
Unravelling the Diversity of Microorganisms in Ticks from Australian Wildlife

 ,
, 
 ,
,  , , , and
, , , and 
Abstract
:1. Introduction
2. Materials and Methods
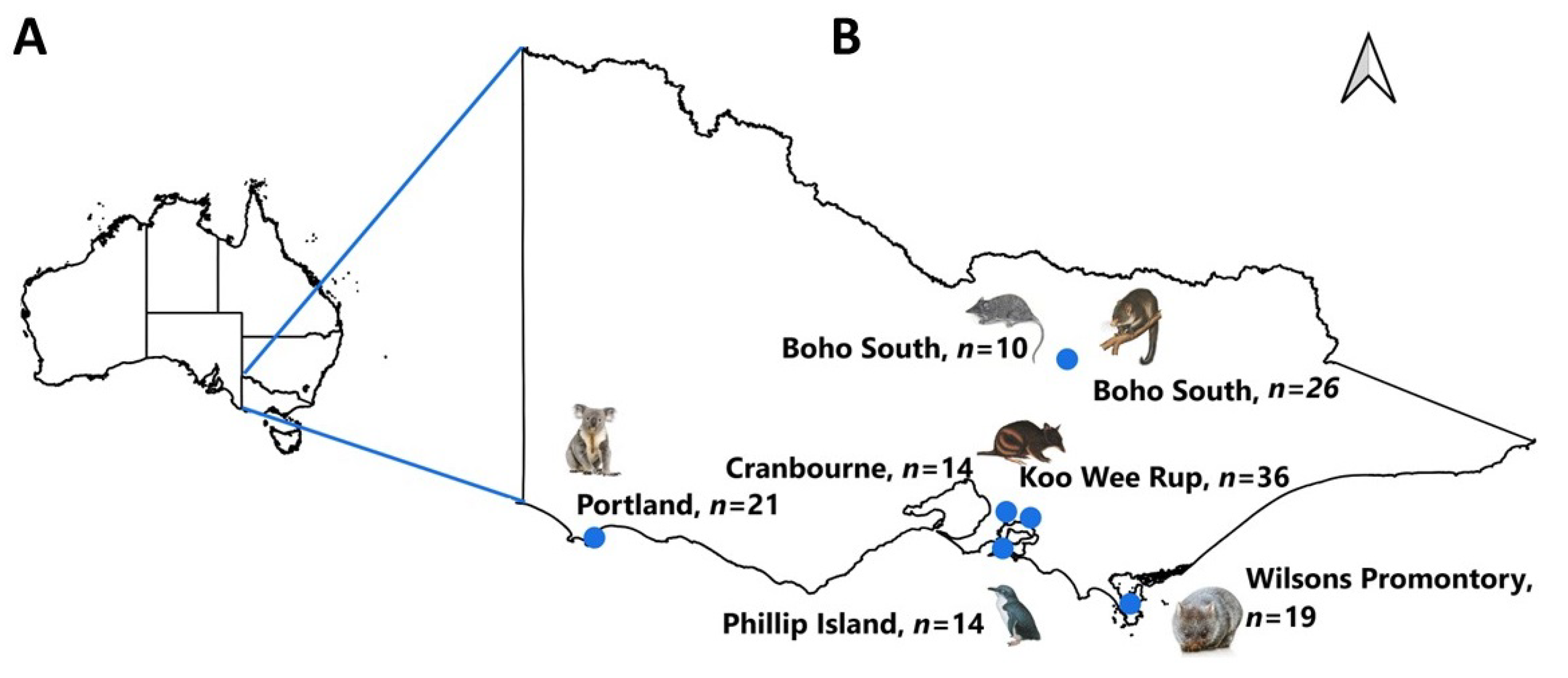
2.1. Study Area and Tick Samples
2.2. Morphological Identification of Ticks
2.3. Molecular Identification of Ticks
2.4. Microfluidic Detection of Microorganisms in Tick DNA Samples
2.5. qPCR Detection of Rickettsia spp.
