
Potent Bile Acid Microbial Metabolites Modulate Clostridium perfringens Virulence


Abstract
:1. Introduction
2. Materials and Methods
2.1. Strains, Media, Growth Condition, and Mariner-Based Transposon Mutagenesis
2.2. Bile Acid Inhibition of C. perfringens Growth Assay
2.3. Bacterial RNA Extraction and Gene Expression Using Real-Time PCR
2.4. H2S Detection Using Lead Acetate Assay
2.5. Statistical Analysis
3. Results
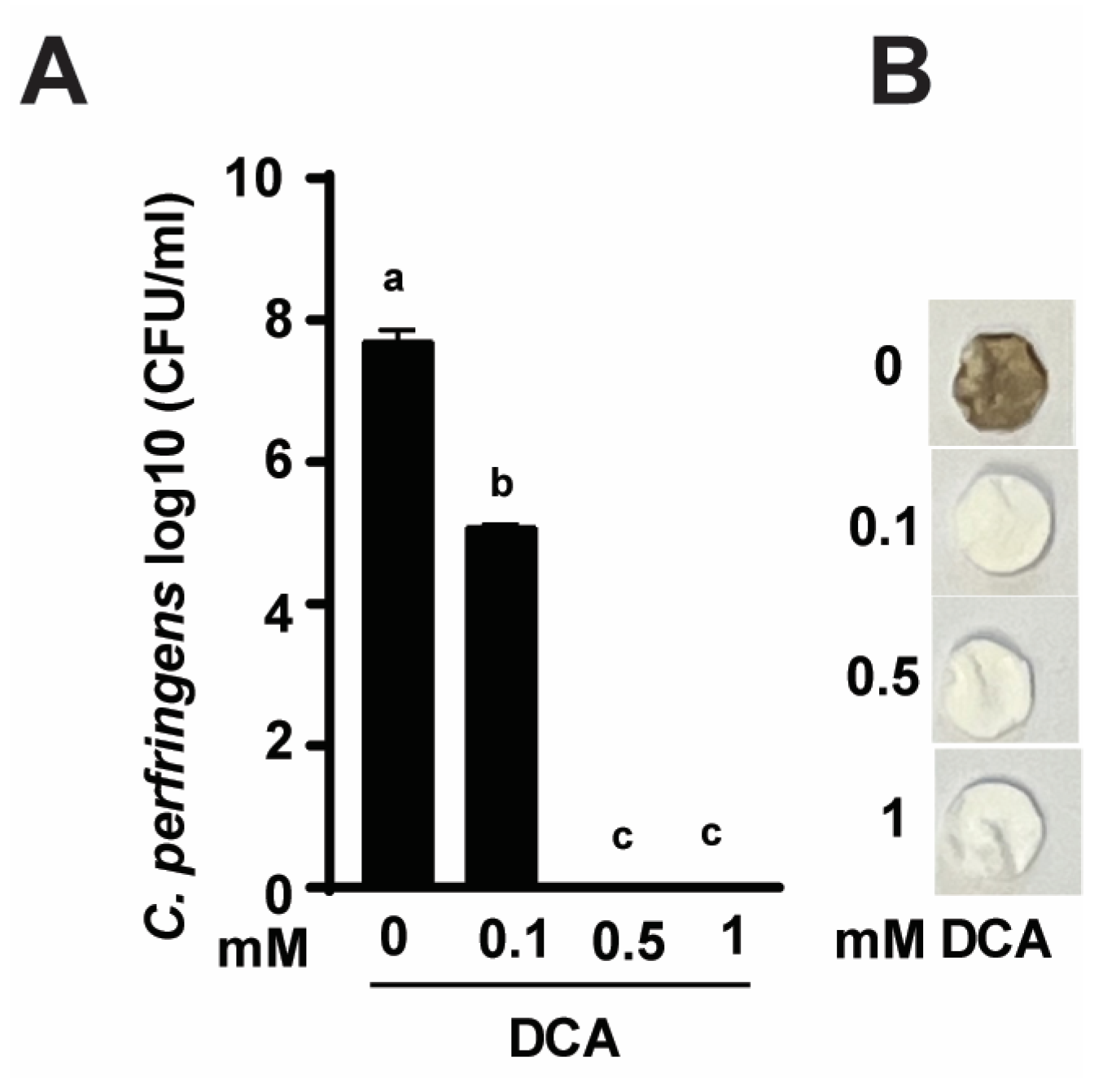
3.1. DCA Reduces C. perfringens Strain CP1 Growth and H2S Production
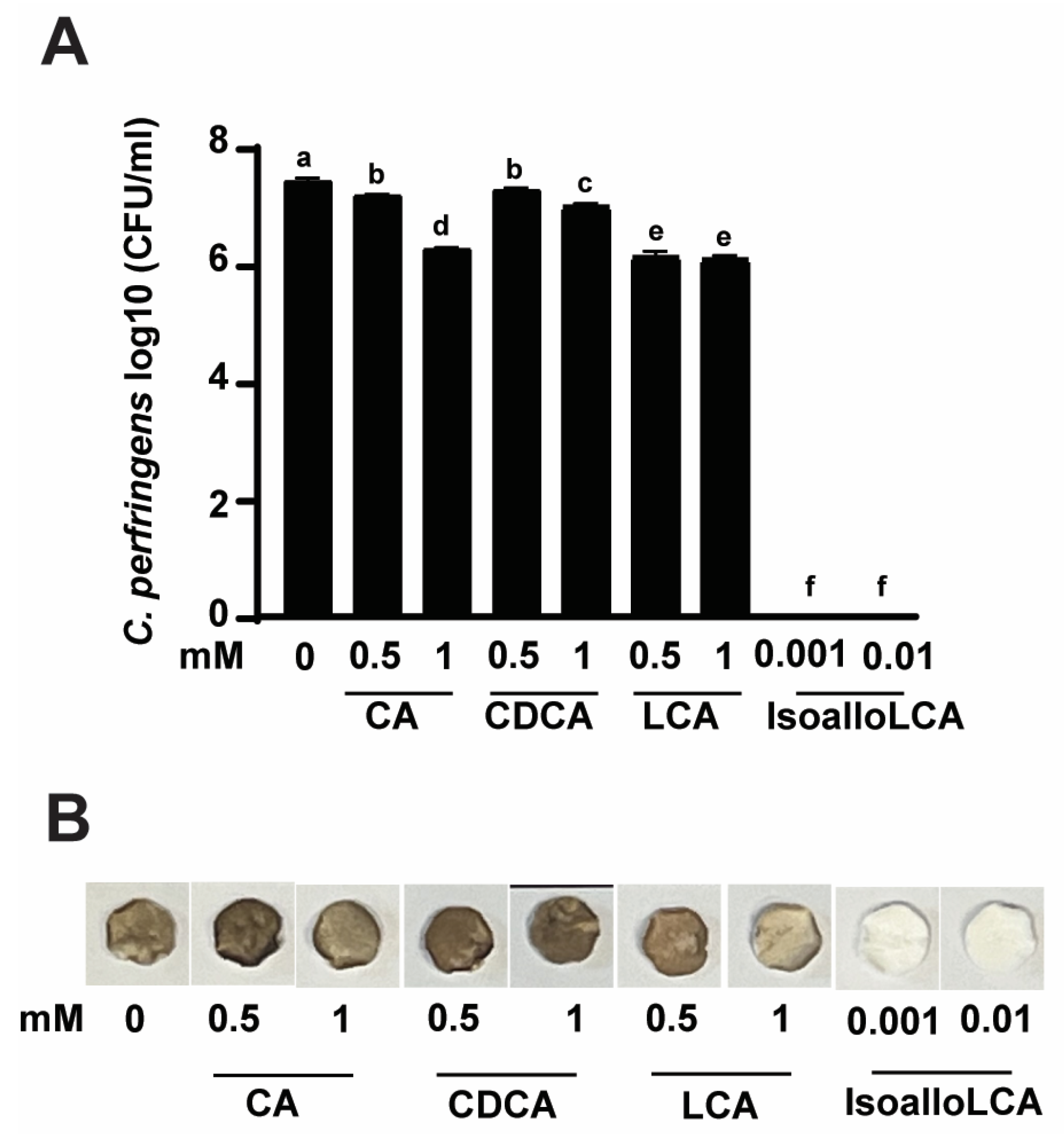
3.2. IsoalloLCA Reduces C. perfringens CP1 Growth and H2S Production More Effectively Compared to Other Bile Acids
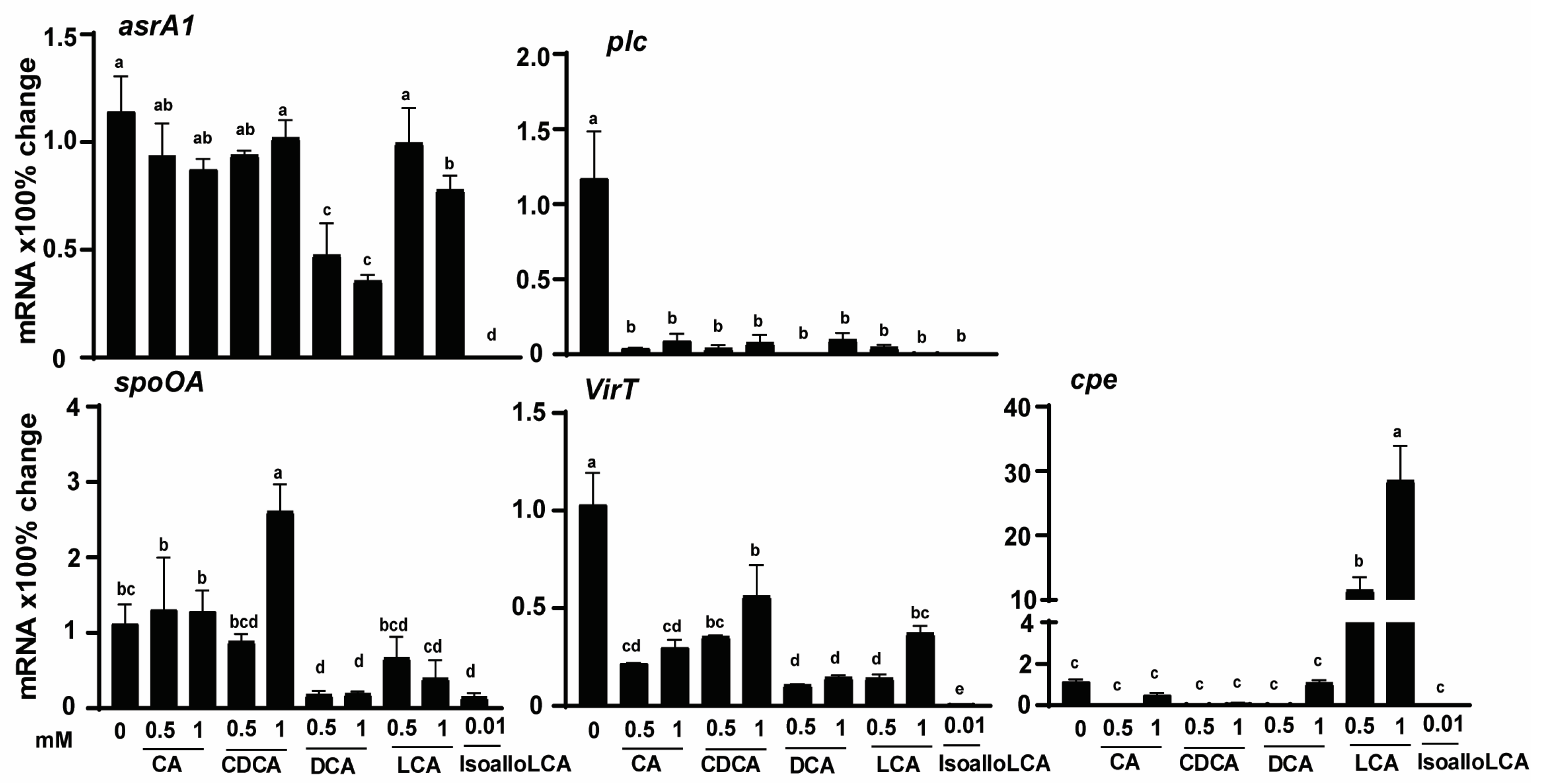
3.3. Bile Acids Reduce C. perfringens CP1 Virulence Gene Expression
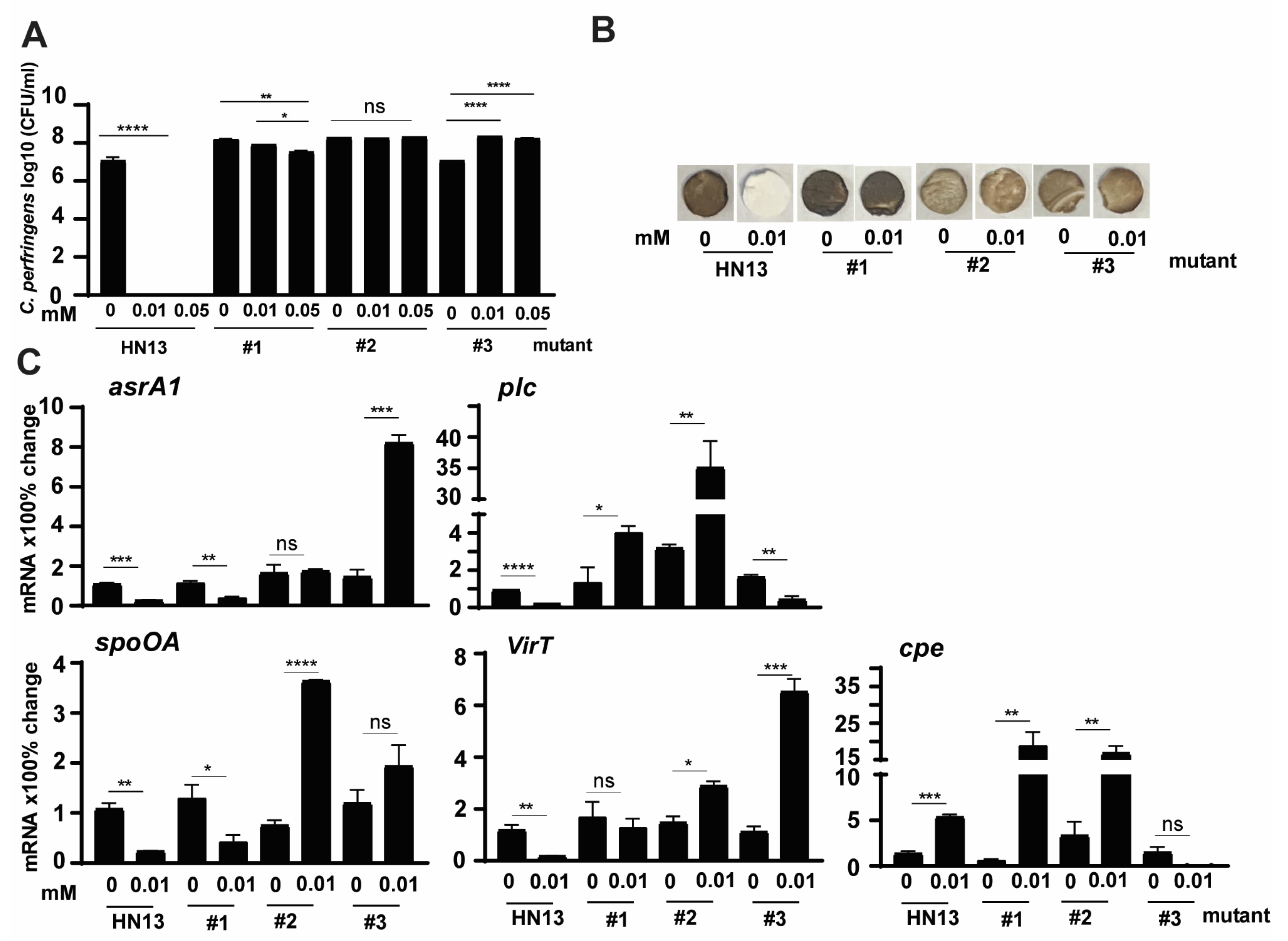
3.4. C. perfringens HN13 Mutants Produce H2S in the Presence of DCA
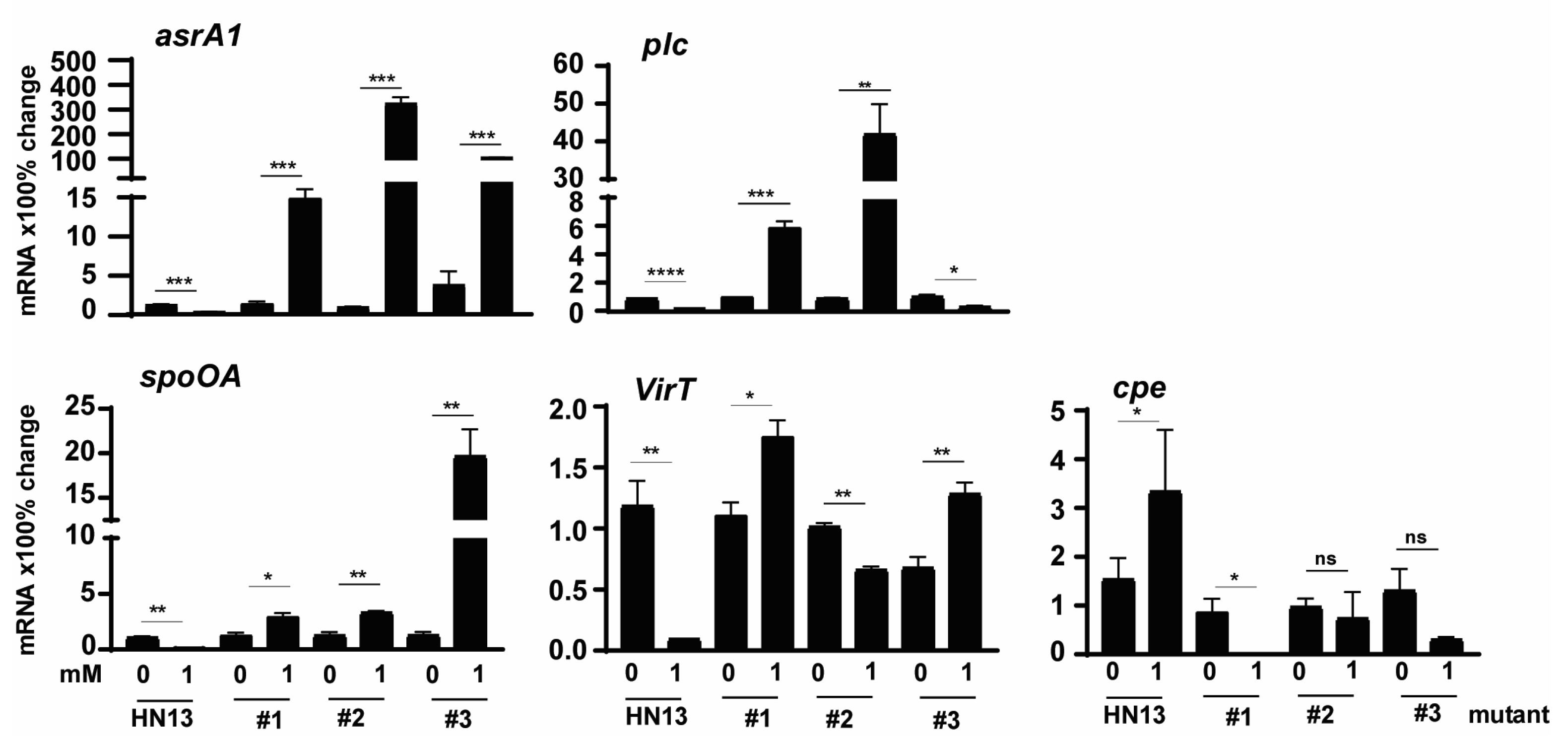
3.5. C. perfringens HN13 Mutants Express Virulence Genes in the Presence of DCA
3.6. Effects of IsoalloLCA on Growth and Virulence of C. perfringens HN13 Mutants
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Fu, Y.; Alenezi, T.; Sun, X. Clostridium perfringens-Induced Necrotic Diseases: An Overview. Immuno 2022, 2, 387–407. [Google Scholar] [CrossRef]
- Rood, J.I.; Keyburn, A.L.; Moore, R.J. NetB and necrotic enteritis: The hole movable story. Avian Pathol. 2016, 45, 295–301. [Google Scholar] [CrossRef] [PubMed]
- Kaldhusdal, M.; Benestad, S.L.; Lovland, A. Epidemiologic aspects of necrotic enteritis in broiler chickens - disease occurrence and production performance. Avian Pathol. 2016, 45, 271–274. [Google Scholar] [CrossRef] [PubMed]
- Williams, R.B. Intercurrent coccidiosis and necrotic enteritis of chickens: Rational, integrated disease management by maintenance of gut integrity. Avian Pathol. 2005, 34, 159–180. [Google Scholar] [CrossRef]
- Latorre, J.D.; Adhikari, B.; Park, S.H.; Teague, K.D.; Graham, L.E.; Mahaffey, B.D.; Baxter, M.F.A.; Hernandez-Velasco, X.; Kwon, Y.M.; Ricke, S.C.; et al. Evaluation of the Epithelial Barrier Function and Ileal Microbiome in an Established Necrotic Enteritis Challenge Model in Broiler Chickens. Front. Vet. Sci. 2018, 5, 199. [Google Scholar] [CrossRef]
- Rood, J.I.; Adams, V.; Lacey, J.; Lyras, D.; McClane, B.A.; Melville, S.B.; Moore, R.J.; Popoff, M.R.; Sarker, M.R.; Songer, J.G.; et al. Expansion of the Clostridium perfringens toxin-based typing scheme. Anaerobe 2018, 53, 5–10. [Google Scholar] [CrossRef]
- Fernandes da Costa, S.P.; Mot, D.; Bokori-Brown, M.; Savva, C.G.; Basak, A.K.; Van Immerseel, F.; Titball, R.W. Protection against avian necrotic enteritis after immunisation with NetB genetic or formaldehyde toxoids. Vaccine 2013, 31, 4003–4008. [Google Scholar] [CrossRef]
- Titball, R.W.; Naylor, C.E.; Basak, A.K. The Clostridium perfringens alpha-toxin. Anaerobe 1999, 5, 51–64. [Google Scholar] [CrossRef]
