

Role of Innate Interferon Responses at the Ocular Surface in Herpes Simplex Virus-1-Induced Herpetic Stromal Keratitis



Abstract
:1. Introduction
2. HSV-1 Entry, Replication, Assembly, and Egress
3. HSV-1 Recognition by the Host Immune System
4. Type I and III IFN Responses
Type I and Type III IFN-Mediated Signaling
5. Anti-HSV-1 ISGs
6. HSV-1 Evasion Strategies to Control Innate IFN Responses
6.1. Evasion of PRR Signaling to Suppress Innate IFN Production
6.2. Evasion of IFN-Mediated Signaling

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Host Responses | Host Target Molecule | HSV-1 Molecule | Mechanism of Action | References |
|---|---|---|---|---|
| TLR signaling | MyD88 | ICP0 | ICP0 degrades TLR adaptor proteins (MyD88 and Mal) to inhibit type I IFN production | [237,238,239,240] |
| TLR2 | US3 | US3 reduces TRAF6 polyubiquitination to inhibit TLR2-mediated NF-kB activation | [242] | |
| TLR3 | US3 suppresses TLR3-mediated type I IFN production | [243] | ||
| p65 | ICP0 | ICP0 mediates USP7 translocation to cytoplasm, which induces deubiquitination of TRAF6 and IKKγ to terminate NF-κB activation | [241] | |
| VP16 | VP16 interacts with p65 subunit to block the NF-kB activation and type I IFN production | [245] | ||
| UL24 | UL24 binds to p65 subunit to inhibit NF-kB mediated type I IFN production | [246] | ||
| IRF3 | US3 | US3 phosphorylates IRF3 to inhibit IFN-β production | [244] | |
| VP16 | VP16 interacts with IRF3 and its coactivator CREB-BP to inhibit type I IFN production | [245] | ||
| UL42 | UL42 inhibits IRF3 phosphorylation to reduce IFN-β gene expression | [248] | ||
| TRAF3 | UL36USP | UL36USP induces TRAF3 deubiquitination and destabilizes the polyubiquitin scaffold to suppress IFN-β production | [247] | |
| dsDNA sensors | cGAS | UL24 | Inhibits cGAS activation to inhibit IFN-β and IL-6 production | [246] |
| vhs (UL41) | vhs targets cGAS mRNA for degradation | [249] | ||
| vhs selectively degrades host antiviral effector molecule production | [249,254] | |||
| UL56 | Binds with cGAS to inhibit its dsDNA-binding and enzymatic activity | [250] | ||
| VP22 | Inhibits the binding of dsDNA to cGAS and suppresses type I IFN production | [251,252] | ||
| UL37 | UL37 promotes deamidation of cGAS to inhibit cGAMP and IFN production | [253] | ||
| UL36USP | Inhibits cGAS–STING-mediated IFN-β promoter activation and blocks NF-kB activation | [255] | ||
| STING | UL36USP | Deubiquitinates STING inhibiting IRF3 activation and type I IFN production | [256] | |
| UL46 | Prevents STING activation to suppress IFN production | [257] | ||
| Inhibits TBK1 dimerization to suppress IRF3 activation and type I IFN production | [259] | |||
| ICP34.5 | ICP34.5 blocks STING translocation from ER to Golgi to prevent its antiviral responses | [258] | ||
| ICP27 | ICP27 interacts with the STING-activated TBK1 to suppress type I IFN production | [260] | ||
| IFI16 | ICP0 | Targets IFI16 for degradation to inhibit sensing viral DNA and IRF3 activation | [261,262] | |
| RNA sensors | RIG-I | US11 | US11 interacts with RIG-I to affect MAVS and IFN-β production | [264] |
| UL37 | UL37 deamidates RIG-I which affects its ability to sense dsRNA and inhibit antiviral immune responses | [265] | ||
| Type I IFN signaling | JAK1 | vhs (UL41) | vhs reduces expression of IFNAR, JAK1 and STAT-2 to suppress ISGF3 formation | [266] |
| UL36USP | UL36USP binds to IFNAR2 to block the recruitment of JAK1 and suppresses activation of STATs and the ISRE promoter | [267] | ||
| STAT1 | ICP0 | ICP0 inhibits the STAT-dependent antiviral responses downstream of IFN signaling | [268] | |
| ICP27 | ICP27 inhibits STAT1 phosphorylation and its nuclear translocation to suppress ISGs | [269] | ||
| ISGs | Viperin | vhs (UL41) | vhs reduces viperin mRNA accumulation to abrogate its antiviral effects and suppress viral replication | [219,220] |
| Tetherin | vhs (UL41) | vhs depletes tetherin mRNA and protein in infected host cells to evade innate immune response | [224] | |
| OAS | US11 | US11 dsRNA-binding domain blocks OAS synthesis and activation | [214] |
