685. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.-H.; Zhang, K.; Wang, N.; Qiu, X.-M.; Wang, Y.-B.; He, P. Involvement of phosphatidylinositol 3-kinase/Akt signaling pathway in β1 integrin-mediated internalization of Staphylococcus aureus by alveolar epithelial cells. J. Microbiol. 2013, 51, 644–650. [Google Scholar] [CrossRef]
- Josse, J.; Laurent, F.; Diot, A. Staphylococcal adhesion and host cell invasion: Fibronectin-binding and other mechanisms. Front. Microbiol. 2017, 8, 2433. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bosi, E.; Monk, J.M.; Aziz, R.K.; Fondi, M.; Nizet, V.; Palsson, B.Ø. Comparative genome-scale modelling of staphylococcus aureus strains identifies strain-specific metabolic capabilities linked to pathogenicity. Proc. Natl. Acad. Sci. USA 2016, 113, E3801–E3809. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lebughe, M.; Phaku, P.; Niemann, S.; Mumba, D.; Peters, G.; Muyembe-Tamfum, J.-J.; Mellmann, A.; Strauß, L.; Schaumburg, F. The impact of the Staphylococcus aureus Virulome on infection in a developing country: A cohort study. Front. Microbiol. 2017, 8, 1662. [Google Scholar] [CrossRef] [PubMed]
- Karki, A.B.; Neyaz, L.; Fakhr, M.K. Comparative genomics of plasmid-bearing staphylococcus aureus strains isolated from various retail meats. Front. Microbiol. 2020, 11, 574923. [Google Scholar] [CrossRef] [PubMed]
- Naushad, S.; Nobrega, D.B.; Naqvi, S.A.; Barkema, H.W.; De Buck, J. Genomic analysis of bovine Staphylococcus aureus isolates from milk to elucidate diversity and determine the distributions of antimicrobial and virulence genes and their association with mastitis. mSystems 2020, 5, e00063-20. [Google Scholar] [CrossRef] [PubMed]
- Fuchs, S.; Mehlan, H.; Bernhardt, J.; Hennig, A.; Michalik, S.; Surmann, K.; Pané-Farré, J.; Giese, A.; Weiss, S.; Backert, L.; et al. AureoWiki ̵ The repository of the Staphylococcus aureus research and annotation community. Int. J. Med. Microbiol. 2018, 6, 558–568. [Google Scholar] [CrossRef]
- Albrecht, T.; Raue, S.; Rosenstein, R.; Nieselt, K.; Gotz, F. Phylogeny of the staphylococcal major Autolysin and its use in genus and species typing. J. Bacteriol. 2012, 194, 2630–2636. [Google Scholar] [CrossRef]
- Manara, S.; Pasolli, E.; Dolce, D.; Ravenni, N.; Campana, S.; Armanini, F.; Asnicar, F.; Mengoni, A.; Galli, L.; Montagnani, C.; et al. Whole-genome epidemiology, characterisation, and phylogenetic reconstruction of Staphylococcus aureus strains in a paediatric hospital. Genome Med. 2018, 10, 82. [Google Scholar] [CrossRef]
- Hussain, M.; von Eiff, C.; Sinha, B.; Joost, I.; Herrmann, M.; Peters, G.; Becker, K. eap Gene as novel target for specific identification of Staphylococcus aureus. J. Clin. Microbiol. 2008, 46, 470–476. [Google Scholar] [CrossRef] [Green Version]
- Amissah, N.A.; Chlebowicz, M.A.; Ablordey, A.; Tetteh, C.S.; Prah, I.; van der werf, T.S.; Friedrich, A.W.; van Dijl, J.M.; Stienstra, Y.; Rossen, J.W. Virulence potential of Staphylococcus aureus isolates from Buruli ulcer patients. Int. J. Med. Microbiol. 2017, 307, 223–232. [Google Scholar] [CrossRef]
- Rollin, G.; Tan, X.; Tros, F.; Dupuis, M.; Nassif, X.; Charbit, A.; Coureuil, M. Intracellular Survival of Staphylococcus aureus in Endothelial Cells: A Matter of Growth or Persistence. Front. Microbiol. 2017, 8, 1354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tuchscherr, L.; Medina, E.; Hussain, M.; Völker, W.; Heitmann, V.; Niemann, S.; Holzinger, D.; Roth, J.; Proctor, R.A.; Becker, K.; et al. Staphylococcus aureus phenotype switching: An effective bacterial strategy to escape host immune response and establish a chronic infection. EMBO Mol. Med. 2011, 3, 129–141. [Google Scholar] [CrossRef] [PubMed]
- Flannagan, R.S.; Heit, B.; Heinrichs, D.E. Intracellular replication of Staphylococcus aureus in mature phagolysosomes in macrophages precedes host cell death, and bacterial escape and dissemination. Cell Microbiol. 2016, 18, 514–535. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Grosz, M.; Kolter, J.; Paprotka, K.; Winkler, A.-C.; Schäfer, D.; Chatterjee, S.S.; Geiger, T.; Wolz, C.; Ohlsen, K.; Otto, M.; et al. Cytoplasmic Replication of Staphylococcus aureus upon phagosomal escape triggered by phenol-soluble modulin α. Cell Microbiol. 2014, 16, 451–465. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lacoma, A.; Cano, V.; Moranta, D.; Regueiro, V.; Domínguez-Villanueva, D.; Laabei, M.; González-Nicolau, M.; Ausina, V.; Prat, C.; Bengoechea, J.A. Investigating intracellular persistence of Staphylococcus aureus within a murine alveolar macrophage cell line. Virulence 2017, 8, 1761–1775. [Google Scholar] [CrossRef] [Green Version]
- Tranchemontagne, Z.R.; Camire, R.B.; O’Donnell, V.J.; Baugh, J.; Burkholder, K.M. Staphylococcus aureus strain USA300 perturbs acquisition of lysosomal enzymes and requires phagosomal acidification for survival inside macrophages. Infect. Immun. 2015, 84, 241–253. [Google Scholar] [CrossRef] [Green Version]