- Marius, N.; Manoilescu, I.; Velnic, A.-A.; Ioan, B. Gas Gangrene: Case Presentation and Literature Data. Jurnalul Chir. 2017, 13. [Google Scholar] [CrossRef]
- Hickey, M.J.; Kwan, R.Y.; Awad, M.M.; Kennedy, C.L.; Young, L.F.; Hall, P.; Cordner, L.M.; Lyras, D.; Emmins, J.J.; Rood, J.I. Molecular and cellular basis of microvascular perfusion deficits induced by Clostridium perfringens and Clostridium septicum. PLoS Pathog. 2008, 4, e1000045. [Google Scholar] [CrossRef]
- Ba-Thein, W.; Lyristis, M.; Ohtani, K.; Nisbet, I.T.; Hayashi, H.; Rood, J.I.; Shimizu, T. The virR/virS locus regulates the transcription of genes encoding extracellular toxin production in Clostridium perfringens. J. Bacteriol. 1996, 178, 2514–2520. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, T.; Yaguchi, H.; Ohtani, K.; Banu, S.; Hayashi, H. Clostridial VirR/VirS regulon involves a regulatory RNA molecule for expression of toxins. Mol. Microbiol. 2002, 43, 257–265. [Google Scholar] [CrossRef]
- Hiscox, T.J.; Chakravorty, A.; Choo, J.M.; Ohtani, K.; Shimizu, T.; Cheung, J.K.; Rood, J.I. Regulation of virulence by the RevR response regulator in Clostridium perfringens. Infect. Immun. 2011, 79, 2145–2153. [Google Scholar] [CrossRef] [PubMed]
- Okumura, K.; Ohtani, K.; Hayashi, H.; Shimizu, T. Characterization of genes regulated directly by the VirR/VirS system in Clostridium perfringens. J. Bacteriol. 2008, 190, 7719–7727. [Google Scholar] [CrossRef] [PubMed]
- Czeczulin, J.R.; Hanna, P.C.; McClane, B.A. Cloning, nucleotide sequencing, and expression of the Clostridium perfringens enterotoxin gene in Escherichia coli. Infect. Immun. 1993, 61, 3429–3439. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Bansal, M.; Alenezi, T.; Almansour, A.; Wang, H.; Sun, X. Vaccines Using Clostridium perfringens Sporulation Proteins Reduce Necrotic Enteritis in Chickens. Microorganisms 2022, 10, 1110. [Google Scholar] [CrossRef] [PubMed]
- Abdel-Glil, M. In Silico Genome Analysis and Molecular Typing of Clostridium Perfringens. Doctoral Dissertation, Mensch und Buch Verlag, Berlin, Germany, 2019. [Google Scholar]
- Xiao, Y. Clostridium perfringens Sporulation, Germination and Outgrowth in Food: A Functional Genomics Approach; Wageningen University and Research: Wageningen, The Netherlands, 2014. [Google Scholar]
- Ohtani, K.; Hirakawa, H.; Paredes-Sabja, D.; Tashiro, K.; Kuhara, S.; Sarker, M.R.; Shimizu, T. Unique regulatory mechanism of sporulation and enterotoxin production in Clostridium perfringens. J. Bacteriol. 2013, 195, 2931–2936. [Google Scholar] [CrossRef]
- Mi, E.; Li, J.; McClane, B.A. NanR Regulates Sporulation and Enterotoxin Production by Clostridium perfringens Type F Strain F4969. Infect. Immun. 2018, 86, 10–1128. [Google Scholar] [CrossRef]
- Ethapa, T.; Leuzzi, R.; Ng, Y.K.; Baban, S.T.; Adamo, R.; Kuehne, S.A.; Scarselli, M.; Minton, N.P.; Serruto, D.; Unnikrishnan, M. Multiple factors modulate biofilm formation by the anaerobic pathogen Clostridium difficile. J. Bacteriol. 2013, 195, 545–555. [Google Scholar] [CrossRef]