6.3. Evasion of ISG Responses
7. Corneal HSV-1 Infection and Induction of Type I and III IFN Responses
7.1. Type I IFNs after Corneal HSV-1 Infection
7.2. Type III IFNs after Corneal HSV-1 Infection
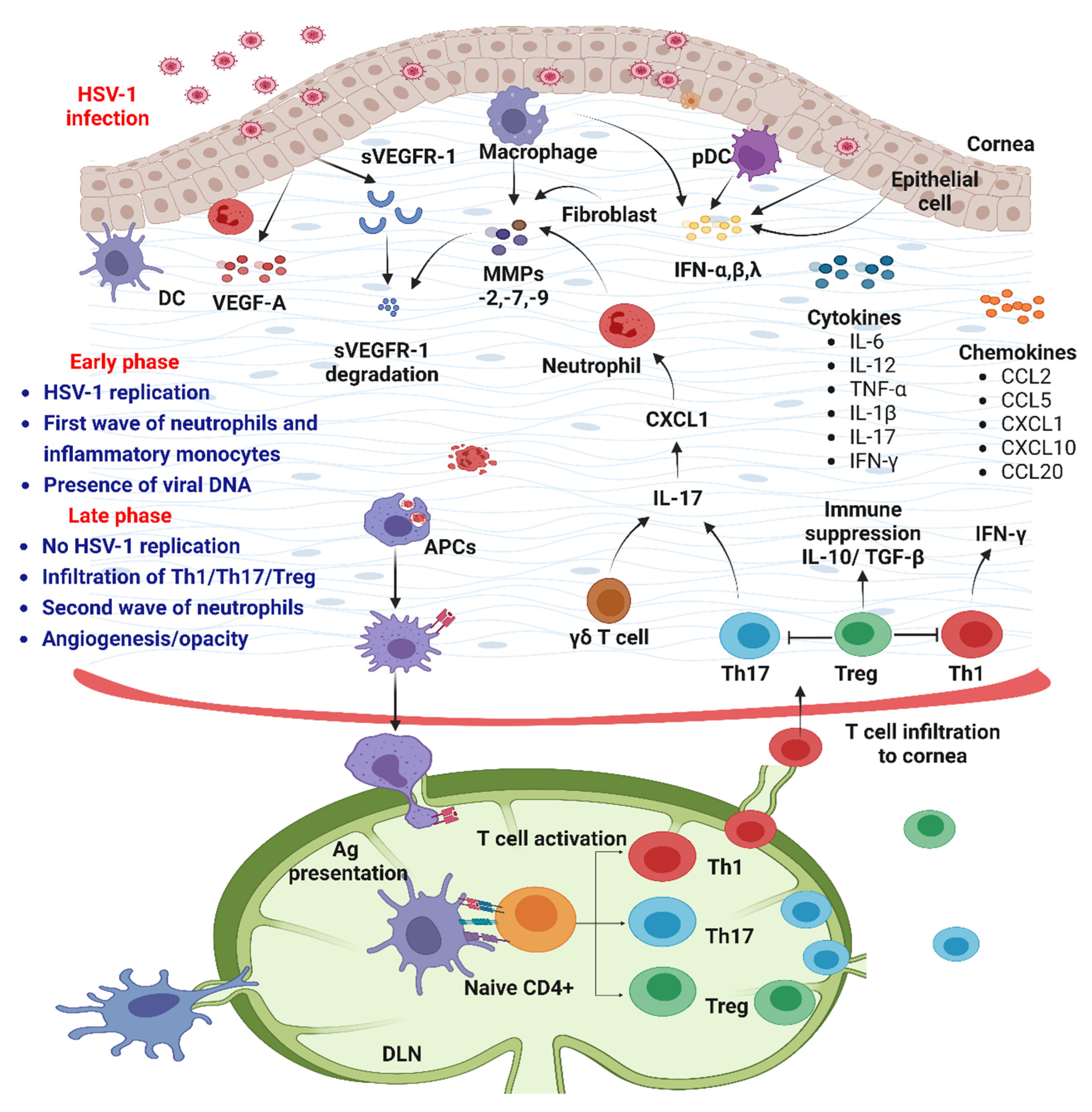
8. Pathogenesis of Corneal HSV-1 Infection
9. HSK Therapeutic Strategies and Challenges
9.1. Alternate Experimental Approaches to Control HSV-1 Infection and HSK
9.2. Is IFN-λ-Based Therapy a Better Approach to Suppress Both HSV-1 Replication and Inflammation?
10. Concluding Remarks
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Farooq, A.V.; Shukla, D. Herpes simplex epithelial and stromal keratitis: An epidemiologic update. Surv. Ophthalmol. 2012, 57, 448–462. [Google Scholar] [CrossRef] [Green Version]
- James, C.; Harfouche, M.; Welton, N.J.; Turner, K.M.; Abu-Raddad, L.J.; Gottlieb, S.L.; Looker, K.J. Herpes simplex virus: Global infection prevalence and incidence estimates, 2016. Bull. World Health Organ. 2020, 98, 315–329. [Google Scholar] [CrossRef]
- Spicknall, I.H.; Flagg, E.W.; Torrone, E.A. Estimates of the Prevalence and Incidence of Genital Herpes, United States, 2018. Sex Transm. Dis. 2021, 48, 260–265. [Google Scholar] [CrossRef]
- Xu, F.; Sternberg, M.R.; Kottiri, B.J.; McQuillan, G.M.; Lee, F.K.; Nahmias, A.J.; Berman, S.M.; Markowitz, L.E. Trends in herpes simplex virus type 1 and type 2 seroprevalence in the United States. JAMA 2006, 296, 964–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Marcocci, M.E.; Napoletani, G.; Protto, V.; Kolesova, O.; Piacentini, R.; Li Puma, D.D.; Lomonte, P.; Grassi, C.; Palamara, A.T.; De Chiara, G. Herpes Simplex Virus-1 in the Brain: The Dark Side of a Sneaky Infection. Trends Microbiol. 2020, 28, 808–820. [Google Scholar] [CrossRef] [PubMed]
- Zhu, S.; Viejo-Borbolla, A. Pathogenesis and virulence of herpes simplex virus. Virulence 2021, 12, 2670–2702. [Google Scholar] [CrossRef] [PubMed]
- Verzosa, A.L.; McGeever, L.A.; Bhark, S.-J.; Delgado, T.; Salazar, N.; Sanchez, E.L. Herpes Simplex Virus 1 Infection of Neuronal and Non-Neuronal Cells Elicits Specific Innate Immune Responses and Immune Evasion Mechanisms. Front. Immunol. 2021, 12, 644664. [Google Scholar] [CrossRef]
- Rouse, B.T.; Schmid, D.S. Fraternal Twins: The Enigmatic Role of the Immune System in Alphaherpesvirus Pathogenesis and Latency and Its Impacts on Vaccine Efficacy. Viruses 2022, 14, 862. [Google Scholar] [CrossRef]
- Wang, L.; Wang, R.; Xu, C.; Zhou, H. Pathogenesis of Herpes Stromal Keratitis: Immune Inflammatory Response Mediated by Inflammatory Regulators. Front. Immunol. 2020, 11, 766. [Google Scholar] [CrossRef]
- Koujah, L.; Suryawanshi, R.K.; Shukla, D. Pathological processes activated by herpes simplex virus-1 (HSV-1) infection in the cornea. Cell. Mol. Life Sci. 2019, 76, 405–419. [Google Scholar] [CrossRef]
- Holland, E.J.; Schwartz, G.S. Classification of herpes simplex virus keratitis. Cornea 1999, 18, 144–154. [Google Scholar] [CrossRef] [PubMed]
- Liesegang, T.J. Classification of herpes simplex virus keratitis and anterior uveitis. Cornea 1999, 18, 127–143. [Google Scholar] [CrossRef] [PubMed]
- Knickelbein, J.E.; Hendricks, R.L.; Charukamnoetkanok, P. Management of herpes simplex virus stromal keratitis: An evidence-based review. Surv. Ophthalmol. 2009, 54, 226–234. [Google Scholar] [CrossRef] [PubMed]
- Liesegang, T.J. Herpes simplex virus epidemiology and ocular importance. Cornea 2001, 20, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Liesegang, T.J.; Melton, L.J., 3rd; Daly, P.J.; Ilstrup, D.M. Epidemiology of ocular herpes simplex. Incidence in Rochester, Minn, 1950 through 1982. Arch. Ophthalmol. 1989, 107, 1155–1159. [Google Scholar] [CrossRef]
- Stuart, P.M.; Keadle, T.L. Recurrent herpetic stromal keratitis in mice: A model for studying human HSK. Clin. Dev. Immunol. 2012, 2012, 728480. [Google Scholar] [CrossRef] [Green Version]
- Lobo, A.M.; Agelidis, A.M.; Shukla, D. Pathogenesis of herpes simplex keratitis: The host cell response and ocular surface sequelae to infection and inflammation. Ocul. Surf. 2019, 17, 40–49. [Google Scholar] [CrossRef]
- Biswas, P.S.; Rouse, B.T. Early events in HSV keratitis--setting the stage for a blinding disease. Microbes Infect. 2005, 7, 799–810. [Google Scholar] [CrossRef]
- Hamrah, P.; Cruzat, A.; Dastjerdi, M.H.; Zheng, L.; Shahatit, B.M.; Bayhan, H.A.; Dana, R.; Pavan-Langston, D. Corneal sensation and subbasal nerve alterations in patients with herpes simplex keratitis: An in vivo confocal microscopy study. Ophthalmology 2010, 117, 1930–1936. [Google Scholar] [CrossRef] [Green Version]
- Chucair-Elliott, A.J.; Zheng, M.; Carr, D.J. Degeneration and regeneration of corneal nerves in response to HSV-1 infection. Investig. Ophthalmol. Vis. Sci. 2015, 56, 1097–1107. [Google Scholar] [CrossRef] [Green Version]
- Antony, F.; Pundkar, C.; Sandey, M.; Jaiswal, A.K.; Mishra, A.; Kumar, A.; Channappanavar, R.; Suryawanshi, A. IFN-lambda Regulates Neutrophil Biology to Suppress Inflammation in Herpes Simplex Virus-1-Induced Corneal Immunopathology. J. Immunol. 2021, 206, 1866–1877. [Google Scholar] [CrossRef]
- Conrady, C.D.; Jones, H.; Zheng, M.; Carr, D.J. A Functional Type I Interferon Pathway Drives Resistance to Cornea Herpes Simplex Virus Type 1 Infection by Recruitment of Leukocytes. J. Biomed. Res. 2011, 25, 111–119. [Google Scholar] [CrossRef]
- Chew, T.; Taylor, K.E.; Mossman, K.L. Innate and Adaptive Immune Responses to Herpes Simplex Virus. Viruses 2009, 1, 979–1002. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hendricks, R.L.; Tumpey, T.M. Contribution of virus and immune factors to herpes simplex virus type I-induced corneal pathology. Investig. Ophthalmol. Vis. Sci. 1990, 31, 1929–1939. [Google Scholar]
- Mercadal, C.M.; Martin, S.; Rouse, B.T. Apparent requirement for CD4+ T cells in primary anti-herpes simplex virus cytotoxic T-lymphocyte induction can be overcome by optimal antigen presentation. Viral Immunol. 1991, 4, 177–186. [Google Scholar] [CrossRef]