- Flannagan, R.S.; Kuiack, R.C.; McGavin, M.J.; Heinrichs, D.E. Staphylococcus aureus uses the GRAXRS regulatory system to sense and adapt to the acidified phagolysosome in macrophages. Mbio 2018, 9, e01143-18. [Google Scholar] [CrossRef] [Green Version]
- Zhou, C.; Bhinderwala, F.; Lehman, M.K.K.; Thomas, V.C.; Chaudhari, S.S.; Yamada, K.J.; Foster, K.W.; Powers, R.; Kielian, T.; Fey, P.D. Urease is an essential component of the acid response network of staphylococcus aureus and is required for a persistent murine kidney infection. PLoS Pathog. 2019, 15, e1007538. [Google Scholar] [CrossRef]
- Jubrail, J.; Morris, P.; Bewley, M.A.; Stoneham, S.; Johnston, S.A.; Foster, S.J.; Peden, A.A.; Read, R.C.; Marriott, H.M.; Dockrell, D.H. Inability to sustain intraphagolysosomal killing of Staphylococcus aureus predisposes to bacterial persistence in macrophages. Cell Microbiol. 2016, 18, 80–96. [Google Scholar] [CrossRef] [Green Version]
- Kubica, M.; Guzik, K.; Koziel, J.; Zarebski, M.; Richter, W.; Gajkowska, B.; Golda, A.; Maciag-Gudowska, A.; Brix, K.; Shaw, L.; et al. A potential new pathway for Staphylococcus aureus dissemination: The silent survival of S. aureus phagocytosed by human monocyte-derived macrophages. PLoS ONE 2008, 3, e1409. [Google Scholar] [CrossRef] [Green Version]
- Flannagan, R.; Heit, B.; Heinrichs, D. Antimicrobial mechanisms of macrophages and the immune evasion strategies of Staphylococcus aureus. Pathogens 2015, 4, 826–868. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lehar, S.M.; Pillow, T.; Xu, M.; Staben, L.; Kajihara, K.K.; Vandlen, R.; DePalatis, L.; Raab, H.; Hazenbos, W.L.; Hiroshi Morisaki, J.; et al. Novel antibody–antibiotic conjugate eliminates intracellular S. aureus. Nature 2015, 527, 323–328. [Google Scholar] [CrossRef] [PubMed]
- Surewaard, B.G.J.; Deniset, J.F.; Zemp, F.J.; Amrein, M.; Otto, M.; Conly, J.; Omri, A.; Yates, R.M.; Kubes, P. Identification and treatment of the Staphylococcus aureus reservoir in vivo. J. Exp. Med. 2016, 213, 1141–1151. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pollitt, E.J.; Szkuta, P.T.; Burns, N.; Foster, S.J. Staphylococcus aureus infection dynamics. PLoS Pathog. 2018, 14, e1007112. [Google Scholar] [CrossRef] [Green Version]
- Giese, B.; Glowinski, F.; Paprotka, K.; Dittmann, S.; Steiner, T.; Sinha, B.; Fraunholz, M.J. Expression of δ-toxin by Staphylococcus aureus mediates escape from phago-endosomes of human epithelial and endothelial cells in the presence of β-toxin. Cell Microbiol. 2011, 13, 316–329. [Google Scholar] [CrossRef]
- Chi, C.-Y.; Lin, C.-C.; Liao, I.-C.; Yao, Y.-C.; Shen, F.-C.; Liu, C.-C.; Lin, C.-F. Panton-Valentine Leukocidin facilitates the escape of Staphylococcus aureus from human keratinocyte endosomes and induces apoptosis. J. Infect. Dis. 2014, 209, 224–235. [Google Scholar] [CrossRef]
- Huitema, L.; Phillips, T.; Alexeev, V.; Tomic-Canic, M.; Pastar, I.; Igoucheva, O. Intracellular escape strategies of Staphylococcus aureus in persistent cutaneous infections. Exp. Dermatol. 2020, 30, 1428–1439. [Google Scholar] [CrossRef]
- Blättner, S.; Das, S.; Paprotka, K.; Eilers, U.; Krischke, M.; Kretschmer, D.; Remmele, C.W.; Dittrich, M.; Müller, T.; Schuelein-Voelk, C.; et al. Staphylococcus aureus exploits a non-ribosomal cyclic dipeptide to modulate survival within epithelial cells and phagocytes. PLoS Pathog. 2016, 12, e1005857. [Google Scholar] [CrossRef]
- Wang, R.; Braughton, K.R.; Kretschmer, D.; Bach, T.-H.L.; Queck, S.Y.; Li, M.; Kennedy, A.D.; Dorward, D.W.; Klebanoff, S.J.; Peschel, A.; et al. Identification of novel cytolytic peptides as key virulence determinants for community-associated MRSA. Nat. Med. 2007, 13, 1510–1514. [Google Scholar] [CrossRef]
- Chatterjee, S.S.; Joo, H.-S.; Duong, A.C.; Dieringer, T.D.; Tan, V.Y.; Song, Y.; Fischer, E.R.; Cheung, G.Y.; Li, M.; Otto, M. Essential Staphylococcus aureus toxin export system. Nat. Med. 2013, 19, 364–367. [Google Scholar] [CrossRef]
- Giese, B.; Dittmann, S.; Paprotka, K.; Levin, K.; Weltrowski, A.; Biehler, D.; Lâm, T.T.; Sinha, B.; Fraunholz, M.J. Staphylococcal alpha-toxin is not sufficient to mediate escape from phagolysosomes in upper-airway epithelial cells. Infect. Immun. 2009, 77, 3611–3625. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Münzenmayer, L.; Geiger, T.; Daiber, E.; Schulte, B.; Autenrieth, S.E.; Fraunholz, M.; Wolz, C. Influence of Sae-regulated and Agr-regulated factors on the escape of Staphylococcus aureus from human macrophages. Cell. Microbiol. 2016, 18, 1172–1183. [Google Scholar] [CrossRef] [PubMed]
- Truong-Bolduc, Q.C.; Khan, N.S.; Vyas, J.M.; Hooper, D.C. Tet38 efflux pump affects Staphylococcus aureus internalization by epithelial cells through interaction with CD36 and contributes to bacterial escape from acidic and nonacidic phagolysosomes. Infec. Immun. 2017, 85, e00862-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Deretic, V.; Saitoh, T.; Akira, S. Autophagy in infection, inflammation and immunity. Nat. Rev. Immunol. 2013, 13, 722–737. [Google Scholar] [CrossRef] [PubMed]