2.6. DNA Sequencing and Phylogenetic Analyses
3. Results
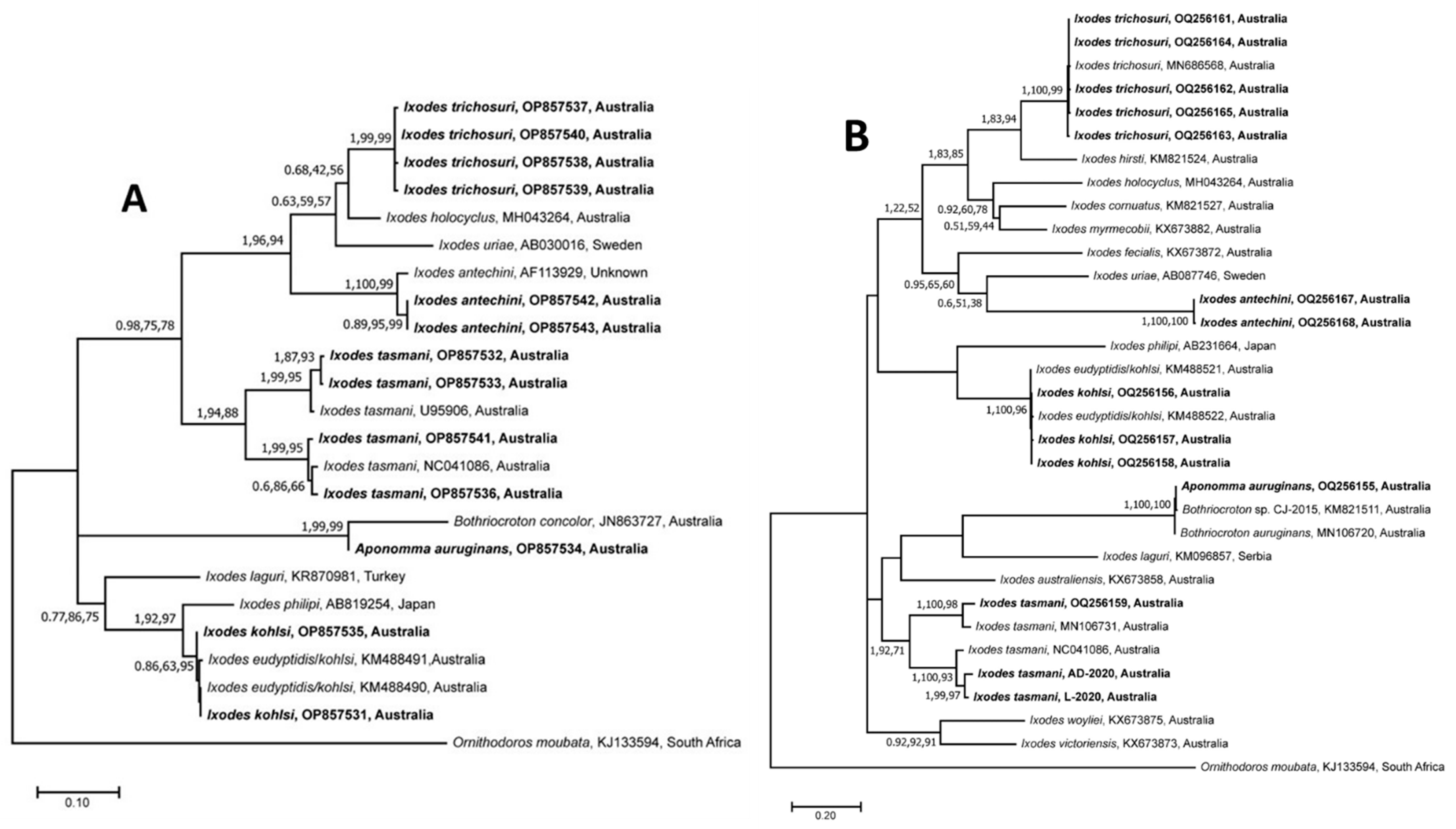
3.1. Morphological and Molecular Characterisation of Ticks
3.2. Sequence and Phylogenetic Analyses of Nucleotide Sequences of Ticks
3.3. Microfluidic Detection of Tick-Borne Microorganisms
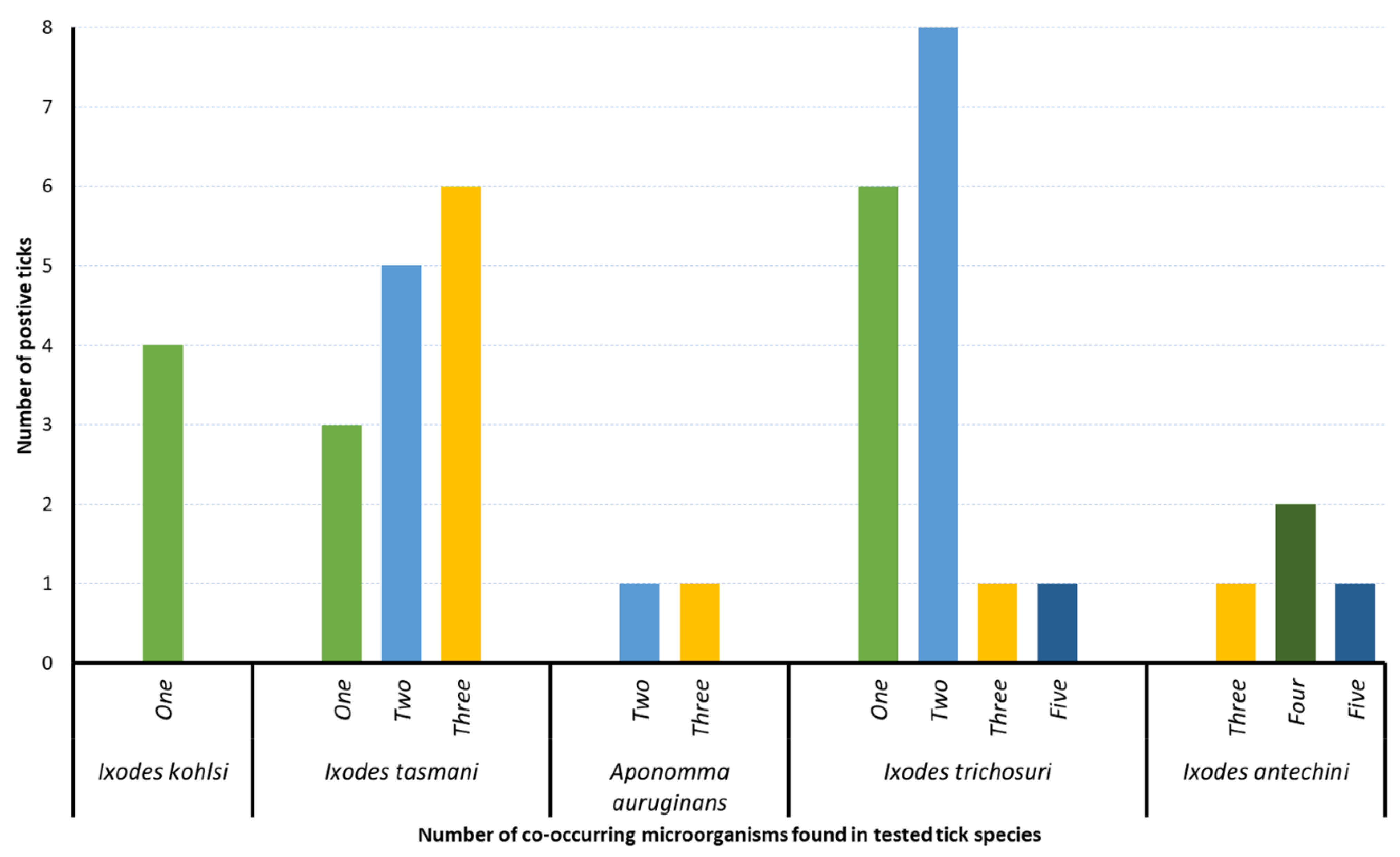
3.4. Co-Occurrence of Microorganisms
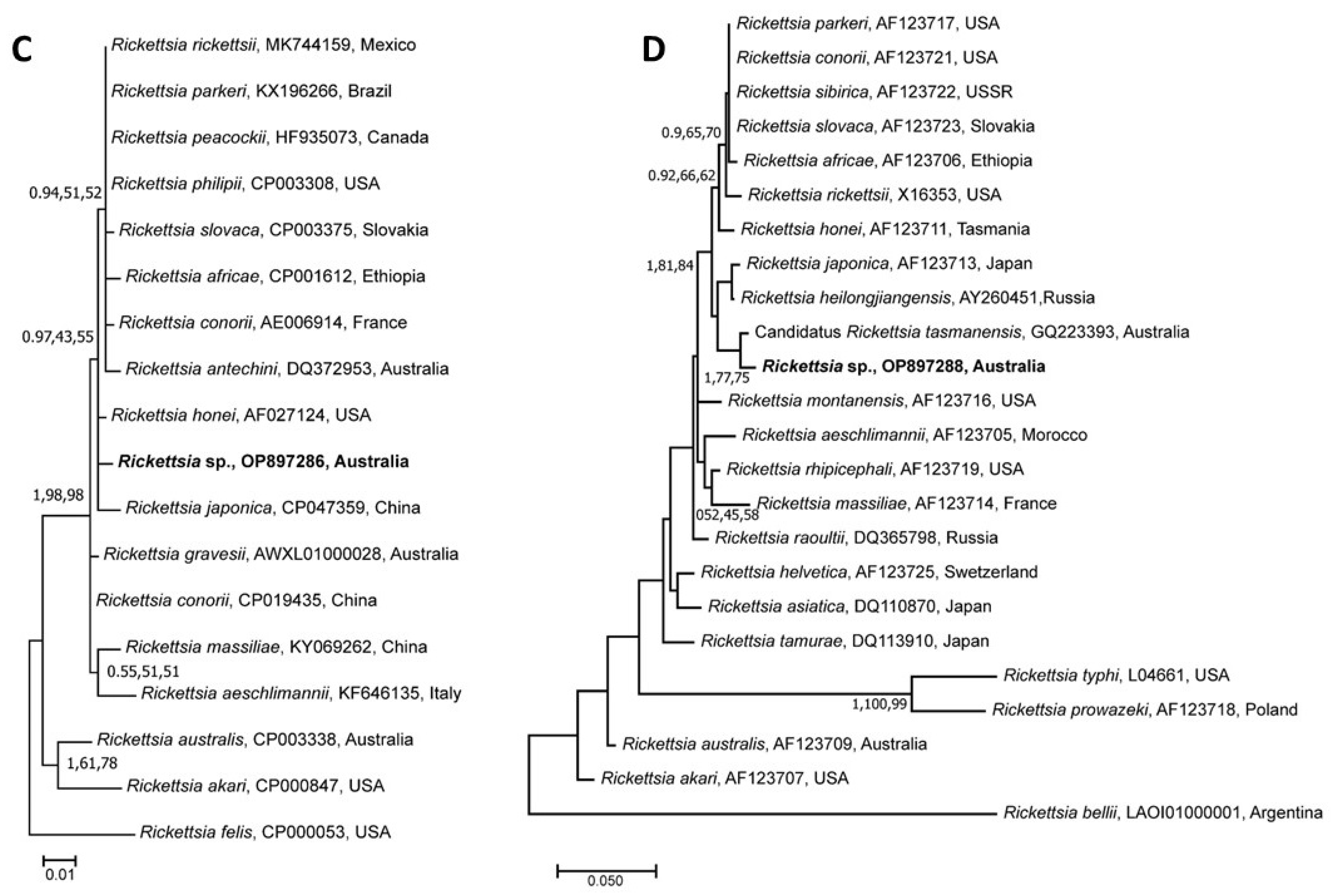
3.5. Genetic Relationship of Rickettsia Species
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Chapman, A.D. Number of Living Species in Australia and the World, 2nd ed.; Report for the Australian Biological Resources Study; Department of the Environment, Water, Heritage and the Arts: Canberra, Australia, 2009. [Google Scholar]
- Anderson, J.F.; Magnarelli, L.A. Biology of ticks. Infect. Dis. Clin. N. Am. 2008, 22, 195–215. [Google Scholar] [CrossRef] [PubMed]
- Jongejan, F.; Uilenberg, G. The global importance of ticks. Parasitology 2004, 129, S3–S14. [Google Scholar] [CrossRef] [PubMed]
- Rinker, D.C.; Pitts, R.J.; Zwiebel, L.J. Disease vectors in the era of next generation sequencing. Genome Biol. 2016, 17, 95. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wikel, S. Ticks and tick-borne infections: Complex ecology, agents, and host interactions. Vet. Sci. 2018, 5, 60. [Google Scholar] [CrossRef] [Green Version]
- Gilbert, L. The impacts of climate change on ticks and tick-borne disease risk. Annu. Rev. Entomol. 2021, 66, 373–388. [Google Scholar] [CrossRef]
- Colwell, D.D.; Dantas-Torres, F.; Otranto, D. Vector-borne parasitic zoonoses: Emerging scenarios and new perspectives. Vet. Parasitol. 2011, 182, 14–21. [Google Scholar] [CrossRef]
- Ostfeld, R.S.; Brunner, J.L. Climate change and Ixodes tick-borne diseases of humans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 2015, 370, 20140051. [Google Scholar] [CrossRef] [Green Version]
- Ferrell, A.; Brinkerhoff, R. Using landscape analysis to test hypotheses about drivers of tick abundance and infection prevalence with Borrelia burgdorferi. Int. J. Environ. Res. Public Health 2018, 15, 737. [Google Scholar] [CrossRef] [Green Version]
- Vilcins, I.-M.; Old, J.M.; Deane, E.M. The impact of ticks and tick-borne diseases on native animal species in Australia. Microbiol. Aust. 2005, 26, 76. [Google Scholar] [CrossRef]
- Allan, B.F.; Keesing, F.; Ostfeld, R.S. Effect of forest fragmentation on Lyme Disease risk. Conserv. Biol. 2003, 17, 267–272. [Google Scholar] [CrossRef]
- Gofton, A.W.; Blasdell, K.; Taylor, C.; Banks, P.B.; Michie, M.; Roy-Dufresne, E.; Poldy, J.; Wang, J.; Dunn, M.; Tachedjian, M. Metatranscriptomic profiling reveals diverse tick-borne bacteria, protozoans, and viruses in ticks and wildlife from Australia. Transbound. Emerg. Dis. 2022, 69, e2389–e2407. [Google Scholar] [CrossRef]