- Rowe, A.M.; St Leger, A.J.; Jeon, S.; Dhaliwal, D.K.; Knickelbein, J.E.; Hendricks, R.L. Herpes keratitis. Prog. Retin. Eye Res. 2013, 32, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Le Bon, A.; Tough, D.F. Links between innate and adaptive immunity via type I interferon. Curr. Opin. Immunol. 2002, 14, 432–436. [Google Scholar] [CrossRef]
- Simmons, D.P.; Wearsch, P.A.; Canaday, D.H.; Meyerson, H.J.; Liu, Y.C.; Wang, Y.; Boom, W.H.; Harding, C.V. Type I IFN drives a distinctive dendritic cell maturation phenotype that allows continued class II MHC synthesis and antigen processing. J. Immunol. 2012, 188, 3116–3126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hu, K.; Harris, D.L.; Yamaguchi, T.; von Andrian, U.H.; Hamrah, P. A Dual Role for Corneal Dendritic Cells in Herpes Simplex Keratitis: Local Suppression of Corneal Damage and Promotion of Systemic Viral Dissemination. PLoS ONE 2015, 10, e0137123. [Google Scholar] [CrossRef] [Green Version]
- Yin, Y.; Favoreel, H.W. Herpesviruses and the Type III Interferon System. Virol. Sin. 2021, 36, 577–587. [Google Scholar] [CrossRef]
- Danastas, K.; Miranda-Saksena, M.; Cunningham, A.L. Herpes Simplex Virus Type 1 Interactions with the Interferon System. Int. J. Mol. Sci. 2020, 21, 5150. [Google Scholar] [CrossRef]
- Rasmussen, S.B.; Sørensen, L.N.; Malmgaard, L.; Ank, N.; Baines, J.D.; Chen, Z.J.; Paludan, S.R. Type I interferon production during herpes simplex virus infection is controlled by cell-type-specific viral recognition through Toll-like receptor 9, the mitochondrial antiviral signaling protein pathway, and novel recognition systems. J. Virol. 2007, 81, 13315–13324. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McNab, F.; Mayer-Barber, K.; Sher, A.; Wack, A.; O’Garra, A. Type I interferons in infectious disease. Nat. Rev. Immunol. 2015, 15, 87–103. [Google Scholar] [CrossRef]
- Lazear, H.M.; Schoggins, J.W.; Diamond, M.S. Shared and Distinct Functions of Type I and Type III Interferons. Immunity 2019, 50, 907–923. [Google Scholar] [CrossRef]
- Koganti, R.; Yadavalli, T.; Naqvi, R.A.; Shukla, D.; Naqvi, A.R. Pathobiology and treatment of viral keratitis. Exp. Eye Res. 2021, 205, 108483. [Google Scholar] [CrossRef]
- Tough, D.F. Type I interferon as a link between innate and adaptive immunity through dendritic cell stimulation. Leuk. Lymphoma 2004, 45, 257–264. [Google Scholar] [CrossRef]
- Casrouge, A.; Zhang, S.-Y.; Eidenschenk, C.; Jouanguy, E.; Puel, A.; Yang, K.; Alcais, A.; Picard, C.; Mahfoufi, N.; Nicolas, N. Herpes simplex virus encephalitis in human UNC-93B deficiency. Science 2006, 314, 308–312. [Google Scholar] [CrossRef] [PubMed]
- Zhang, S.-Y.; Jouanguy, E.; Ugolini, S.; Smahi, A.; Elain, G.; Romero, P.; Segal, D.; Sancho-Shimizu, V.; Lorenzo, L.; Puel, A. TLR3 deficiency in patients with herpes simplex encephalitis. Science 2007, 317, 1522–1527. [Google Scholar] [CrossRef]
- Rosato, P.C.; Leib, D.A. Neuronal interferon signaling is required for protection against herpes simplex virus replication and pathogenesis. PLoS Pathog. 2015, 11, e1005028. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Zheng, C. The Race between Host Antiviral Innate Immunity and the Immune Evasion Strategies of Herpes Simplex Virus 1. Microbiol. Mol. Biol. Rev. 2020, 84, e00099-20. [Google Scholar] [CrossRef]
- Su, C.; Zhan, G.; Zheng, C. Evasion of host antiviral innate immunity by HSV-1, an update. Virol. J. 2016, 13, 38. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greenan, E.; Gallagher, S.; Khalil, R.; Murphy, C.C.; Ni Gabhann-Dromgoole, J. Advancing Our Understanding of Corneal Herpes Simplex Virus-1 Immune Evasion Mechanisms and Future Therapeutics. Viruses 2021, 13, 1856. [Google Scholar] [CrossRef] [PubMed]
- Heming, J.D.; Conway, J.F.; Homa, F.L. Herpesvirus Capsid Assembly and DNA Packaging. Adv. Anat. Embryol. Cell Biol. 2017, 223, 119–142. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McGeoch, D.J.; Rixon, F.J.; Davison, A.J. Topics in herpesvirus genomics and evolution. Virus Res. 2006, 117, 90–104. [Google Scholar] [CrossRef] [PubMed]
- Agelidis, A.M.; Shukla, D. Cell entry mechanisms of HSV: What we have learned in recent years. Future Virol. 2015, 10, 1145–1154. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yoon, M.; Zago, A.; Shukla, D.; Spear, P.G. Mutations in the N termini of herpes simplex virus type 1 and 2 gDs alter functional interactions with the entry/fusion receptors HVEM, nectin-2, and 3-O-sulfated heparan sulfate but not with nectin-1. J. Virol. 2003, 77, 9221–9231. [Google Scholar] [CrossRef] [Green Version]
- Gianni, T.; Salvioli, S.; Chesnokova, L.S.; Hutt-Fletcher, L.M.; Campadelli-Fiume, G. αvβ6- and αvβ8-integrins serve as interchangeable receptors for HSV gH/gL to promote endocytosis and activation of membrane fusion. PLoS Pathog. 2013, 9, e1003806. [Google Scholar] [CrossRef] [Green Version]
- Cooper, R.S.; Georgieva, E.R.; Borbat, P.P.; Freed, J.H.; Heldwein, E.E. Structural basis for membrane anchoring and fusion regulation of the herpes simplex virus fusogen gB. Nat. Struct. Mol. Biol. 2018, 25, 416–424. [Google Scholar] [CrossRef]
- Madavaraju, K.; Koganti, R.; Volety, I.; Yadavalli, T.; Shukla, D. Herpes Simplex Virus Cell Entry Mechanisms: An Update. Front. Cell. Infect. Microbiol. 2021, 10, 617578. [Google Scholar] [CrossRef]
- Arii, J.; Goto, H.; Suenaga, T.; Oyama, M.; Kozuka-Hata, H.; Imai, T.; Minowa, A.; Akashi, H.; Arase, H.; Kawaoka, Y.; et al. Non-muscle myosin IIA is a functional entry receptor for herpes simplex virus-1. Nature 2010, 467, 859–862. [Google Scholar] [CrossRef]
- Satoh, T.; Arii, J.; Suenaga, T.; Wang, J.; Kogure, A.; Uehori, J.; Arase, N.; Shiratori, I.; Tanaka, S.; Kawaguchi, Y.; et al. PILRalpha is a herpes simplex virus-1 entry coreceptor that associates with glycoprotein B. Cell 2008, 132, 935–944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schelhaas, M.; Jansen, M.; Haase, I.; Knebel-Mörsdorf, D. Herpes simplex virus type 1 exhibits a tropism for basal entry in polarized epithelial cells. J. Gen. Virol. 2003, 84, 2473–2484. [Google Scholar] [CrossRef]