- Galluzzi, L.; Pietrocola, F.; Levine, B.; Kroemer, G. Metabolic control of Autophagy. Cell 2014, 159, 1263–1276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mostowy, S. Autophagy and bacterial clearance: A not so clear picture. Cell. Microbiol. 2012, 15, 395–402. [Google Scholar] [CrossRef] [Green Version]
- Fang, L.; Wu, H.-M.; Ding, P.-S.; Liu, R.-Y. TLR2 mediates phagocytosis and autophagy through JNK signaling pathway in Staphylococcus aureus-stimulated raw264.7 cells. Cell. Signal. 2014, 26, 806–814. [Google Scholar] [CrossRef]
- Neumann, Y.; Bruns, S.A.; Rohde, M.; Prajsnar, T.K.; Foster, S.J.; Schmitz, I. Intracellular Staphylococcus aureus eludes selective autophagy by activating a host cell kinase. Autophagy 2016, 12, 2069–2084. [Google Scholar] [CrossRef] [Green Version]
- O’Keeffe, K.M.; Wilk, M.M.; Leech, J.M.; Murphy, A.G.; Laabei, M.; Monk, I.R.; Massey, R.C.; Lindsay, J.A.; Foster, T.J.; Geoghegan, J.A.; et al. Manipulation of autophagy in phagocytes facilitates Staphylococcus aureus bloodstream infection. Infect. Immun. 2015, 83, 3445–3457. [Google Scholar] [CrossRef] [Green Version]
- Herb, M.; Gluschko, A.; Schramm, M. LC3-associated phagocytosis—the highway to hell for phagocytosed microbes. Semin. Cell Dev. Biol. 2020, 101, 68–76. [Google Scholar] [CrossRef]
- Prajsnar, T.K.; Serba, J.J.; Dekker, B.M.; Gibson, J.F.; Masud, S.; Fleming, A.; Johnston, S.A.; Renshaw, S.A.; Meijer, A.H. The autophagic response to Staphylococcus aureus provides an intracellular niche in neutrophils. Autophagy 2020, 17, 888–902. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mestre, M.B.; Fader, C.M.; Sola, C.; Colombo, M.I. A-hemolysin is required for the activation of the autophagic pathway in Staphylococcus aureus infected cells. Autophagy 2010, 6, 110–125. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schnaith, A.; Kashkar, H.; Leggio, S.A.; Addicks, K.; Krönke, M.; Krut, O. Staphylococcus aureus subvert autophagy for induction of caspase-independent host cell death. J. Biol. Chem. 2007, 282, 2695–2706. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ma, J.; Gulbins, E.; Edwards, M.J.; Caldwell, C.C.; Fraunholz, M.; Becker, K.A. Staphylococcus aureus alpha-toxin induces inflammatory cytokines via lysosomal acid sphingomyelinase and ceramides. Cell. Physiol. Biochem. 2017, 43, 2170–2184. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Becker, K.A.; Fahsel, B.; Kemper, H.; Mayeres, J.; Li, C.; Wilker, B.; Keitsch, S.; Soddemann, M.; Sehl, C.; Kohnen, M.; et al. Staphylococcus aureus alpha-toxin disrupts endothelial-cell tight junctions via acid sphingomyelinase and ceramide. Infect. Immun. 2018, 86, e00606–e00617. [Google Scholar] [CrossRef] [Green Version]
- Bronesky, D.; Wu, Z.; Marzi, S.; Walter, P.; Geissmann, T.; Moreau, K.; Vandenesch, F.; Caldelari, I.; Romby, P. Staphylococcus aureus RNAIII and its regulon link quorum sensing, stress responses, metabolic adaptation, and regulation of virulence gene expression. Annu. Rev. Microbiol. 2016, 70, 299–316. [Google Scholar] [CrossRef]
- Soong, G.; Paulino, F.; Wachtel, S.; Parker, D.; Wickersham, M.; Zhang, D.; Brown, A.; Lauren, C.; Dowd, M.; West, E.; et al. Methicillin-resistant Staphylococcus aureus adaptation to human keratinocytes. Mbio 2015, 6, e00289-15. [Google Scholar] [CrossRef] [Green Version]
- Mulcahy, M.E.; O’Brien, E.C.; O’Keeffe, K.M.; Vozza, E.G.; Leddy, N.; McLoughlin, R.M. Manipulation of autophagy and apoptosis facilitates intracellular survival of Staphylococcus aureus in human neutrophils. Front. Immunol. 2020, 11, 565545. [Google Scholar] [CrossRef]
- Bravo-Santano, N.; Ellis, J.K.; Mateos, L.M.; Calle, Y.; Keun, H.C.; Behrends, V.; Letek, M. Intracellular Staphylococcus aureus modulates host central carbon metabolism to activate autophagy. Msphere 2018, 3, e00374-18. [Google Scholar] [CrossRef] [Green Version]
- López de Armentia, M.M.; Gauron, M.C.; Colombo, M.I. Staphylococcus aureus alpha-toxin induces the formation of dynamic tubules labeled with LC3 within host cells in a Rab7 and rab1b-dependent manner. Front. Cell. Infect. Microbiol. 2017, 7, 431. [Google Scholar] [CrossRef]
- Ashida, H.; Suzuki, T.; Sasakawa, C. Shigella infection and host cell death: A double-edged sword for the host and pathogen survival. Curr. Opin. Microbiol. 2021, 59, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Jorgensen, I.; Rayamajhi, M.; Miao, E.A. Programmed cell death as a defence against infection. Nat. Rev. Immunol. 2017, 17, 151–164. [Google Scholar] [CrossRef] [PubMed]
- Missiakas, D.; Winstel, V. Selective host cell death by Staphylococcus aureus: A strategy for bacterial persistence. Front. Immunol. 2021, 11, 621733. [Google Scholar] [CrossRef] [PubMed]