- Chalada, M.J.; Stenos, J.; Bradbury, R.S. Is there a Lyme-like disease in Australia? Summary of the findings to date. One Health 2016, 2, 42–54. [Google Scholar] [CrossRef] [Green Version]
- Andrew, R.; Bonnin, J.; Williams, S. Tick typhus in North Queensland. Med. J. Aust. 1946, 2, 253–258. [Google Scholar] [CrossRef]
- Stewart, A.; Armstrong, M.; Graves, S.; Hajkowicz, K. Epidemiology and characteristics of Rickettsia australis (Queensland tick typhus) infection in hospitalized patients in North Brisbane, Australia. Trop. Med. Infect. Dis. 2017, 2, 10. [Google Scholar] [CrossRef] [Green Version]
- Graves, S.R.; Stewart, L.; Stenos, J.; Stewart, R.S.; Schmidt, E.; Hudson, S.; Banks, J.; Huang, Z.; Dwyer, B. Spotted fever group rickettsial infection in south-eastern Australia: Isolation of Rickettsiae. Comp. Immunol. Microbiol. Infect. Dis. 1993, 16, 223–233. [Google Scholar] [CrossRef]
- Unsworth, N.B.; Stenos, J.; Graves, S.R.; Faa, A.G.; Cox, G.E.; Dyer, J.R.; Boutlis, C.S.; Lane, A.M.; Shaw, M.D.; Robson, J. Flinders Island spotted fever rickettsioses caused by “marmionii” strain of Rickettsia honei, Eastern Australia. Emerg. Infect. Dis. 2007, 13, 566. [Google Scholar] [CrossRef]
- Tadepalli, M.; Vincent, G.; Hii, S.F.; Watharow, S.; Graves, S.; Stenos, J. Molecular evidence of novel spotted fever group Rickettsia species in Amblyomma albolimbatum ticks from the shingleback skink (Tiliqua rugosa) in southern western Australia. Pathogens 2021, 10, 35. [Google Scholar] [CrossRef]
- Vilcins, I.M.; Kosoy, M.; Old, J.M.; Deane, E.M. Bartonella-like DNA detected in Ixodes tasmani ticks (Acari: Ixodida) infesting Koalas (Phascolarctos cinereus) in Victoria, Australia. Vector Borne Zoonotic Dis. 2009, 9, 499–503. [Google Scholar] [CrossRef]
- Kaewmongkol, G.; Kaewmongkol, S.; Owen, H.; Fleming, P.A.; Adams, P.J.; Ryan, U.; Irwin, P.J.; Fenwick, S.G. Candidatus Bartonella antechini: A novel Bartonella species detected in fleas and ticks from the yellow-footed antechinus (Antechinus flavipes), an Australian marsupial. Vet. Microbiol. 2011, 149, 517–521. [Google Scholar] [CrossRef] [Green Version]
- Dehhaghi, M.; Kazemi Shariat Panahi, H.; Holmes, E.C.; Hudson, B.J.; Schloeffel, R.; Guillemin, G.J. Human tick-borne diseases in Australia. Front. Cell Infect. Microbiol. 2019, 9, 3. [Google Scholar] [CrossRef]
- Greay, T.L.; Zahedi, A.; Krige, A.-S.; Owens, J.M.; Rees, R.L.; Ryan, U.M.; Oskam, C.L.; Irwin, P.J. Endemic, exotic and novel apicomplexan parasites detected during a national study of ticks from companion animals in Australia. Parasit. Vectors 2018, 11, 197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Loh, S.-M.; Egan, S.; Gillett, A.; Banks, P.B.; Ryan, U.M.; Irwin, P.J.; Oskam, C.L. Molecular surveillance of piroplasms in ticks from small and medium-sized urban and peri-urban mammals in Australia. Int. J. Parasitol. Parasites Wildl. 2018, 7, 197–203. [Google Scholar] [CrossRef] [PubMed]