- Karaba, A.H.; Kopp, S.J.; Longnecker, R. Herpesvirus entry mediator and nectin-1 mediate herpes simplex virus 1 infection of the murine cornea. J. Virol. 2011, 85, 10041–10047. [Google Scholar] [CrossRef] [Green Version]
- Radtke, K.; Kieneke, D.; Wolfstein, A.; Michael, K.; Steffen, W.; Scholz, T.; Karger, A.; Sodeik, B. Plus- and minus-end directed microtubule motors bind simultaneously to herpes simplex virus capsids using different inner tegument structures. PLoS Pathog. 2010, 6, e1000991. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schipke, J.; Pohlmann, A.; Diestel, R.; Binz, A.; Rudolph, K.; Nagel, C.H.; Bauerfeind, R.; Sodeik, B. The C terminus of the large tegument protein pUL36 contains multiple capsid binding sites that function differently during assembly and cell entry of herpes simplex virus. J. Virol. 2012, 86, 3682–3700. [Google Scholar] [CrossRef] [Green Version]
- Zaichick, S.V.; Bohannon, K.P.; Hughes, A.; Sollars, P.J.; Pickard, G.E.; Smith, G.A. The herpesvirus VP1/2 protein is an effector of dynein-mediated capsid transport and neuroinvasion. Cell Host Microbe 2013, 13, 193–203. [Google Scholar] [CrossRef] [Green Version]
- Krautwald, M.; Fuchs, W.; Klupp, B.G.; Mettenleiter, T.C. Translocation of Incoming Pseudorabies Virus Capsids to the Cell Nucleus Is Delayed in the Absence of Tegument Protein pUL37. J. Virol. 2009, 83, 3389–3396. [Google Scholar] [CrossRef] [Green Version]
- Kelly, B.J.; Bauerfeind, R.; Binz, A.; Sodeik, B.; Laimbacher, A.S.; Fraefel, C.; Diefenbach, R.J. The interaction of the HSV-1 tegument proteins pUL36 and pUL37 is essential for secondary envelopment during viral egress. Virology 2014, 454–455, 67–77. [Google Scholar] [CrossRef] [Green Version]
- Rode, K.; Döhner, K.; Binz, A.; Glass, M.; Strive, T.; Bauerfeind, R.; Sodeik, B. Uncoupling Uncoating of Herpes Simplex Virus Genomes from Their Nuclear Import and Gene Expression. J. Virol. 2011, 85, 4271–4283. [Google Scholar] [CrossRef] [Green Version]
- Birkenheuer, C.H.; Danko, C.G.; Baines, J.D. Herpes Simplex Virus 1 Dramatically Alters Loading and Positioning of RNA Polymerase II on Host Genes Early in Infection. J. Virol. 2018, 92, e02184-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.F.; Wagner, E.K. The kinetics of expression of individual herpes simplex virus type 1 transcripts. Virus Genes 1987, 1, 49–60. [Google Scholar] [CrossRef]
- Packard, J.E.; Dembowski, J.A. HSV-1 DNA Replication-Coordinated Regulation by Viral and Cellular Factors. Viruses 2021, 13, 2015. [Google Scholar] [CrossRef] [PubMed]
- Hay, J.; Ruyechan, W.T. Regulation of herpes simplex virus type 1 gene expression. Curr. Top Microbiol. Immunol. 1992, 179, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Weir, J.P. Regulation of herpes simplex virus gene expression. Gene 2001, 271, 117–130. [Google Scholar] [CrossRef] [PubMed]
- Arvin, A.; Campadelli-Fiume, G.; Mocarski, E.; Moore, P.S.; Roizman, B.; Whitley, R.; Yamanishi, K. (Eds.) Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis; Cambridge University Press: Cambridge, UK, 2007. [Google Scholar]
- Pesola, J.M.; Zhu, J.; Knipe, D.M.; Coen, D.M. Herpes simplex virus 1 immediate-early and early gene expression during reactivation from latency under conditions that prevent infectious virus production. J. Virol. 2005, 79, 14516–14525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dremel, S.E.; DeLuca, N.A. Genome replication affects transcription factor binding mediating the cascade of herpes simplex virus transcription. Proc. Natl. Acad. Sci. USA 2019, 116, 3734–3739. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gruffat, H.; Marchione, R.; Manet, E. Herpesvirus Late Gene Expression: A Viral-Specific Pre-initiation Complex Is Key. Front. Microbiol. 2016, 7, 869. [Google Scholar] [CrossRef]
- Fan, D.; Wang, M.; Cheng, A.; Jia, R.; Yang, Q.; Wu, Y.; Zhu, D.; Zhao, X.; Chen, S.; Liu, M.; et al. The Role of VP16 in the Life Cycle of Alphaherpesviruses. Front. Microbiol. 2020, 11, 1910. [Google Scholar] [CrossRef]
- Döhner, K.; Ramos-Nascimento, A.; Bialy, D.; Anderson, F.; Hickford-Martinez, A.; Rother, F.; Koithan, T.; Rudolph, K.; Buch, A.; Prank, U.; et al. Importin α1 is required for nuclear import of herpes simplex virus proteins and capsid assembly in fibroblasts and neurons. PLoS Pathog. 2018, 14, e1006823. [Google Scholar] [CrossRef]
- Mettenleiter, T.C. Herpesvirus Assembly and Egress. J. Virol. 2002, 76, 1537–1547. [Google Scholar] [CrossRef] [Green Version]
- Johnson, D.C.; Baines, J.D. Herpesviruses remodel host membranes for virus egress. Nat. Rev. Microbiol. 2011, 9, 382–394. [Google Scholar] [CrossRef] [PubMed]
- Roller, R.J. Nuclear egress of herpesviruses. Virol. Sin. 2008, 23, 406–415. [Google Scholar] [CrossRef]
- Maric, M.; Haugo, A.C.; Dauer, W.; Johnson, D.; Roller, R.J. Nuclear envelope breakdown induced by herpes simplex virus type 1 involves the activity of viral fusion proteins. Virology 2014, 460–461, 128–137. [Google Scholar] [CrossRef] [Green Version]
- Funk, C.; Ott, M.; Raschbichler, V.; Nagel, C.H.; Binz, A.; Sodeik, B.; Bauerfeind, R.; Bailer, S.M. The Herpes Simplex Virus Protein pUL31 Escorts Nucleocapsids to Sites of Nuclear Egress, a Process Coordinated by Its N-Terminal Domain. PLoS Pathog. 2015, 11, e1004957. [Google Scholar] [CrossRef] [Green Version]
- Owen, D.J.; Crump, C.M.; Graham, S.C. Tegument Assembly and Secondary Envelopment of Alphaherpesviruses. Viruses 2015, 7, 5084–5114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sandbaumhüter, M.; Döhner, K.; Schipke, J.; Binz, A.; Pohlmann, A.; Sodeik, B.; Bauerfeind, R. Cytosolic herpes simplex virus capsids not only require binding inner tegument protein pUL36 but also pUL37 for active transport prior to secondary envelopment. Cell Microbiol. 2013, 15, 248–269. [Google Scholar] [CrossRef]