- Watkins, K.E.; Unnikrishnan, M. Evasion of host defenses by intracellular Staphylococcus aureus. Adv. Appl. Microbiol. 2020, 112, 105–141. [Google Scholar] [CrossRef] [PubMed]
- Korea, C.G.; Balsamo, G.; Pezzicoli, A.; Merakou, C.; Tavarini, S.; Bagnoli, F.; Serruto, D.; Unnikrishnan, M. Staphylococcal ESX proteins modulate apoptosis and release of intracellular Staphylococcus aureus during infection in epithelial cells. Infect. Immun. 2014, 82, 4144–4153. [Google Scholar] [CrossRef] [Green Version]
- Kitur, K.; Parker, D.; Nieto, P.; Ahn, D.S.; Cohen, T.S.; Chung, S.; Wachtel, S.; Bueno, S.; Prince, A. Toxin-induced necroptosis is a major mechanism of Staphylococcus aureus lung damage. PLoS Pathog. 2015, 11, e1004820. [Google Scholar] [CrossRef]
- Kitur, K.; Wachtel, S.; Brown, A.; Wickersham, M.; Paulino, F.; Peñaloza, H.F.; Soong, G.; Bueno, S.; Parker, D.; Prince, A. Necroptosis promotes Staphylococcus aureus clearance by inhibiting excessive inflammatory signaling. Cell Rep. 2016, 16, 2219–2230. [Google Scholar] [CrossRef] [Green Version]
- Otto, M. Staphylococcus aureus toxins. Curr. Opin. Microbiol. 2014, 17, 32–37. [Google Scholar] [CrossRef] [Green Version]
- Al Kindi, A.; Alkahtani, A.M.; Nalubega, M.; El-Chami, C.; O’Neill, C.; Arkwright, P.D.; Pennock, J.L. Staphylococcus aureus internalized by skin keratinocytes evade antibiotic killing. Front. Microbiol. 2019, 10, 2242. [Google Scholar] [CrossRef]
- Stelzner, K.; Winkler, A.-C.; Liang, C.; Boyny, A.; Ade, C.P.; Dandekar, T.; Fraunholz, M.J.; Rudel, T. Intracellular Staphylococcus aureus perturbs the host cell Ca2+ homeostasis to promote cell death. Mbio 2020, 11, e02250-20. [Google Scholar] [CrossRef]
- DuMont, A.L.; Yoong, P.; Surewaard, B.G.; Benson, M.A.; Nijland, R.; van Strijp, J.A.; Torres, V.J. Staphylococcus aureus elaborates leukocidin ab to mediate escape from within human neutrophils. Infect. Immun. 2013, 81, 1830–1841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melehani, J.H.; James, D.B.; DuMont, A.L.; Torres, V.J.; Duncan, J.A. Staphylococcus aureus Leukocidin A/B (LukAB) kills human monocytes via host NLRP3 and ASC when extracellular, but not intracellular. PLoS Pathog. 2015, 11, e1004970. [Google Scholar] [CrossRef] [PubMed]
- Ventura, C.L.; Malachowa, N.; Hammer, C.H.; Nardone, G.A.; Robinson, M.A.; Kobayashi, S.D.; DeLeo, F.R. Identification of a novel Staphylococcus aureus two-component leukotoxin using cell surface proteomics. PLoS ONE 2010, 5, e11634. [Google Scholar] [CrossRef] [PubMed]
- Kobayashi, S.D.; Braughton, K.R.; Palazzolo-Ballance, A.M.; Kennedy, A.D.; Sampaio, E.; Kristosturyan, E.; Whitney, A.R.; Sturdevant, D.E.; Dorward, D.W.; Holland, S.M.; et al. Rapid neutrophil destruction following phagocytosis of Staphylococcus aureus. J. Innate Immun. 2010, 2, 560–575. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Koziel, J.; Maciag-Gudowska, A.; Mikolajczyk, T.; Bzowska, M.; Sturdevant, D.E.; Whitney, A.R.; Shaw, L.N.; DeLeo, F.R.; Potempa, J. Phagocytosis of Staphylococcus aureus by macrophages exerts cytoprotective effects manifested by the upregulation of antiapoptotic factors. PLoS ONE 2009, 4, e5210. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cruciani, M.; Etna, M.P.; Camilli, R.; Giacomini, E.; Percario, Z.A.; Severa, M.; Sandini, S.; Rizzo, F.; Brandi, V.; Balsamo, G.; et al. Staphylococcus aureus ESX factors control human dendritic cell functions conditioning th1/th17 response. Front. Cell. Infect. Microbiol. 2017, 7, 330. [Google Scholar] [CrossRef] [PubMed]
- Kerr, K.G.; Snelling, A.M. Pseudomonas aeruginosa: A formidable and ever-present adversary. J. Hosp. Infect. 2009, 73, 338–344. [Google Scholar] [CrossRef] [PubMed]
- Sause, W.E.; Buckley, P.T.; Strohl, W.R.; Lynch, A.S.; Torres, V.J. Antibody-based biologics and their promise to combat Staphylococcus aureus infections. Trends Pharm. Sci. 2016, 37, 231–241. [Google Scholar] [CrossRef] [Green Version]
- Wang-Lin, S.X.; Balthasar, J.P. Pharmacokinetic and pharmacodynamic considerations for the use of monoclonal antibodies in the treatment of bacterial infections. Antibodies 2018, 7, 5. [Google Scholar] [CrossRef] [Green Version]
- Yaeger, L.N.; Coles, V.E.; Chan, D.C.K.; Burrows, L.L. How to kill Pseudomonas—Emerging therapies for a challenging pathogen. Ann. N. Y. Acad. Sci. 2021, 1496, 59–81. [Google Scholar] [CrossRef]
- DiGiandomenico, A.; Keller, A.E.; Gao, C.; Rainey, G.J.; Warrener, P.; Camara, M.M.; Bonnell, J.; Fleming, R.; Bezabeh, B.; Dimasi, N.; et al. A multifunctional bispecific antibody protects against Pseudomonas aeruginosa. Sci. Transl. Med. 2014, 6, 262ra155. [Google Scholar] [CrossRef] [PubMed]
- AstraZeneca. Clinical Trials Appendix Q1 2020 Results Update. Available online: https://www.astrazeneca.com/content/dam/az/PDF/2020/q1-2020/Q1_2020_results_clinical_trials_appendix.pdf (accessed on 28 September 2021).