- Storey-Lewis, B.; Mitrovic, A.; McParland, B. Molecular detection and characterisation of Babesia and Theileria in Australian hard ticks. Ticks Tick Borne Dis. 2018, 9, 471–478. [Google Scholar] [CrossRef] [PubMed]
- Beard, D.; Stannard, H.J.; Old, J.M. Morphological identification of ticks and molecular detection of tick-borne pathogens from bare-nosed wombats (Vombatus ursinus). Parasit. Vectors 2021, 14, 60. [Google Scholar] [CrossRef] [PubMed]
- Panetta, J.L.; Šíma, R.; Calvani, N.E.; Hajdušek, O.; Chandra, S.; Panuccio, J.; Šlapeta, J. Reptile-associated Borrelia species in the goanna tick (Bothriocroton undatum) from Sydney, Australia. Parasit. Vectors 2017, 10, 616. [Google Scholar] [CrossRef] [Green Version]
- Gofton, A.W.; Waudby, H.P.; Petit, S.; Greay, T.L.; Ryan, U.M.; Irwin, P.J. Detection and phylogenetic characterisation of novel Anaplasma and Ehrlichia species in Amblyomma triguttatum subsp. from four allopatric populations in Australia. Ticks Tick Borne Dis. 2017, 8, 749–756. [Google Scholar] [CrossRef]
- Gofton, A.W.; Loh, S.-M.; Barbosa, A.D.; Paparini, A.; Gillett, A.; Macgregor, J.; Oskam, C.L.; Ryan, U.M.; Irwin, P.J. A novel Ehrlichia species in blood and Ixodes ornithorhynchi ticks from platypuses (Ornithorhynchus anatinus) in Queensland and Tasmania, Australia. Ticks Tick Borne Dis. 2018, 9, 435–442. [Google Scholar] [CrossRef]
- Egan, S.L.; Loh, S.-M.; Banks, P.B.; Gillett, A.; Ahlstrom, L.; Ryan, U.M.; Irwin, P.J.; Oskam, C.L. Bacterial community profiling highlights complex diversity and novel organisms in wildlife ticks. Ticks Tick Borne Dis. 2020, 11, 101407. [Google Scholar] [CrossRef]
- Gofton, A.W.; Doggett, S.; Ratchford, A.; Ryan, U.; Irwin, P. Phylogenetic characterisation of two novel Anaplasmataceae from Australian Ixodes holocyclus ticks: ‘Candidatus Neoehrlichia australis’ and ‘Candidatus Neoehrlichia arcana’. Int. J. Syst. Evol. Microbiol. 2016, 66, 4256–4261. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Hall-Mendelin, S.; Hobson-Peters, J.; Deliyannis, G.; Allen, A.; Lew-Tabor, A.; Rodriguez-Valle, M.; Barker, D.; Barker, S.C.; Hall, R.A. Discovery of a novel iflavirus sequence in the eastern paralysis tick Ixodes holocyclus. Arch. Virol. 2018, 163, 2451–2457. [Google Scholar] [CrossRef]
- Harvey, E.; Rose, K.; Eden, J.-S.; Lo, N.; Abeyasuriya, T.; Shi, M.; Doggett, S.L.; Holmes, E.C. Extensive diversity of RNA viruses in Australian ticks. J. Virol. 2019, 93, e01358-18. [Google Scholar] [CrossRef] [Green Version]
- Moutailler, S.; Valiente Moro, C.; Vaumourin, E.; Michelet, L.; Tran, F.H.; Devillers, E.; Cosson, J.-F.; Gasqui, P.; Van, V.T.; Mavingui, P.; et al. Co-infection of ticks: The rule rather than the exception. PLoS Negl. Trop. Dis. 2016, 10, e0004539. [Google Scholar] [CrossRef] [Green Version]