- Avitabile, E.; Di Gaeta, S.; Torrisi, M.R.; Ward, P.L.; Roizman, B.; Campadelli-Fiume, G. Redistribution of microtubules and Golgi apparatus in herpes simplex virus-infected cells and their role in viral exocytosis. J. Virol. 1995, 69, 7472–7482. [Google Scholar] [CrossRef] [Green Version]
- Sharma, P.; Kapoor, D.; Shukla, D. Role of Heparanase and Syndecan-1 in HSV-1 Release from Infected Cells. Viruses 2022, 14, 2156. [Google Scholar] [CrossRef]
- Banerjee, A.; Kulkarni, S.; Mukherjee, A. Herpes Simplex Virus: The Hostile Guest That Takes Over Your Home. Front. Microbiol. 2020, 11, 733. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, I.; Wilson, D.W. HSV-1 Cytoplasmic Envelopment and Egress. Int. J. Mol. Sci. 2020, 21, 5969. [Google Scholar] [CrossRef]
- Knipe, D.M.; Cliffe, A. Chromatin control of herpes simplex virus lytic and latent infection. Nat. Rev. Microbiol. 2008, 6, 211–221. [Google Scholar] [CrossRef] [PubMed]
- Bloom, D.C.; Giordani, N.V.; Kwiatkowski, D.L. Epigenetic regulation of latent HSV-1 gene expression. Biochim. Biophys. Acta 2010, 1799, 246–256. [Google Scholar] [CrossRef] [Green Version]
- Catez, F.; Picard, C.; Held, K.; Gross, S.; Rousseau, A.; Theil, D.; Sawtell, N.; Labetoulle, M.; Lomonte, P. HSV-1 genome subnuclear positioning and associations with host-cell PML-NBs and centromeres regulate LAT locus transcription during latency in neurons. PLoS Pathog. 2012, 8, e1002852. [Google Scholar] [CrossRef] [PubMed]
- Maillet, S.; Naas, T.; Crepin, S.; Roque-Afonso, A.M.; Lafay, F.; Efstathiou, S.; Labetoulle, M. Herpes simplex virus type 1 latently infected neurons differentially express latency-associated and ICP0 transcripts. J. Virol. 2006, 80, 9310–9321. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Valerio, G.S.; Lin, C.C. Ocular manifestations of herpes simplex virus. Curr. Opin. Ophthalmol. 2019, 30, 525–531. [Google Scholar] [CrossRef]
- Yi, X.; Wang, Y.; Yu, F.S. Corneal epithelial tight junctions and their response to lipopolysaccharide challenge. Investig. Ophthalmol. Vis. Sci. 2000, 41, 4093–4100. [Google Scholar]
- Mantelli, F.; Mauris, J.; Argueso, P. The ocular surface epithelial barrier and other mechanisms of mucosal protection: From allergy to infectious diseases. Curr. Opin. Allergy Clin. Immunol. 2013, 13, 563–568. [Google Scholar] [CrossRef] [Green Version]
- Marchiando, A.M.; Graham, W.V.; Turner, J.R. Epithelial barriers in homeostasis and disease. Annu. Rev. Pathol. 2010, 5, 119–144. [Google Scholar] [CrossRef]
- Moens, E.; Veldhoen, M. Epithelial barrier biology: Good fences make good neighbours. Immunology 2012, 135, 1–8. [Google Scholar] [CrossRef]
- Duarte, L.F.; Reyes, A.; Farías, M.A.; Riedel, C.A.; Bueno, S.M.; Kalergis, A.M.; González, P.A. Crosstalk Between Epithelial Cells, Neurons and Immune Mediators in HSV-1 Skin Infection. Front. Immunol. 2021, 12, 662234. [Google Scholar] [CrossRef]
- Yasin, B.; Pang, M.; Turner, J.S.; Cho, Y.; Dinh, N.N.; Waring, A.J.; Lehrer, R.I.; Wagar, E.A. Evaluation of the inactivation of infectious Herpes simplex virus by host-defense peptides. Eur. J. Clin. Microbiol. Infect. Dis. 2000, 19, 187–194. [Google Scholar] [CrossRef] [PubMed]
- Yasin, B.; Wang, W.; Pang, M.; Cheshenko, N.; Hong, T.; Waring, A.J.; Herold, B.C.; Wagar, E.A.; Lehrer, R.I. Theta defensins protect cells from infection by herpes simplex virus by inhibiting viral adhesion and entry. J. Virol. 2004, 78, 5147–5156. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mantelli, F.; Argueso, P. Functions of ocular surface mucins in health and disease. Curr. Opin. Allergy Clin. Immunol. 2008, 8, 477–483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gordon, Y.J.; Huang, L.C.; Romanowski, E.G.; Yates, K.A.; Proske, R.J.; McDermott, A.M. Human cathelicidin (LL-37), a multifunctional peptide, is expressed by ocular surface epithelia and has potent antibacterial and antiviral activity. Curr. Eye Res. 2005, 30, 385–394. [Google Scholar] [CrossRef]
- Diamond, G.; Molchanova, N.; Herlan, C.; Fortkort, J.A.; Lin, J.S.; Figgins, E.; Bopp, N.; Ryan, L.K.; Chung, D.; Adcock, R.S.; et al. Potent Antiviral Activity against HSV-1 and SARS-CoV-2 by Antimicrobial Peptoids. Pharmaceuticals 2021, 14, 304. [Google Scholar] [CrossRef]
- Jamali, A.; Hu, K.; Sendra, V.G.; Blanco, T.; Lopez, M.J.; Ortiz, G.; Qazi, Y.; Zheng, L.; Turhan, A.; Harris, D.L.; et al. Characterization of Resident Corneal Plasmacytoid Dendritic Cells and Their Pivotal Role in Herpes Simplex Keratitis. Cell Rep. 2020, 32, 108099. [Google Scholar] [CrossRef]
- Paludan, S.R.; Bowie, A.G.; Horan, K.A.; Fitzgerald, K.A. Recognition of herpesviruses by the innate immune system. Nat. Rev. Immunol. 2011, 11, 143–154. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. The role of pattern-recognition receptors in innate immunity: Update on Toll-like receptors. Nat. Immunol. 2010, 11, 373–384. [Google Scholar] [CrossRef]
- Kawai, T.; Akira, S. Toll-like receptors and their crosstalk with other innate receptors in infection and immunity. Immunity 2011, 34, 637–650. [Google Scholar] [CrossRef] [Green Version]
- Takeuchi, O.; Akira, S. Pattern recognition receptors and inflammation. Cell 2010, 140, 805–820. [Google Scholar] [CrossRef] [Green Version]
- Li, D.; Wu, M. Pattern recognition receptors in health and diseases. Signal Transduct. Target. Ther. 2021, 6, 291. [Google Scholar] [CrossRef] [PubMed]
- Moretti, J.; Blander, J.M. Insights into phagocytosis-coupled activation of pattern recognition receptors and inflammasomes. Curr. Opin. Immunol. 2014, 26, 100–110. [Google Scholar] [CrossRef] [Green Version]
- Ma, Y.; He, B. Recognition of herpes simplex viruses: Toll-like receptors and beyond. J. Mol. Biol. 2014, 426, 1133–1147. [Google Scholar] [CrossRef] [Green Version]
- Akira, S.; Uematsu, S.; Takeuchi, O. Pathogen Recognition and Innate Immunity. Cell 2006, 124, 783–801. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol. 2004, 4, 499–511. [Google Scholar] [CrossRef] [PubMed]