- Kajihara, K.K.; Pantua, H.; Hernandez-Barry, H.; Hazen, M.; Deshmukh, K.; Chiang, N.; Ohri, R.; Castellanos, E.R.; Martin, L.; Matsumoto, M.L.; et al. Potent killing of Pseudomonas aeruginosa by an antibody-antibiotic conjugate. mBio 2021, 12, e00202-21. [Google Scholar] [CrossRef] [PubMed]
- Evans, M.D.; Sammelson, R.; McDowell, S. Differential effects of cotreatment of the antibiotic rifampin with host-directed therapeutics in reducing intracellular Staphylococcus aureus infection. PeerJ 2020, 8, e10330. [Google Scholar] [CrossRef] [PubMed]
- Xiao, Y.; Zhang, T.-S.; Li, Y.-H.; Liu, C.-F.; Yang, S.-J.; Zeng, L.-T.; Huang, S.-H.; Deng, X.-Y.; Peng, L. Memantine promotes bactericidal effect of neutrophils against Infection with Pseudomonas aeruginosa and its drug-resistant strain, by improving reactive oxygen species generation. Microb. Drug Resist. 2022, 28, 7–17. [Google Scholar] [CrossRef]
- Bravo-Santano, N.; Stölting, H.; Cooper, F.; Bileckaja, N.; Majstorovic, A.; Ihle, N.; Mateos, L.M.; Calle, Y.; Behrends, V.; Letek, M. Host-directed kinase inhibitors act as novel therapies against intracellular Staphylococcus aureus. Sci. Rep. 2019, 9, 4876. [Google Scholar] [CrossRef]
- Bravo-Santano, N.; Capilla-Lasheras, P.; Mateos, L.M.; Calle, Y.; Behrends, V.; Letek, M. Identification of novel targets for host-directed therapeutics against intracellular Staphylococcus aureus. Sci. Rep. 2019, 9, 15435. [Google Scholar] [CrossRef] [Green Version]
- Alphonse, M.P.; Rubens, J.H.; Ortines, R.V.; Orlando, N.A.; Patel, A.M.; Dikeman, D.; Wang, Y.; Vuong, I.; Joyce, D.P.; Zhang, J.; et al. Pan-caspase inhibition as a potential host-directed immunotherapy against MRSA and other bacterial skin infections. Sci. Transl. Med. 2021, 13, eabe9887. [Google Scholar] [CrossRef]
- Poerio, N.; De Santis, F.; Rossi, A.; Ranucci, S.; De Fino, I.; Henriquez, A.; D’Andrea, M.M.; Ciciriello, F.; Lucidi, V.; Nisini, R.; et al. Liposomes loaded with phosphatidylinositol 5-phosphate improve the antimicrobial response to Pseudomonas aeruginosa in impaired macrophages from cystic fibrosis patients and limit airway inflammatory response. Front. Immunol. 2020, 11, 532225. [Google Scholar] [CrossRef]
- Roberts, S.C.; Zembower, T.R. Global increases in antibiotic consumption: A concerning trend for WHO targets. Lancet Infect. Dis. 2021, 21, 10–11. [Google Scholar] [CrossRef]
- Klein, E.Y.; Milkowska-Shibata, M.; Tseng, K.K.; Sharland, M.; Gandra, S.; Pulcini, C.; Laxminarayan, R. Assessment of WHO antibiotic consumption and access targets in 76 countries, 2000–2015: An analysis of pharmaceutical sales data. Lancet Infect. Dis. 2021, 21, 107–115. [Google Scholar] [CrossRef]
- National Health Commission of the People’s Republic of China. China Antimicrobial Resistance Surveillance System (in Chinese). Available online: http://www.carss.cn/ (accessed on 20 October 2021).