- Hamon, M.A.; Lazazzera, B.A. The sporulation transcription factor Spo0A is required for biofilm development in Bacillus subtilis. Mol. Microbiol. 2001, 42, 1199–1209. [Google Scholar] [CrossRef]
- Hu, W.S.; Woo, D.U.; Kang, Y.J.; Koo, O.K. Biofilm and Spore Formation of Clostridium perfringens and Its Resistance to Disinfectant and Oxidative Stress. Antibiotics 2021, 10, 396. [Google Scholar] [CrossRef]
- Linden, D.R. Hydrogen sulfide signaling in the gastrointestinal tract. Antioxid. Redox Signal 2014, 20, 818–830. [Google Scholar] [CrossRef] [PubMed]
- Yang, G.; Sun, X.; Wang, R. Hydrogen sulfide-induced apoptosis of human aorta smooth muscle cells via the activation of mitogen-activated protein kinases and caspase-3. FASEB J. 2004, 18, 1782–1784. [Google Scholar] [CrossRef] [PubMed]
- Yurinskaya, M.M.; Krasnov, G.S.; Kulikova, D.A.; Zatsepina, O.G.; Vinokurov, M.G.; Chuvakova, L.N.; Rezvykh, A.P.; Funikov, S.Y.; Morozov, A.V.; Evgen’ev, M.B. H(2)S counteracts proinflammatory effects of LPS through modulation of multiple pathways in human cells. Inflamm. Res. 2020, 69, 481–495. [Google Scholar] [CrossRef] [PubMed]
- Xavier, K.B.; Bassler, B.L. LuxS quorum sensing: More than just a numbers game. Curr. Opin. Microbiol. 2003, 6, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Andre, G.; Haudecoeur, E.; Monot, M.; Ohtani, K.; Shimizu, T.; Dupuy, B.; Martin-Verstraete, I. Global regulation of gene expression in response to cysteine availability in Clostridium perfringens. BMC Microbiol. 2010, 10, 234. [Google Scholar] [CrossRef] [PubMed]
- Funabashi, M.; Grove, T.L.; Wang, M.; Varma, Y.; McFadden, M.E.; Brown, L.C.; Guo, C.; Higginbottom, S.; Almo, S.C.; Fischbach, M.A. A metabolic pathway for bile acid dehydroxylation by the gut microbiome. Nature 2020, 582, 566–570. [Google Scholar] [CrossRef]
- Sato, Y.; Atarashi, K.; Plichta, D.R.; Arai, Y.; Sasajima, S.; Kearney, S.M.; Suda, W.; Takeshita, K.; Sasaki, T.; Okamoto, S.; et al. Novel bile acid biosynthetic pathways are enriched in the microbiome of centenarians. Nature 2021, 599, 458–464. [Google Scholar] [CrossRef]
- Maruyama, T.; Miyamoto, Y.; Nakamura, T.; Tamai, Y.; Okada, H.; Sugiyama, E.; Nakamura, T.; Itadani, H.; Tanaka, K. Identification of membrane-type receptor for bile acids (M-BAR). Biochem. Biophys. Res. Commun. 2002, 298, 714–719. [Google Scholar] [CrossRef]
- Kawamata, Y.; Fujii, R.; Hosoya, M.; Harada, M.; Yoshida, H.; Miwa, M.; Fukusumi, S.; Habata, Y.; Itoh, T.; Shintani, Y.; et al. A G protein-coupled receptor responsive to bile acids. J. Biol. Chem. 2003, 278, 9435–9440. [Google Scholar] [CrossRef]
- Kumar, D.P.; Asgharpour, A.; Mirshahi, F.; Park, S.H.; Liu, S.; Imai, Y.; Nadler, J.L.; Grider, J.R.; Murthy, K.S.; Sanyal, A.J. Activation of Transmembrane Bile Acid Receptor TGR5 Modulates Pancreatic Islet alpha Cells to Promote Glucose Homeostasis. J. Biol. Chem. 2016, 291, 6626–6640. [Google Scholar] [CrossRef] [PubMed]