- Chang, L.; Karin, M. Mammalian MAP kinase signalling cascades. Nature 2001, 410, 37–40. [Google Scholar] [CrossRef] [PubMed]
- Hoebe, K.; Janssen, E.M.; Kim, S.O.; Alexopoulou, L.; Flavell, R.A.; Han, J.; Beutler, B. Upregulation of costimulatory molecules induced by lipopolysaccharide and double-stranded RNA occurs by Trif-dependent and Trif-independent pathways. Nat. Immunol. 2003, 4, 1223–1229. [Google Scholar] [CrossRef]
- Honda, K.; Sakaguchi, S.; Nakajima, C.; Watanabe, A.; Yanai, H.; Matsumoto, M.; Ohteki, T.; Kaisho, T.; Takaoka, A.; Akira, S.; et al. Selective contribution of IFN-alpha/beta signaling to the maturation of dendritic cells induced by double-stranded RNA or viral infection. Proc. Natl. Acad. Sci. USA 2003, 100, 10872–10877. [Google Scholar] [CrossRef] [Green Version]
- Kawasaki, T.; Kawai, T. Toll-Like Receptor Signaling Pathways. Front. Immunol. 2014, 5, 461. [Google Scholar] [CrossRef] [Green Version]
- Kenny, E.F.; O’Neill, L.A. Signalling adaptors used by Toll-like receptors: An update. Cytokine 2008, 43, 342–349. [Google Scholar] [CrossRef]
- Carty, M.; Goodbody, R.; Schröder, M.; Stack, J.; Moynagh, P.N.; Bowie, A.G. The human adaptor SARM negatively regulates adaptor protein TRIF-dependent Toll-like receptor signaling. Nat. Immunol. 2006, 7, 1074–1081. [Google Scholar] [CrossRef]
- Jacobs, M.D.; Harrison, S.C. Structure of an IkappaBalpha/NF-kappaB complex. Cell 1998, 95, 749–758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hinz, M.; Scheidereit, C. The IκB kinase complex in NF-κB regulation and beyond. EMBO Rep. 2014, 15, 46–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blasius, A.L.; Beutler, B. Intracellular toll-like receptors. Immunity 2010, 32, 305–315. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leifer, C.A.; Kennedy, M.N.; Mazzoni, A.; Lee, C.; Kruhlak, M.J.; Segal, D.M. TLR9 is localized in the endoplasmic reticulum prior to stimulation. J. Immunol. 2004, 173, 1179–1183. [Google Scholar] [CrossRef] [Green Version]
- Honda, K.; Takaoka, A.; Taniguchi, T. Type I Inteferon Gene Induction by the Interferon Regulatory Factor Family of Transcription Factors. Immunity 2006, 25, 349–360. [Google Scholar] [CrossRef] [Green Version]
- Lukhele, S.; Boukhaled, G.M.; Brooks, D.G. Type I interferon signaling, regulation and gene stimulation in chronic virus infection. Semin. Immunol. 2019, 43, 101277. [Google Scholar] [CrossRef]
- Zhao, J.; Qin, C.; Liu, Y.; Rao, Y.; Feng, P. Herpes Simplex Virus and Pattern Recognition Receptors: An Arms Race. Front. Immunol. 2020, 11, 613799. [Google Scholar] [CrossRef]
- Zheng, W.; Xu, Q.; Zhang, Y.; E, X.; Gao, W.; Zhang, M.; Zhai, W.; Rajkumar, R.S.; Liu, Z. Toll-like receptor-mediated innate immunity against herpesviridae infection: A current perspective on viral infection signaling pathways. Virol. J. 2020, 17, 192. [Google Scholar] [CrossRef]
- Jin, X.; Qin, Q.; Chen, W.; Qu, J. Expression of toll-like receptors in the healthy and herpes simplex virus-infected cornea. Cornea 2007, 26, 847–852. [Google Scholar] [CrossRef]
- Johnson, A.C.; Heinzel, F.P.; Diaconu, E.; Sun, Y.; Hise, A.G.; Golenbock, D.; Lass, J.H.; Pearlman, E. Activation of Toll-Like Receptor (TLR)2, TLR4, and TLR9 in the Mammalian Cornea Induces MyD88-Dependent Corneal Inflammation. Investig. Ophthalmol. Vis. Sci. 2005, 46, 589–595. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pearlman, E.; Johnson, A.; Adhikary, G.; Sun, Y.; Chinnery, H.R.; Fox, T.; Kester, M.; McMenamin, P.G. Toll-like receptors at the ocular surface. Ocul. Surf. 2008, 6, 108–116. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leoni, V.; Gianni, T.; Salvioli, S.; Campadelli-Fiume, G. Herpes simplex virus glycoproteins gH/gL and gB bind Toll-like receptor 2, and soluble gH/gL is sufficient to activate NF-κB. J. Virol. 2012, 86, 6555–6562. [Google Scholar] [CrossRef] [Green Version]
- Sarangi, P.P.; Kim, B.; Kurt-Jones, E.; Rouse, B.T. Innate recognition network driving herpes simplex virus-induced corneal immunopathology: Role of the toll pathway in early inflammatory events in stromal keratitis. J. Virol. 2007, 81, 11128–11138. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Welner, R.S.; Pelayo, R.; Nagai, Y.; Garrett, K.P.; Wuest, T.R.; Carr, D.J.; Borghesi, L.A.; Farrar, M.A.; Kincade, P.W. Lymphoid precursors are directed to produce dendritic cells as a result of TLR9 ligation during herpes infection. Blood 2008, 112, 3753–3761. [Google Scholar] [CrossRef]
- Zheng, M.; Klinman, D.M.; Gierynska, M.; Rouse, B.T. DNA containing CpG motifs induces angiogenesis. Proc. Natl. Acad. Sci. USA 2002, 99, 8944–8949. [Google Scholar] [CrossRef] [Green Version]
- Wuest, T.; Austin, B.A.; Uematsu, S.; Thapa, M.; Akira, S.; Carr, D.J. Intact TRL 9 and type I interferon signaling pathways are required to augment HSV-1 induced corneal CXCL9 and CXCL10. J. Neuroimmunol. 2006, 179, 46–52. [Google Scholar] [CrossRef] [Green Version]
- Zheng, C. Evasion of Cytosolic DNA-Stimulated Innate Immune Responses by Herpes Simplex Virus 1. J. Virol. 2018, 92, e00099-17. [Google Scholar] [CrossRef] [Green Version]
- Sun, L.; Wu, J.; Du, F.; Chen, X.; Chen, Z.J. Cyclic GMP-AMP synthase is a cytosolic DNA sensor that activates the type I interferon pathway. Science 2013, 339, 786–791. [Google Scholar] [CrossRef] [Green Version]
- Iqbal, J.; Ansari, M.A.; Kumar, B.; Dutta, D.; Roy, A.; Chikoti, L.; Pisano, G.; Dutta, S.; Vahedi, S.; Veettil, M.V.; et al. Histone H2B-IFI16 Recognition of Nuclear Herpesviral Genome Induces Cytoplasmic Interferon-β Responses. PLoS Pathog. 2016, 12, e1005967. [Google Scholar] [CrossRef] [Green Version]