- Hu, F.; Guo, Y.; Zhu, D.; Wang, F.; Jiang, X.; Xu, Y. Antimicrobial resistance profile of clinical isolates in hospitals across China: Report from the CHINET surveillance program, 2017 (in Chinese). Chin. J. Infect. Chemother. 2018, 18, 241–251. [Google Scholar]
- Wang, J.; Zhou, M.; Huang, G.; Guo, Z.; Sauser, J.; Metsini, A.; Pittet, D.; Zingg, W. Antimicrobial resistance in southern China: Results of prospective surveillance in Dongguan city, 2017. J. Hosp. Infect. 2020, 105, 188–196. [Google Scholar] [CrossRef] [PubMed]
- Wang, J. The challenges of antimicrobial resistance surveillance in China. Am. J. Infect. Control 2019, 47, 1403–1404. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Taylor, M.W. Vaccines against viral infections. In Viruses and Man: A History of Interactions; Springer: Cham, Switzerland, 2014; pp. 355–377. [Google Scholar] [CrossRef]
- Jackson, L.A.; Anderson, E.J.; Rouphael, N.G.; Roberts, P.C.; Makhene, M.; Coler, R.N.; McCullough, M.P.; Chappell, J.D.; Denison, M.R.; Stevens, L.J.; et al. An mRNA vaccine against SARS-CoV-2—Preliminary report. N. Eng. J. Med. 2020, 383, 1920–1931. [Google Scholar] [CrossRef] [PubMed]
- Polack, F.P.; Thomas, S.J.; Kitchin, N.; Absalon, J.; Gurtman, A.; Lockhart, S.; Perez, J.L.; Pérez Marc, G.; Moreira, E.D.; Zerbini, C.; et al. Safety and efficacy of the BNT162b2 mRNA Covid-19 vaccine. N. Eng. J. Med. 2020, 383, 2603–2615. [Google Scholar] [CrossRef]
- Syed, K.A.; Saluja, T.; Cho, H.; Hsiao, A.; Shaikh, H.; Wartel, T.A.; Mogasale, V.; Lynch, J.; Kim, J.H.; Excler, J.L.; et al. Review on the recent advances on Typhoid vaccine development and challenges ahead. Clin. Infect. Dis. 2020, 71, S141–S150. [Google Scholar] [CrossRef]
- Lopez, A.L.; Gonzales, M.L.; Aldaba, J.G.; Nair, G.B. Killed oral cholera vaccines: History, development and implementation challenges. Ther. Adv. Vaccine 2014, 2, 123–136. [Google Scholar] [CrossRef] [Green Version]
- Cherry, J.D. The 112-year odyssey of pertussis and pertussis vaccines-mistakes made and implications for the future. J. Pediat. Infect. Dis Soc. 2019, 8, 334–341. [Google Scholar] [CrossRef]
- Dolstad, H.A.; Franke, M.F.; Vissieres, K.; Jerome, J.G.; Ternier, R.; Ivers, L.C. Factors associated with diarrheal disease among children aged 1-5 years in a cholera epidemic in rural Haiti. PLoS Negl. Trop. Dis. 2021, 15, e0009726. [Google Scholar] [CrossRef]
- Zwerling, A.; Behr, M.A.; Verma, A.; Brewer, T.F.; Menzies, D.; Pai, M. The BCG World Atlas: A database of global BCG vaccination policies and practices. PLoS Med. 2011, 8, e1001012. [Google Scholar] [CrossRef] [Green Version]
- Germanier, R.; Fürer, E. Characteristics of the attenuated oral vaccine strain “S. typhi” Ty 21a. Dev. Biol Stand. 1983, 53, 3–7. [Google Scholar] [PubMed]
- Xu, D.; Cisar, J.O.; Poly, F.; Yang, J.; Albanese, J.; Dharmasena, M.; Wai, T.; Guerry, P.; Kopecko, D.J. Genome sequence of Salmonella enterica serovar Typhi oral vaccine strain Ty21a. Gen. Announ. 2013, 1, e00650-13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, W.; Singh, A.K. Plague vaccine: Recent progress and prospects. NPJ Vaccine 2019, 4, 11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gupta, S.S.; Bharati, K.; Sur, D.; Khera, A.; Ganguly, N.K.; Nair, G.B. Why is the oral cholera vaccine not considered an option for prevention of cholera in India? Analysis of possible reasons. Ind. J. Med. Res. 2016, 143, 545–551. [Google Scholar] [CrossRef]
- Lundgren, A.; Bourgeois, L.; Carlin, N.; Clements, J.; Gustafsson, B.; Hartford, M.; Holmgren, J.; Petzold, M.; Walker, R.; Svennerholm, A.M. Safety and immunogenicity of an improved oral inactivated multivalent enterotoxigenic Escherichia coli (ETEC) vaccine administered alone and together with dmLT adjuvant in a double-blind, randomized, placebo-controlled Phase I study. Vaccine 2014, 32, 7077–7084. [Google Scholar] [CrossRef] [Green Version]