- Bonnet, S.I.; Pollet, T. Update on the intricate tango between tick microbiomes and tick-borne pathogens. Parasite Immunol. 2021, 43, e12813. [Google Scholar] [CrossRef]
- Michelet, L.; Delannoy, S.; Devillers, E.; Umhang, G.; Aspan, A.; Juremalm, M.; Chirico, J.; van der Wal, F.J.; Sprong, H.; Boye Pihl, T.P. High-throughput screening of tick-borne pathogens in Europe. Front. Cell Infect. Microbiol. 2014, 4, 103. [Google Scholar] [CrossRef]
- Liu, J.; Hansen, C.; Quake, S.R. Solving the "world-to-chip" interface problem with a microfluidic matrix. Anal. Chem. 2003, 75, 4718–4723. [Google Scholar] [CrossRef]
- Ghafar, A.; Cabezas-Cruz, A.; Galon, C.; Obregon, D.; Gasser, R.B.; Moutailler, S.; Jabbar, A. Bovine ticks harbour a diverse array of microorganisms in Pakistan. Parasit. Vectors 2020, 13, 1. [Google Scholar] [CrossRef] [Green Version]
- Ghafar, A.; Khan, A.; Cabezas-Cruz, A.; Gauci, C.G.; Niaz, S.; Ayaz, S.; Mateos-Hernández, L.; Galon, C.; Nasreen, N.; Moutailler, S. An assessment of the molecular diversity of ticks and tick-borne microorganisms of small ruminants in Pakistan. Microorganisms 2020, 8, 1428. [Google Scholar] [CrossRef]
- Barker, S.C.; Walker, A.R. Ticks of Australia. The species that infest domestic animals and humans. Zootaxa 2014, 3816, 1–144. [Google Scholar] [CrossRef]
- Roberts, F.H.S. Australian Ticks; Commonwealth Scientific and Industrial Research Organization: Melbourne, Australia, 1970. [Google Scholar]
- Black, W.C.; Piesman, J. Phylogeny of hard- and soft-tick taxa (Acari: Ixodida) based on mitochondrial 16S rDNA sequences. Proc. Natl. Acad. Sci. USA 1994, 91, 10034–10038. [Google Scholar] [CrossRef] [Green Version]
- Vrijenhoek, R. DNA primers for amplification of mitochondrial cytochrome c oxidase subunit I from diverse metazoan invertebrates. Mol. Mar. Biol. Biotechnol. 1994, 3, 294–299. [Google Scholar]
- Fournier, P.-E.; Dumler, J.S.; Greub, G.; Zhang, J.; Wu, Y.; Raoult, D. Gene sequence-based criteria for identification of new Rickettsia isolates and description of Rickettsia heilongjiangensis sp. nov. J. Clin. Microbiol. 2003, 41, 5456–5465. [Google Scholar] [CrossRef] [PubMed]
- Hall, T.; Biosciences, I.; Carlsbad, C. BioEdit: An important software for molecular biology. GERF Bull. Biosci. 2011, 2, 60–61. [Google Scholar]
- Tamura, K.; Stecher, G.; Kumar, S. MEGA11: Molecular Evolutionary Genetics Analysis version 11. Mol. Biol. Evol. 2021, 38, 3022–3027. [Google Scholar] [CrossRef] [PubMed]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Huelsenbeck, J.P.; Ronquist, F. MRBAYES: Bayesian inference of phylogenetic trees. Bioinformatics 2001, 17, 754–755. [Google Scholar] [CrossRef] [Green Version]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K. Estimation of the number of nucleotide substitutions when there are strong transition-transversion and G+ C-content biases. Mol. Biol. Evol. 1992, 9, 678–687. [Google Scholar]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Kimura, M. A simple method for estimating evolutionary rates of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef]