- Ishikawa, H.; Ma, Z.; Barber, G.N. STING regulates intracellular DNA-mediated, type I interferon-dependent innate immunity. Nature 2009, 461, 788–792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishikawa, H.; Barber, G.N. STING is an endoplasmic reticulum adaptor that facilitates innate immune signalling. Nature 2008, 455, 674–678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Decout, A.; Katz, J.D.; Venkatraman, S.; Ablasser, A. The cGAS–STING pathway as a therapeutic target in inflammatory diseases. Nat. Rev. Immunol. 2021, 21, 548–569. [Google Scholar] [CrossRef]
- Ablasser, A.; Goldeck, M.; Cavlar, T.; Deimling, T.; Witte, G.; Röhl, I.; Hopfner, K.-P.; Ludwig, J.; Hornung, V. cGAS produces a 2′-5′-linked cyclic dinucleotide second messenger that activates STING. Nature 2013, 498, 380–384. [Google Scholar] [CrossRef] [Green Version]
- Royer, D.J.; Carr, D.J. A STING-dependent innate-sensing pathway mediates resistance to corneal HSV-1 infection via upregulation of the antiviral effector tetherin. Mucosal. Immunol. 2016, 9, 1065–1075. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tan, T.C.; Xia, L.K. TRIM21 Aggravates Herpes Simplex Virus Epithelial Keratitis by Attenuating STING-IRF3-Mediated Type I Interferon Signaling. Front. Microbiol. 2020, 11, 703. [Google Scholar] [CrossRef] [Green Version]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; Xiao, T.S.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef] [Green Version]
- Howard, T.R.; Lum, K.K.; Kennedy, M.A.; Cristea, I.M. The Nuclear DNA Sensor IFI16 Indiscriminately Binds to and Diminishes Accessibility of the HSV-1 Genome to Suppress Infection. mSystems 2022, 7, e0019822. [Google Scholar] [CrossRef] [PubMed]
- Johnson, K.E.; Bottero, V.; Flaherty, S.; Dutta, S.; Singh, V.V.; Chandran, B. IFI16 restricts HSV-1 replication by accumulating on the hsv-1 genome, repressing HSV-1 gene expression, and directly or indirectly modulating histone modifications. PLoS Pathog. 2014, 10, e1004503. [Google Scholar] [CrossRef]
- Conrady, C.D.; Zheng, M.; Fitzgerald, K.A.; Liu, C.; Carr, D.J. Resistance to HSV-1 infection in the epithelium resides with the novel innate sensor, IFI-16. Mucosal. Immunol. 2012, 5, 173–183. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.S.; Vidhyasagar, V.; Yang, S.; Arna, A.B.; Yadav, M.; Aggarwal, A.; Aguilera, A.N.; Shinriki, S.; Bhanumathy, K.K.; Pandey, K.; et al. DDX41 is required for cGAS-STING activation against DNA virus infection. Cell Rep. 2022, 39, 110856. [Google Scholar] [CrossRef]
- Takaoka, A.; Wang, Z.; Choi, M.K.; Yanai, H.; Negishi, H.; Ban, T.; Lu, Y.; Miyagishi, M.; Kodama, T.; Honda, K.; et al. DAI (DLM-1/ZBP1) is a cytosolic DNA sensor and an activator of innate immune response. Nature 2007, 448, 501–505. [Google Scholar] [CrossRef]
- Pham, T.H.; Kwon, K.M.; Kim, Y.E.; Kim, K.K.; Ahn, J.H. DNA sensing-independent inhibition of herpes simplex virus 1 replication by DAI/ZBP1. J. Virol. 2013, 87, 3076–3086. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chan, C.P.; Jin, D.Y. Cytoplasmic RNA sensors and their interplay with RNA-binding partners in innate antiviral response: Theme and variations. RNA 2022, 28, 449–477. [Google Scholar] [CrossRef]
- Rehwinkel, J.; Gack, M.U. RIG-I-like receptors: Their regulation and roles in RNA sensing. Nat. Rev. Immunol. 2020, 20, 537–551. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.G.; Hur, S. Cellular origins of dsRNA, their recognition and consequences. Nat. Rev. Mol. Cell Biol. 2022, 23, 286–301. [Google Scholar] [CrossRef] [PubMed]
- Kell, A.M.; Gale, M., Jr. RIG-I in RNA virus recognition. Virology 2015, 479–480, 110–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Melchjorsen, J.; Rintahaka, J.; Søby, S.; Horan, K.A.; Poltajainen, A.; Østergaard, L.; Paludan, S.R.; Matikainen, S. Early innate recognition of herpes simplex virus in human primary macrophages is mediated via the MDA5/MAVS-dependent and MDA5/MAVS/RNA polymerase III-independent pathways. J. Virol. 2010, 84, 11350–11358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chiang, J.J.; Sparrer, K.M.J.; van Gent, M.; Lässig, C.; Huang, T.; Osterrieder, N.; Hopfner, K.P.; Gack, M.U. Viral unmasking of cellular 5S rRNA pseudogene transcripts induces RIG-I-mediated immunity. Nat. Immunol. 2018, 19, 53–62. [Google Scholar] [CrossRef]
- Chiu, Y.-H.; MacMillan, J.B.; Chen, Z.J. RNA Polymerase III Detects Cytosolic DNA and Induces Type I Interferons through the RIG-I Pathway. Cell 2009, 138, 576–591. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Goulet, M.L.; Sze, A.; Hadj, S.B.; Belgnaoui, S.M.; Lababidi, R.R.; Zheng, C.; Fritz, J.H.; Olagnier, D.; Lin, R. RIG-I-Mediated STING Upregulation Restricts Herpes Simplex Virus 1 Infection. J. Virol. 2016, 90, 9406–9419. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Li, R.; Li, Y.; Chen, J.; Feng, F.; Sun, C. Broadly Antiviral Activities of TAP1 through Activating the TBK1-IRF3-Mediated Type I Interferon Production. Int. J. Mol. Sci. 2021, 22, 4668. [Google Scholar] [CrossRef]
- Lee, A.J.; Ashkar, A.A. The Dual Nature of Type I and Type II Interferons. Front. Immunol. 2018, 9, 2061. [Google Scholar] [CrossRef] [Green Version]
- Mesev, E.V.; LeDesma, R.A.; Ploss, A. Decoding type I and III interferon signalling during viral infection. Nat. Microbiol. 2019, 4, 914–924. [Google Scholar] [CrossRef]
- Ivashkiv, L.B.; Donlin, L.T. Regulation of type I interferon responses. Nat. Rev. Immunol. 2014, 14, 36–49. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pestka, S.; Krause, C.D.; Walter, M.R. Interferons, interferon-like cytokines, and their receptors. Immunol. Rev. 2004, 202, 8–32. [Google Scholar] [CrossRef]
- Lazear, H.M.; Nice, T.J.; Diamond, M.S. Interferon-lambda: Immune Functions at Barrier Surfaces and Beyond. Immunity 2015, 43, 15–28. [Google Scholar] [CrossRef] [Green Version]
- Kotenko, S.V.; Gallagher, G.; Baurin, V.V.; Lewis-Antes, A.; Shen, M.; Shah, N.K.; Langer, J.A.; Sheikh, F.; Dickensheets, H.; Donnelly, R.P. IFN-lambdas mediate antiviral protection through a distinct class II cytokine receptor complex. Nat. Immunol. 2003, 4, 69–77. [Google Scholar] [CrossRef] [PubMed]