- Chakraborty, S.; Harro, C.; DeNearing, B.; Bream, J.; Bauers, N.; Dally, L.; Flores, J.; Van de Verg, L.; Sack, D.A.; Walker, R. Evaluation of the safety, tolerability, and immunogenicity of an oral, inactivated whole-cell Shigella flexneri 2a vaccine in healthy adult subjects. Clin. Vaccine Immunol. 2016, 23, 315–325. [Google Scholar] [CrossRef] [Green Version]
- Plotkin, S.A.; Orenstein, W.A.; Offit, P.A. (Eds.) Vaccines, 6th ed.; Elsevier: Philadelphia, PA, USA, 2013. [Google Scholar]
- Bonner, K.; Welch, E.; Elder, K.; Cohn, J. Impact of pneumococcal conjugate vaccine administration in pediatric older age groups in low and middle income countries: A systematic review. PLoS ONE 2015, 10, e0135270. [Google Scholar] [CrossRef]
- Thumburu, K.K.; Singh, M.; Das, R.R.; Jaiswal, N.; Agarwal, A.; Kumar, A.; Kaur, H. Two or three primary dose regime for Haemophilus influenzae type b conjugate vaccine: Meta-analysis of randomized controlled trials. Ther. Adv. Vaccine 2015, 3, 31–40. [Google Scholar] [CrossRef] [Green Version]
- Henriques-Normark, B.; Normark, S. Bacterial vaccines and antibiotic resistance. Ups J. Med. Sci. 2014, 119, 205–208. [Google Scholar] [CrossRef]
- Priebe, G.P.; Goldberg, J.B. Vaccines for Pseudomonas aeruginosa: A long and winding road. Expert Rev. Vaccine 2014, 13, 507–519. [Google Scholar] [CrossRef] [Green Version]
- López-Siles, M.; Corral-Lugo, A.; McConnell, M.J. Vaccines for multidrug resistant Gram negative bacteria: Lessons from the past for guiding future success. FEMS. Microbiol. Rev. 2021, 45, fuaa054. [Google Scholar] [CrossRef] [PubMed]
- McIntosh, E. Development of vaccines against the sexually transmitted infections gonorrhoea, syphilis, Chlamydia, herpes simplex virus, human immunodeficiency virus and Zika virus. Ther. Adv. Vaccine Immunother. 2020, 8, 2515135520923887. [Google Scholar] [CrossRef] [PubMed]
- Baker, S.; Thomson, N.; Weill, F.X.; Holt, K.E. Genomic insights into the emergence and spread of antimicrobial-resistant bacterial pathogens. Science 2018, 360, 733–738. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sharma, N.; Singh, V.; Shyma, K.P. Role of parasitic vaccines in integrated control of parasitic diseases in livestock. Vet. World 2015, 8, 590–598. [Google Scholar] [CrossRef] [PubMed]
- Oliveira, L.; Wang, R.; Specht, C.A.; Levitz, S.M. Vaccines for human fungal diseases: Close but still a long way to go. NPJ Vaccine 2021, 6, 33. [Google Scholar] [CrossRef] [PubMed]
- Sacks, D.L. Vaccines against tropical parasitic diseases: A persisting answer to a persisting problem. Nat. Immunol. 2014, 15, 403–405. [Google Scholar] [CrossRef]
- World Health Organization. Sixty-Eighth World Health Assembly (WHA68.7); Agenda Item 15.1, Global Action Plan on Antimicrobial Resistance; World Health Organization: Geneva, Switzerland, 2015. [Google Scholar]
- Mulani, M.S.; Kamble, E.E.; Kumkar, S.N.; Tawre, M.S.; Pardesi, K.R. Emerging strategies to combat ESKAPE pathogens in the era of antimicrobial resistance: A review. Front. Microbiol. 2019, 10, 539. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
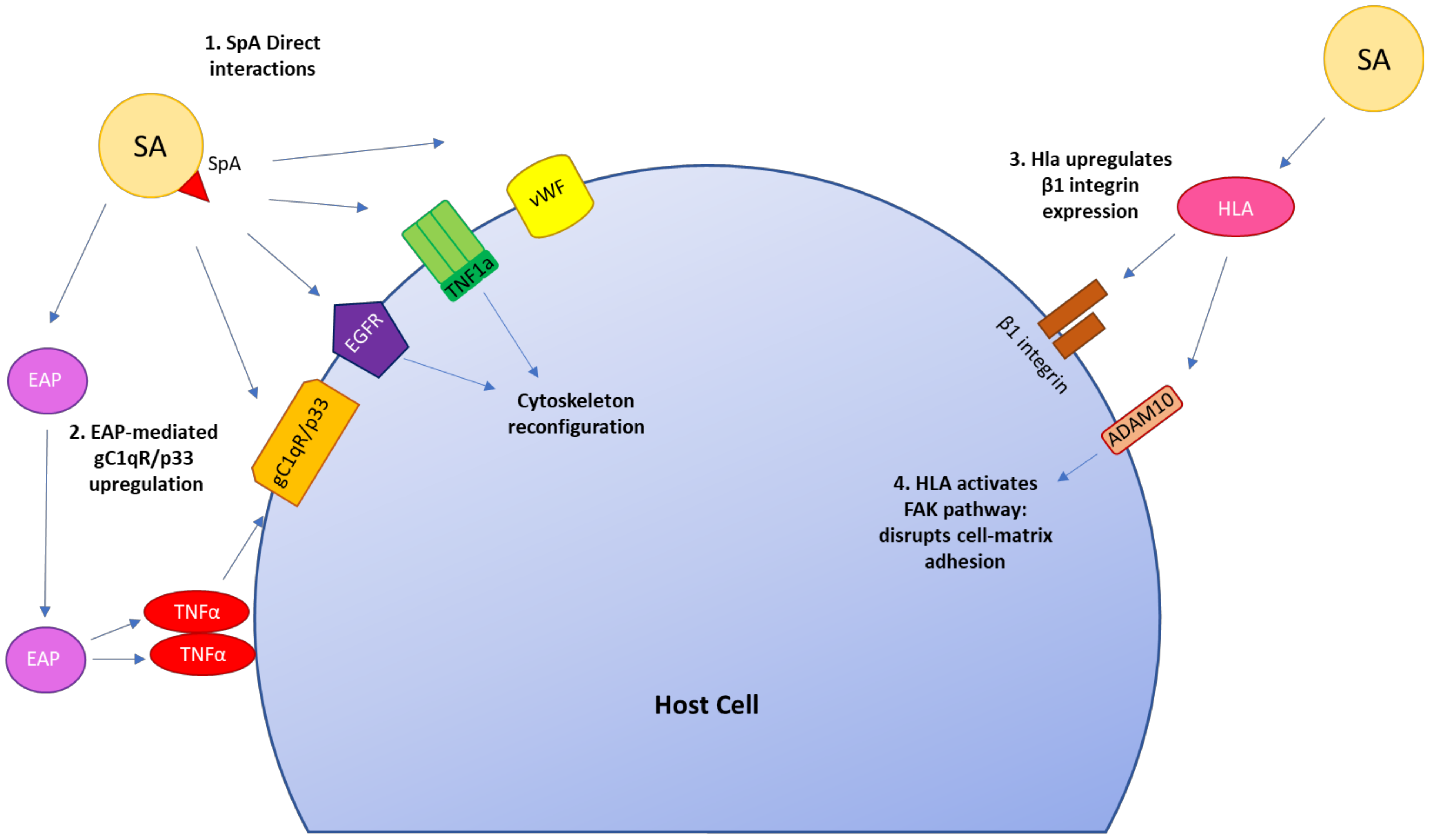{kind=link}
| Antibiotic Class | Mode of Action | Host Cell Permeable [25,26,27] |
|---|---|---|
| Aminoglycosides | Inhibit protein synthesis | Yes, some antibiotics in this class enter host cells via endocytosis |
| Ansamycins | Inhibit RNA synthesis | Yes, rifamycin enters via passive uptake (diffusion) |
| β-Lactams | Inhibit cell wall synthesis | Yes, small molecules via diffusion, larger molecules possibly via endocytosis |
| Chloramphenicol | Inhibits protein synthesis | No, requires modification for enhanced entry into host cells |
| Glycopeptides | Inhibit cell wall synthesis | No, requires modification for enhanced entry into host cells |
| Lipopeptides | Disrupt cell membrane functions | No/Unknown |
| Macrolides | Inhibit protein synthesis | Yes, diffusion and partly active uptake |