- Rajagopal, S.; Kumar, D.P.; Mahavadi, S.; Bhattacharya, S.; Zhou, R.; Corvera, C.U.; Bunnett, N.W.; Grider, J.R.; Murthy, K.S. Activation of G protein-coupled bile acid receptor, TGR5, induces smooth muscle relaxation via both Epac- and PKA-mediated inhibition of RhoA/Rho kinase pathway. Am. J. Physiol. -Gastrointest. Liver Physiol. 2013, 304, G527–G535. [Google Scholar] [CrossRef] [PubMed]
- Lew, J.L.; Zhao, A.; Yu, J.; Huang, L.; De Pedro, N.; Pelaez, F.; Wright, S.D.; Cui, J. The farnesoid X receptor controls gene expression in a ligand- and promoter-selective fashion. J. Biol. Chem. 2004, 279, 8856–8861. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Lo, J.L.; Huang, L.; Zhao, A.; Metzger, E.; Adams, A.; Meinke, P.T.; Wright, S.D.; Cui, J. Lithocholic acid decreases expression of bile salt export pump through farnesoid X receptor antagonist activity. J. Biol. Chem. 2002, 277, 31441–31447. [Google Scholar] [CrossRef]
- Oberkampf, M.; Hamiot, A.; Altamirano-Silva, P.; Belles-Sancho, P.; Tremblay, Y.D.N.; DiBenedetto, N.; Seifert, R.; Soutourina, O.; Bry, L.; Dupuy, B.; et al. c-di-AMP signaling is required for bile salt resistance, osmotolerance, and long-term host colonization by Clostridioides difficile. Sci. Signal. 2022, 15, eabn8171. [Google Scholar] [CrossRef]
- Francis, M.B.; Allen, C.A.; Shrestha, R.; Sorg, J.A. Bile acid recognition by the Clostridium difficile germinant receptor, CspC, is important for establishing infection. PLoS Pathog. 2013, 9, e1003356. [Google Scholar] [CrossRef]
- Shen, A. Clostridioides difficile Spore Formation and Germination: New Insights and Opportunities for Intervention. Annu. Rev. Microbiol. 2020, 74, 545–566. [Google Scholar] [CrossRef]
- Gopal-Srivastava, R.; Hylemon, P.B. Purification and characterization of bile salt hydrolase from Clostridium perfringens. J. Lipid Res. 1988, 29, 1079–1085. [Google Scholar] [CrossRef]
- Macdonald, I.A.; Hutchison, D.M.; Forrest, T.P.; Bokkenheuser, V.D.; Winter, J.; Holdeman, L.V. Metabolism of primary bile acids by Clostridium perfringens. J. Steroid Biochem. 1983, 18, 97–104. [Google Scholar] [CrossRef]
- Hickey, C.S.; Johnson, M.G. Effects of pH shifts, bile salts, and glucose on sporulation of Clostridium perfringens NCTC 8798. Appl. Environ. Microbiol. 1981, 41, 124–129. [Google Scholar] [CrossRef]
- Heredia, N.L.; Labbe, R.G.; Rodriguez, M.A.; Garcia-Alvarado, J.S. Growth, sporulation and enterotoxin production by Clostridium perfringens type A in the presence of human bile salts. FEMS Microbiol. Lett. 1991, 68, 15–21. [Google Scholar] [CrossRef]
- Yasugi, M.; Okuzaki, D.; Kuwana, R.; Takamatsu, H.; Fujita, M.; Sarker, M.R.; Miyake, M. Transcriptional Profile during Deoxycholate-Induced Sporulation in a Clostridium perfringens Isolate Causing Foodborne Illness. Appl. Environ. Microbiol. 2016, 82, 2929–2942. [Google Scholar] [CrossRef]
- Wang, H.; Latorre, J.D.; Bansal, M.; Abraha, M.; Al-Rubaye, B.; Tellez-Isaias, G.; Hargis, B.; Sun, X. Microbial metabolite deoxycholic acid controls Clostridium perfringens-induced chicken necrotic enteritis through attenuating inflammatory cyclooxygenase signaling. Sci. Rep. 2019, 9, 14541. [Google Scholar] [CrossRef]