- Cotté, V.; Bonnet, S.; Cote, M.; Vayssier-Taussat, M. Prevalence of five pathogenic agents in questing Ixodes ricinus ticks from western France. Vector Borne Zoonotic Dis 2010, 10, 723–730. [Google Scholar] [CrossRef]
- Murphy, T.W.; Hsieh, Y.-P.; Zhu, B.; Naler, L.B.; Lu, C. Microfluidic platform for Next-Generation Sequencing library preparation with low-input samples. Anal. Chem. 2020, 92, 2519–2526. [Google Scholar] [CrossRef]
- Kim, H.; Bartsch, M.S.; Renzi, R.F.; He, J.; Van De Vreugde, J.L.; Claudnic, M.R.; Patel, K.D. Automated digital Microfluidic sample preparation for Next-Generation DNA Sequencing. JALA J. Assoc. Lab. Autom. 2011, 16, 405–414. [Google Scholar] [CrossRef]
- Nasseri, B.; Soleimani, N.; Rabiee, N.; Kalbasi, A.; Karimi, M.; Hamblin, M.R. Point-of-care microfluidic devices for pathogen detection. Biosens. Bioelectron. 2018, 117, 112–128. [Google Scholar] [CrossRef]
- Kwak, M.L.; Beveridge, I.; Koehler, A.V.; Malipatil, M.; Gasser, R.B.; Jabbar, A. Phylogenetic analysis of the Australasian paralysis ticks and their relatives (Ixodidae: Ixodes: Sternalixodes). Parasit. Vectors 2017, 10, 122. [Google Scholar] [CrossRef]
- Spencer, A.J.; Canfield, P.J. Age-related changes in the haematology of young koalas (Phascolarctos cinereus) up to one year old. Comp. Haematol. Int. 1994, 4, 146–151. [Google Scholar] [CrossRef]
- Gemmell, R.T.; Cepon, G.; Green, P.E.; Stewart, N.P. Some effects of tick infestations on juvenile northern brown bandicoot (Isoodon macrourus). J. Wildl. Dis. 1991, 27, 269–275. [Google Scholar] [CrossRef] [Green Version]
- Donahoe, S.L.; Peacock, C.S.; Choo, A.Y.L.; Cook, R.W.; O’Donoghue, P.; Crameri, S.; Vogelnest, L.; Gordon, A.N.; Scott, J.L.; Rose, K. A retrospective study of Babesia macropus associated with morbidity and mortality in eastern grey kangaroos (Macropus giganteus) and agile wallabies (Macropus agilis). Int. J. Parasitol. Parasites Wildl. 2015, 4, 268–276. [Google Scholar] [CrossRef] [Green Version]
- Dawood, K.E.; Morgan, J.A.T.; Busfield, F.; Srivastava, M.; Fletcher, T.I.; Sambono, J.; Jackson, L.A.; Venus, B.; Philbey, A.W.; Lew-Tabor, A.E. Observation of a novel Babesia spp. in eastern grey kangaroos (Macropus giganteus) in Australia. Int. J. Parasitol. Parasites Wildl. 2013, 2, 54–61. [Google Scholar] [CrossRef] [Green Version]
- Shaw, S.E.; Day, M.J.; Birtles, R.J.; Breitschwerdt, E.B. Tick-borne infectious diseases of dogs. Trends Parasitol. 2001, 17, 74–80. [Google Scholar] [CrossRef]