| Oxazolidinones | Inhibit protein synthesis | Yes, passive uptake |
| Quinolones | Interfere with bacterial DNA replication | Yes, active and passive cellular uptake, depending on the quinolone |
| Streptogramins | Inhibit protein synthesis | No/Unknown |
| Sulfonamides | Inhibit folic acid synthesis | Yes, active uptake |
| Tetracyclines | Inhibit protein synthesis | Yes, active uptake |
| Strain | ExoS | ExoU |
|---|---|---|
| PAO1 | + | − |
| CF18 | + | − |
| CF27 | + | − |
| PAK | + | − |
| JJ692 | − | + |
| E2 | + | − |
| MSH10 | + | − |
| X13273 | − | + |
| S. aureus Component | Host Component | Bridge | Host Cell Type | References |
|---|---|---|---|---|
| Atl | Heat shock cognate protein 70 | Keratinocytes | [87] | |
| Endothelial cells | [88] | |||
| ClfA | αvβ3 integrins | Fibrinogen | None reported | [89] |
| Vascular endothelial cells | [90] | |||
| Annexin A2 | MAC-T cell | [91] | ||
| Von Willebrand Factor | Von Willebrand binding protein | Endothelial cells | [92,93,94] | |
| ClfB | Plasma fibrinogen | [95] | ||
| Cytokeratin 10 | Desquamated epithelial cells | [96] | ||
| Loricrin | Squamous epithelial cells | [97,98] | ||
| IsdB | β3-containing integrins | Extracellular matrix Vitronectin | HEK-293T, HeLa | [99] |
| αvβ3 integrins | Epithelial/endothelial cells | [100] | ||
| Lpl | Hsp90 | Keratinocytes | [101,102] | |
| SraP | gp340 | A549 cells | [103] | |
| SdrD | Desmoglein 1 | Keratinocytes | [104] | |
| Desquamated nasal cells | [105] |
| S. aureus Component | Gene Prevalence in the Investigated S. aureus Isolates’ Genome | References |
|---|---|---|
| Atl | 100% | [119,120,121,122] |
| ClfA | 100% | [118,119] |
| 87% | [120] | |
| 82% | [121] | |
| 70.4% | [123] | |
| ClfB | 100% | [119,121] |
| 98% | [120] | |
| Eap | 100% | [119,124] |
| 99% | [120] | |
| 45% | [121] | |
| FnBPA | 100% | [119,120] |
| 99% | [121] | |
| FnBPB | 100% | [119] |
| 73% | [121] | |
| 44% | [120] | |
| HLA | 100% | [120,121,125] |
| 96% | [117] | |
| 91.9% | [123] | |
| 90.3% | [118] | |
| IsdB | 97% | [121] |
| 94% | [120] | |
| SpA | 100% | [120,121] |
| 90% | [117] | |
| SraP | 100% | [121] |
| 43% | [120] | |
| SdrD | 91% | [121] |
| 40% | [119] | |
| 36% | [120] |
| Disease(s) | Bacterial Pathogen | Vaccine Type |
|---|---|---|
| diphtheria | Corynebacterium diphtheriae | toxoid |
| tetanus | Clostridium tetani | toxoid |
| whooping cough | Bordetella pertussis | subunit or inactivated |
| meningitis/pneumonia | Haemophilus influenzae | conjugated |
| meningitis/pneumonia | Streptococcus pneumoniae | subunit or conjugated |
| meningitis | Neisseria meningitidis | subunit or conjugated |
| typhoid fever | Salmonella typhi | attenuated or subunit |
| cholera | Vibrio cholerae | inactivated |
| plague | Yersinia pestis | inactivated |
| anthrax | Bacillus anthracis | subunit |
| tuberculosis | Mycobacterium tuberculosis | attenuated |
| tularemia | Francisella tularensis | attenuated |
| typhus | Rickettsia prowazekii | inactivated |
| Q fever | Coxiella burnetii | inactivated |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kember, M.; Grandy, S.; Raudonis, R.; Cheng, Z. Non-Canonical Host Intracellular Niche Links to New Antimicrobial Resistance Mechanism. Pathogens 2022, 11, 220. https://doi.org/10.3390/pathogens11020220
Kember M, Grandy S, Raudonis R, Cheng Z. Non-Canonical Host Intracellular Niche Links to New Antimicrobial Resistance Mechanism. Pathogens. 2022; 11(2):220. https://doi.org/10.3390/pathogens11020220
Chicago/Turabian StyleKember, Michaela, Shannen Grandy, Renee Raudonis, and Zhenyu Cheng. 2022. "Non-Canonical Host Intracellular Niche Links to New Antimicrobial Resistance Mechanism" Pathogens 11, no. 2: 220. https://doi.org/10.3390/pathogens11020220
APA StyleKember, M., Grandy, S., Raudonis, R., & Cheng, Z. (2022). Non-Canonical Host Intracellular Niche Links to New Antimicrobial Resistance Mechanism. Pathogens, 11(2), 220. https://doi.org/10.3390/pathogens11020220