- Bansal, M.; Alenezi, T.; Fu, Y.; Almansour, A.; Wang, H.; Gupta, A.; Liyanage, R.; Graham, D.B.; Hargis, B.M.; Sun, X. Specific Secondary Bile Acids Control Chicken Necrotic Enteritis. Pathogens 2021, 10, 1041. [Google Scholar] [CrossRef] [PubMed]
- Nariya, H.; Miyata, S.; Suzuki, M.; Tamai, E.; Okabe, A. Development and application of a method for counterselectable in-frame deletion in Clostridium perfringens. Appl. Environ. Microbiol. 2011, 77, 1375–1382. [Google Scholar] [CrossRef] [PubMed]
- Liu, H.; Bouillaut, L.; Sonenshein, A.L.; Melville, S.B. Use of a mariner-based transposon mutagenesis system to isolate Clostridium perfringens mutants deficient in gliding motility. J. Bacteriol. 2013, 195, 629–636. [Google Scholar] [CrossRef]
- Bansal, M.; Fu, Y.; Alrubaye, B.; Abraha, M.; Almansour, A.; Gupta, A.; Liyanage, R.; Wang, H.; Hargis, B.; Sun, X. A secondary bile acid from microbiota metabolism attenuates ileitis and bile acid reduction in subclinical necrotic enteritis in chickens. J. Anim. Sci. Biotechnol. 2020, 11, 37. [Google Scholar] [CrossRef] [PubMed]
- Fu, Y.; Almansour, A.; Bansal, M.; Alenezi, T.; Alrubaye, B.; Wang, H.; Sun, X. Microbiota attenuates chicken transmission-exacerbated campylobacteriosis in Il10(-/-) mice. Sci. Rep. 2020, 10, 20841. [Google Scholar] [CrossRef]
- Moschetta, A.; vanBerge-Henegouwen, G.P.; Portincasa, P.; Renooij, W.L.; Groen, A.K.; van Erpecum, K.J. Hydrophilic bile salts enhance differential distribution of sphingomyelin and phosphatidylcholine between micellar and vesicular phases: Potential implications for their effects in vivo. J. Hepatol. 2001, 34, 492–499. [Google Scholar] [CrossRef] [PubMed]
- Hylemon, P.B.; Zhou, H.; Pandak, W.M.; Ren, S.; Gil, G.; Dent, P. Bile acids as regulatory molecules. J. Lipid Res. 2009, 50, 1509–1520. [Google Scholar] [CrossRef]
- Chiang, J.Y. Bile acid regulation of gene expression: Roles of nuclear hormone receptors. Endocr. Rev. 2002, 23, 443–463. [Google Scholar] [CrossRef] [PubMed]
- Xie, W.; Radominska-Pandya, A.; Shi, Y.; Simon, C.M.; Nelson, M.C.; Ong, E.S.; Waxman, D.J.; Evans, R.M. An essential role for nuclear receptors SXR/PXR in detoxification of cholestatic bile acids. Proc. Natl. Acad. Sci. USA 2001, 98, 3375–3380. [Google Scholar] [CrossRef] [PubMed]
- Makishima, M.; Lu, T.T.; Xie, W.; Whitfield, G.K.; Domoto, H.; Evans, R.M.; Haussler, M.R.; Mangelsdorf, D.J. Vitamin D receptor as an intestinal bile acid sensor. Science 2002, 296, 1313–1316. [Google Scholar] [CrossRef]
- Chiang, J.Y. Bile acid metabolism and signaling. Compr. Physiol. 2013, 3, 1191–1212. [Google Scholar] [CrossRef]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control T(H)17 and T(reg) cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef] [PubMed]
- Campbell, C.; McKenney, P.T.; Konstantinovsky, D.; Isaeva, O.I.; Schizas, M.; Verter, J.; Mai, C.; Jin, W.B.; Guo, C.J.; Violante, S.; et al. Bacterial metabolism of bile acids promotes generation of peripheral regulatory T cells. Nature 2020, 581, 475–479. [Google Scholar] [CrossRef] [PubMed]