- Marendy, D.; Baker, K.; Emery, D.; Rolls, P.; Stutchbury, R. Haemaphysalis longicornis: The life-cycle on dogs and cattle, with confirmation of its vector status for Theileria orientalis in Australia. Vet. Parasitol. 2020, 277, 100022. [Google Scholar] [CrossRef] [PubMed]
- Forshaw, D.; Alex, S.; Palmer, D.; Cotter, J.; Roberts, W.; Jenkins, C.; Hair, S. Theileria orientalis Ikeda genotype infection associated with anaemia, abortion and death in beef cattle in Western Australia. Aust. Vet. J. 2020, 98, 290–297. [Google Scholar] [CrossRef] [PubMed]
- McLeod, R.S. Costs of major parasites to the Australian livestock industries. Int. J. Parasitol. 1995, 25, 1363–1367. [Google Scholar] [CrossRef] [PubMed]
- Jonsson, N.N.; Bock, R.E.; Jorgensen, W.K. Productivity and health effects of anaplasmosis and babesiosis on Bos indicus cattle and their crosses, and the effects of differing intensity of tick control in Australia. Vet. Parasitol. 2008, 155, 1–9. [Google Scholar] [CrossRef]
- James, S.P. On a Parasite Found in the White Corpuscles of the Blood of Dogs. Sci. Mem. Offrs. Med. Sanit. Deps. India 1905, 14, 1–12. [Google Scholar]
- Baneth, G.; Samish, M.; Alekseev, E.; Aroch, I.; Shkap, V. Transmission of Hepatozoon canis to dogs by naturally-fed or percutaneously-injected Rhipicephalus sanguineus ticks. J. Parasitol. 2001, 87, 606–611. [Google Scholar] [CrossRef]
- Greay, T.L.; Barbosa, A.D.; Rees, R.L.; Paparini, A.; Ryan, U.M.; Oskam, C.L.; Irwin, P.J. An Australian dog diagnosed with an exotic tick-borne infection: Should Australia still be considered free from Hepatozoon canis? Int. J. Parasitol. 2018, 48, 805–815. [Google Scholar] [CrossRef]
- Majláthová, V.; Hurníková, Z.; Majláth, I.; Peťko, B. Hepatozoon canis infection in Slovakia: Imported or autochthonous? Vector Borne Zoonotic Dis. 2007, 7, 199–202. [Google Scholar] [CrossRef]
- Playford, G.; Whitby, M. Tick-borne diseases in Australia. Aust. Fam. Physician 1996, 25, 1841–1845. [Google Scholar]
- Duron, O.; Noël, V.; McCoy, K.D.; Bonazzi, M.; Sidi-Boumedine, K.; Morel, O.; Vavre, F.; Zenner, L.; Jourdain, E.; Durand, P. The recent evolution of a maternally-inherited endosymbiont of ticks led to the emergence of the Q fever pathogen, Coxiella burnetii. PLoS Pathog. 2015, 11, e1004892. [Google Scholar] [CrossRef] [Green Version]
- Izzard, L.; Graves, S.; Cox, E.; Fenwick, S.; Unsworth, N.; Stenos, J. Novel Rickettsia in ticks, Tasmania, Australia. Emerg. Infect. Dis. 2009, 15, 1654. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host Species | Tick Species | Location (Latitude, Longitude) | Ticks | |
|---|---|---|---|---|
| Collected | Tested | |||
| Bare-nosed wombat (Vombatus ursinus) | Aponomma auruginans | Wilsons Promontory (−39.0080, 146.3895) | 19 | 2 |
| Little penguin (Eudyptula minor) | Ixodes kohlsi | Phillip Island (Nature Parks) (−38.4833314, 145.2333324) | 14 | 6 |