- Elazhary, M.A.; Saheb, S.A.; Roy, R.S.; Lagace, A. A simple procedure for the preliminary identification of aerobic gram negative intestinal bacteria with special reference to the Enterobacteriaceae. Can. J. Comp. Med. 1973, 37, 43–46. [Google Scholar]
- Usui, Y.; Ayibieke, A.; Kamiichi, Y.; Okugawa, S.; Moriya, K.; Tohda, S.; Saito, R. Impact of deoxycholate on Clostridioides difficile growth, toxin production, and sporulation. Heliyon 2020, 6, e03717. [Google Scholar] [CrossRef]
- Wilson, K.H. Efficiency of various bile salt preparations for stimulation of Clostridium difficile spore germination. J. Clin. Microbiol. 1983, 18, 1017–1019. [Google Scholar] [CrossRef]
- Sorg, J.A.; Sonenshein, A.L. Inhibiting the initiation of Clostridium difficile spore germination using analogs of chenodeoxycholic acid, a bile acid. J. Bacteriol. 2010, 192, 4983–4990. [Google Scholar] [CrossRef]
- Sorg, J.A.; Sonenshein, A.L. Bile salts and glycine as cogerminants for Clostridium difficile spores. J. Bacteriol. 2008, 190, 2505–2512. [Google Scholar] [CrossRef] [PubMed]
- Kandell, R.L.; Bernstein, C. Bile salt/acid induction of DNA damage in bacterial and mammalian cells: Implications for colon cancer. Nutr. Cancer 1991, 16, 227–238. [Google Scholar] [CrossRef] [PubMed]
- Prieto, A.I.; Ramos-Morales, F.; Casadesus, J. Bile-induced DNA damage in Salmonella enterica. Genetics 2004, 168, 1787–1794. [Google Scholar] [CrossRef]
- Quillin, S.J.; Schwartz, K.T.; Leber, J.H. The novel Listeria monocytogenes bile sensor BrtA controls expression of the cholic acid efflux pump MdrT. Mol. Microbiol. 2011, 81, 129–142. [Google Scholar] [CrossRef] [PubMed]
- Pope, L.M.; Reed, K.E.; Payne, S.M. Increased protein secretion and adherence to HeLa cells by Shigella spp. following growth in the presence of bile salts. Infect. Immun. 1995, 63, 3642–3648. [Google Scholar] [CrossRef]
- Eade, C.R.; Hung, C.C.; Bullard, B.; Gonzalez-Escobedo, G.; Gunn, J.S.; Altier, C. Bile Acids Function Synergistically to Repress Invasion Gene Expression in Salmonella by Destabilizing the Invasion Regulator HilD. Infect. Immun. 2016, 84, 2198–2208. [Google Scholar] [CrossRef]
- Faherty, C.S.; Redman, J.C.; Rasko, D.A.; Barry, E.M.; Nataro, J.P. Shigella flexneri effectors OspE1 and OspE2 mediate induced adherence to the colonic epithelium following bile salts exposure. Mol. Microbiol. 2012, 85, 107–121. [Google Scholar] [CrossRef]
- van Velkinburgh, J.C.; Gunn, J.S. PhoP-PhoQ-regulated loci are required for enhanced bile resistance in Salmonella spp. Infect. Immun. 1999, 67, 1614–1622. [Google Scholar] [CrossRef]
- Prouty, A.M.; Gunn, J.S. Salmonella enterica serovar Typhimurium invasion is repressed in the presence of bile. Infect. Immun. 2000, 68, 6763–6769. [Google Scholar] [CrossRef]
- Gupta, S.; Chowdhury, R. Bile affects production of virulence factors and motility of Vibrio cholerae. Infect. Immun. 1997, 65, 1131–1134. [Google Scholar] [CrossRef]
- Li, P.; Rivera-Cancel, G.; Kinch, L.N.; Salomon, D.; Tomchick, D.R.; Grishin, N.V.; Orth, K. Bile salt receptor complex activates a pathogenic type III secretion system. Elife 2016, 5, e15718. [Google Scholar] [CrossRef] [PubMed]
- Malik-Kale, P.; Parker, C.T.; Konkel, M.E. Culture of Campylobacter jejuni with sodium deoxycholate induces virulence gene expression. J. Bacteriol. 2008, 190, 2286–2297. [Google Scholar] [CrossRef] [PubMed]
- Hamner, S.; McInnerney, K.; Williamson, K.; Franklin, M.J.; Ford, T.E. Bile salts affect expression of Escherichia coli O157:H7 genes for virulence and iron acquisition, and promote growth under iron limiting conditions. PLoS One 2013, 8, e74647. [Google Scholar] [CrossRef] [PubMed]
- Sharma, N.K.; Choct, M.; Wu, S.B.; Swick, R.A. Necrotic enteritis challenge and high dietary sodium level affect odorant composition or emission from broilers. Poult. Sci. 2018, 97, 39–46. [Google Scholar] [CrossRef]
- Teague, B.; Asiedu, S.; Moore, P.K. The smooth muscle relaxant effect of hydrogen sulphide in vitro: Evidence for a physiological role to control intestinal contractility. Br. J. Pharmacol. 2002, 137, 139–145. [Google Scholar] [CrossRef] [PubMed]
- Gallego, D.; Clave, P.; Donovan, J.; Rahmati, R.; Grundy, D.; Jimenez, M.; Beyak, M.J. The gaseous mediator, hydrogen sulphide, inhibits in vitro motor patterns in the human, rat and mouse colon and jejunum. Neurogastroenterol. Motil. 2008, 20, 1306–1316. [Google Scholar] [CrossRef]
- Quan, X.; Luo, H.; Liu, Y.; Xia, H.; Chen, W.; Tang, Q. Hydrogen sulfide regulates the colonic motility by inhibiting both L-type calcium channels and BKCa channels in smooth muscle cells of rat colon. PLoS ONE 2015, 10, e0121331. [Google Scholar] [CrossRef]
- Pitcher, M.C.; Beatty, E.R.; Cummings, J.H. The contribution of sulphate reducing bacteria and 5-aminosalicylic acid to faecal sulphide in patients with ulcerative colitis. Gut 2000, 46, 64–72. [Google Scholar] [CrossRef]
- Edmond, L.M.; Hopkins, M.J.; Magee, E.A.; Cummings, J.H. The effect of 5-aminosalicylic acid-containing drugs on sulfide production by sulfate-reducing and amino acid-fermenting bacteria. Inflamm. Bowel Dis. 2003, 9, 10–17. [Google Scholar] [CrossRef]
- Li, L.; Bhatia, M.; Zhu, Y.Z.; Zhu, Y.C.; Ramnath, R.D.; Wang, Z.J.; Anuar, F.B.; Whiteman, M.; Salto-Tellez, M.; Moore, P.K. Hydrogen sulfide is a novel mediator of lipopolysaccharide-induced inflammation in the mouse. FASEB J. 2005, 19, 1196–1198. [Google Scholar] [CrossRef]
- Moore, J.; Babidge, W.; Millard, S.; Roediger, W. Colonic luminal hydrogen sulfide is not elevated in ulcerative colitis. Dig. Dis. Sci. 1998, 43, 162–165. [Google Scholar] [CrossRef] [PubMed]
- Mard, S.A.; Neisi, N.; Solgi, G.; Hassanpour, M.; Darbor, M.; Maleki, M. Gastroprotective effect of NaHS against mucosal lesions induced by ischemia-reperfusion injury in rat. Dig. Dis. Sci. 2012, 57, 1496–1503. [Google Scholar] [CrossRef]