| Koala (Phascolarctos cinereus) | Ixodes tasmani | Portland (−38.3333, 141.6000) | 21 | 4 |
| Mountain brushtail possum (Trichosurus cunninghami) | Ixodes trichosuri | Boho South (−36.783, 145.800) | 24 | 10 |
| Ixodes tasmani | 2 | 2 | ||
| Agile antechinus (Antechinus agilis) | Ixodes antechini | 7 | 4 | |
| Ixodes tasmani | 3 | 1 | ||
| Southern brown bandicoot (Isoodon obesulus) | Ixodes tasmani | Koo Wee Rup (−38.198798, 145.489126) | 30 | 8 |
| Ixodes trichosuri | 6 | 1 | ||
| Ixodes trichosuri | Cranbourne (Botanic Gardens) (−38.1298617, 45.2701999) | 14 | 7 | |
| Microorganisms | Little Penguin (Eudyptula minor) | Koala (Phascolarctos cinereus) | Bare-Nosed Wombat (Vombatus ursinus) | Mountain Brushtail Possum (Trichosurus cunninghami) | Agile Antechinus (Antechinus agilis) | Southern Brown Bandicoot (Isoodon obesulus obesulus) | |||
|---|---|---|---|---|---|---|---|---|---|
| Ixodes kohlsi (n = 6) | Ixodes tasmani (n = 4) | Aponomma auruginans (n = 2) | Ixodes trichosuri (n = 10) | Ixodes tasmani (n = 2) | Ixodes antechini (n = 4) | Ixodes tasmani (n = 1) | Ixodes tasmani (n = 8) | Ixodes trichosuri (n = 8) | |
| Apicomplexa sp. | 4 | 3 | 2 | 7 | 2 | 4 | 1 | 8 | 7 |
| Bartonella sp. | - | - | - | 3 | - | 3 | - | 2 | - |
| Coxiella-like sp. | - | - | 2 | 1 | - | - | - | - | - |
| Ehrlichia sp. | - | - | - | - | - | 1 | - | - | - |
| Hepatozoon sp. | - | - | - | - | - | - | - | 1 | - |
| Rickettsia sp. | - | 1 | 1 | 6 | - | 4 | - | 8 | 4 |
| Theileria sp. | - | 1 | - | 2 | - | 4 | 1 | 2 | - |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ghafar, A.; Davies, N.; Tadepalli, M.; Breidahl, A.; Death, C.; Haros, P.; Li, Y.; Dann, P.; Cabezas-Cruz, A.; Moutailler, S.; et al. Unravelling the Diversity of Microorganisms in Ticks from Australian Wildlife. Pathogens 2023, 12, 153. https://doi.org/10.3390/pathogens12020153
Ghafar A, Davies N, Tadepalli M, Breidahl A, Death C, Haros P, Li Y, Dann P, Cabezas-Cruz A, Moutailler S, et al. Unravelling the Diversity of Microorganisms in Ticks from Australian Wildlife. Pathogens. 2023; 12(2):153. https://doi.org/10.3390/pathogens12020153
Chicago/Turabian StyleGhafar, Abdul, Nick Davies, Mythili Tadepalli, Amanda Breidahl, Clare Death, Philip Haros, Yuting Li, Peter Dann, Alejandro Cabezas-Cruz, Sara Moutailler, and et al. 2023. "Unravelling the Diversity of Microorganisms in Ticks from Australian Wildlife" Pathogens 12, no. 2: 153. https://doi.org/10.3390/pathogens12020153
APA StyleGhafar, A., Davies, N., Tadepalli, M., Breidahl, A., Death, C., Haros, P., Li, Y., Dann, P., Cabezas-Cruz, A., Moutailler, S., Foucault-Simonin, A., Gauci, C. G., Stenos, J., Hufschmid, J., & Jabbar, A. (2023). Unravelling the Diversity of Microorganisms in Ticks from Australian Wildlife. Pathogens, 12(2), 153. https://doi.org/10.3390/pathogens12020153