- Wallace, J.L.; Caliendo, G.; Santagada, V.; Cirino, G.; Fiorucci, S. Gastrointestinal safety and anti-inflammatory effects of a hydrogen sulfide-releasing diclofenac derivative in the rat. Gastroenterology 2007, 132, 261–271. [Google Scholar] [CrossRef] [PubMed]
- Zanardo, R.C.; Brancaleone, V.; Distrutti, E.; Fiorucci, S.; Cirino, G.; Wallace, J.L. Hydrogen sulfide is an endogenous modulator of leukocyte-mediated inflammation. FASEB J. 2006, 20, 2118–2120. [Google Scholar] [CrossRef] [PubMed]
- Mariggio, M.A.; Minunno, V.; Riccardi, S.; Santacroce, R.; De Rinaldis, P.; Fumarulo, R. Sulfide enhancement of PMN apoptosis. Immunopharmacol. Immunotoxicol. 1998, 20, 399–408. [Google Scholar] [CrossRef]
- Shatalin, K.; Shatalina, E.; Mironov, A.; Nudler, E. H2S: A universal defense against antibiotics in bacteria. Science 2011, 334, 986–990. [Google Scholar] [CrossRef]
- Jones, R.T.; Thai, L.P.; Silver, R.P. Genetic and molecular characterization of an Escherichia coli plasmid coding for hydrogen sulfide production and drug resistance. Antimicrob. Agents Chemother. 1978, 14, 765–770. [Google Scholar] [CrossRef] [PubMed]
- Shatalin, K.; Nuthanakanti, A.; Kaushik, A.; Shishov, D.; Peselis, A.; Shamovsky, I.; Pani, B.; Lechpammer, M.; Vasilyev, N.; Shatalina, E.; et al. Inhibitors of bacterial H(2)S biogenesis targeting antibiotic resistance and tolerance. Science 2021, 372, 1169–1175. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Primer Name | Sequence |
|---|---|---|
| gyrA | gyrA-F | TGCTTGTTGAC-GGACATGGT |
| gyrA-R | ACAACTGGTTCTTTTTCCTCACC | |
| asrA1 | asrA1-F | ACATCTTCCACATCCTACACACA |
| asrA1-R | TGATGAAATGGCTGGTGGACA | |
| plc | plc- F | TGACACAGGGGAATCACAAA |
| plc- R | CGCTATCAACGGCAGTAACA | |
| spoOA | spoOA- F | ACAGGAATTGCAAAGGATGG |
| spoOA- R | TTTTGTCTTGTCCAACAGCAG | |
| virT | virT- F | TGAAATTGTTCTTTTGGATGAAGA |
| virT- R | GCTTGAAAAGCTCCTGCCTA | |
| cpe | cpe- F | CAACTGCTGGTCCAAATGAA |
| cpe- R | GCATCTTTCGCCAGTTTCAA |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alenezi, T.; Fu, Y.; Alrubaye, B.; Alanazi, T.; Almansour, A.; Wang, H.; Sun, X. Potent Bile Acid Microbial Metabolites Modulate Clostridium perfringens Virulence. Pathogens 2023, 12, 1202. https://doi.org/10.3390/pathogens12101202
Alenezi T, Fu Y, Alrubaye B, Alanazi T, Almansour A, Wang H, Sun X. Potent Bile Acid Microbial Metabolites Modulate Clostridium perfringens Virulence. Pathogens. 2023; 12(10):1202. https://doi.org/10.3390/pathogens12101202
Chicago/Turabian StyleAlenezi, Tahrir, Ying Fu, Bilal Alrubaye, Thamer Alanazi, Ayidh Almansour, Hong Wang, and Xiaolun Sun. 2023. "Potent Bile Acid Microbial Metabolites Modulate Clostridium perfringens Virulence" Pathogens 12, no. 10: 1202. https://doi.org/10.3390/pathogens12101202
APA StyleAlenezi, T., Fu, Y., Alrubaye, B., Alanazi, T., Almansour, A., Wang, H., & Sun, X. (2023). Potent Bile Acid Microbial Metabolites Modulate Clostridium perfringens Virulence. Pathogens, 12(10), 1202. https://doi.org/10.3390/pathogens12101202