- Kotenko, S.V.; Durbin, J.E. Contribution of type III interferons to antiviral immunity: Location, location, location. J. Biol. Chem. 2017, 292, 7295–7303. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zanoni, I.; Granucci, F.; Broggi, A. Interferon (IFN)-lambda Takes the Helm: Immunomodulatory Roles of Type III IFNs. Front. Immunol. 2017, 8, 1661. [Google Scholar] [CrossRef]
- Galani, I.E.; Triantafyllia, V.; Eleminiadou, E.E.; Koltsida, O.; Stavropoulos, A.; Manioudaki, M.; Thanos, D.; Doyle, S.E.; Kotenko, S.V.; Thanopoulou, K.; et al. Interferon-lambda Mediates Non-redundant Front-Line Antiviral Protection against Influenza Virus Infection without Compromising Host Fitness. Immunity 2017, 46, 875–890.e876. [Google Scholar] [CrossRef]
- Ferguson, S.H.; Foster, D.M.; Sherry, B.; Magness, S.T.; Nielsen, D.M.; Gookin, J.L. Interferon-λ3 promotes epithelial defense and barrier function against Cryptosporidium parvum infection. Cell. Mol. Gastroenterol. Hepatol. 2019, 8, 1–20. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wack, A.; Terczyńska-Dyla, E.; Hartmann, R. Guarding the frontiers: The biology of type III interferons. Nat. Immunol. 2015, 16, 802–809. [Google Scholar] [CrossRef] [PubMed]
- Ye, L.; Schnepf, D.; Staeheli, P. Interferon-λ orchestrates innate and adaptive mucosal immune responses. Nat. Rev. Immunol. 2019, 19, 614–625. [Google Scholar] [CrossRef] [PubMed]
- Yin, Z.; Dai, J.; Deng, J.; Sheikh, F.; Natalia, M.; Shih, T.; Lewis-Antes, A.; Amrute, S.B.; Garrigues, U.; Doyle, S. Type III IFNs are produced by and stimulate human plasmacytoid dendritic cells. J. Immunol. 2012, 189, 2735–2745. [Google Scholar] [CrossRef] [Green Version]
- Kelly, A.; Robinson, M.W.; Roche, G.; Biron, C.A.; O’Farrelly, C.; Ryan, E.J. Immune cell profiling of IFN-λ response shows pDCs express highest level of IFN-λR1 and are directly responsive via the JAK-STAT pathway. J. Interferon Cytokine Res. 2016, 36, 671–680. [Google Scholar] [CrossRef] [Green Version]
- Odendall, C.; Kagan, J.C. The unique regulation and functions of type III interferons in antiviral immunity. Curr. Opin. Virol. 2015, 12, 47–52. [Google Scholar] [CrossRef] [Green Version]
- Broggi, A.; Tan, Y.; Granucci, F.; Zanoni, I. IFN-lambda suppresses intestinal inflammation by non-translational regulation of neutrophil function. Nat. Immunol. 2017, 18, 1084–1093. [Google Scholar] [CrossRef]
- Levy, D.E.; Marié, I.J.; Durbin, J.E. Induction and function of type I and III interferon in response to viral infection. Curr. Opin. Virol. 2011, 1, 476–486. [Google Scholar] [CrossRef] [Green Version]
- Crotta, S.; Davidson, S.; Mahlakoiv, T.; Desmet, C.J.; Buckwalter, M.R.; Albert, M.L.; Staeheli, P.; Wack, A. Type I and Type III Interferons Drive Redundant Amplification Loops to Induce a Transcriptional Signature in Influenza-Infected Airway Epithelia. PLoS Pathog. 2013, 9, e1003773. [Google Scholar] [CrossRef] [Green Version]
- Doyle, S.E.; Schreckhise, H.; Khuu-Duong, K.; Henderson, K.; Rosler, R.; Storey, H.; Yao, L.; Liu, H.; Barahmand-pour, F.; Sivakumar, P.; et al. Interleukin-29 uses a type 1 interferon-like program to promote antiviral responses in human hepatocytes. Hepatology 2006, 44, 896–906. [Google Scholar] [CrossRef] [PubMed]
- Marcello, T.; Grakoui, A.; Barba-Spaeth, G.; Machlin, E.S.; Kotenko, S.V.; MacDonald, M.R.; Rice, C.M. Interferons alpha and lambda inhibit hepatitis C virus replication with distinct signal transduction and gene regulation kinetics. Gastroenterology 2006, 131, 1887–1898. [Google Scholar] [CrossRef]
- Bolen, C.R.; Ding, S.Y.; Robek, M.D.; Kleinstein, S.H. Dynamic Expression Profiling of Type I and Type III Interferon-Stimulated Hepatocytes Reveals a Stable Hierarchy of Gene Expression. Hepatology 2014, 59, 1262–1272. [Google Scholar] [CrossRef] [Green Version]
- Jilg, N.; Lin, W.Y.; Hong, J.; Schaefer, E.A.; Wolski, D.; Meixong, J.; Goto, K.; Brisac, C.; Chusri, P.; Fusco, D.N.; et al. Kinetic Differences in the Induction of Interferon Stimulated Genes by Interferon-alpha and Interleukin 28B Are Altered by Infection With Hepatitis C Virus. Hepatology 2014, 59, 1250–1261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kohli, A.; Zhang, X.; Yang, J.; Russell, R.S.; Donnelly, R.P.; Sheikh, F.; Sherman, A.; Young, H.; Imamichi, T.; Lempicki, R.A.; et al. Distinct and overlapping genomic profiles and antiviral effects of Interferon-lambda and -alpha on HCV-infected and noninfected hepatoma cells. J. Viral. Hepatitis. 2012, 19, 843–853. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Bogunovic, D.; Payelle-Brogard, B.; Francois-Newton, V.; Speer, S.D.; Yuan, C.; Volpi, S.; Li, Z.; Sanal, O.; Mansouri, D.; et al. Human intracellular ISG15 prevents interferon-alpha/beta over-amplification and auto-inflammation. Nature 2015, 517, 89–93. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Francois-Newton, V.; Magno de Freitas Almeida, G.; Payelle-Brogard, B.; Monneron, D.; Pichard-Garcia, L.; Piehler, J.; Pellegrini, S.; Uze, G. USP18-based negative feedback control is induced by type I and type III interferons and specifically inactivates interferon alpha response. PLoS ONE 2011, 6, e22200. [Google Scholar] [CrossRef] [Green Version]
- Burke, J.D.; Young, H.A. IFN-γ: A cytokine at the right time, is in the right place. Semin. Immunol. 2019, 43, 101280. [Google Scholar] [CrossRef]
- Ghoreschi, K.; Laurence, A.; O’Shea, J.J. Janus kinases in immune cell signaling. Immunol. Rev. 2009, 228, 273–287. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Platanias, L.C. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat. Rev. Immunol. 2005, 5, 375–386. [Google Scholar] [CrossRef]
- Bigley, N.J. Complexity of Interferon-γ Interactions with HSV-1. Front. Immunol. 2014, 5, 15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schoggins, J.W. Interferon-Stimulated Genes: What Do They All Do? Annu. Rev. Virol. 2019, 6, 567–584. [Google Scholar] [CrossRef]
- Schneider, W.M.; Chevillotte, M.D.; Rice, C.M. Interferon-stimulated genes: A complex web of host defenses. Annu. Rev. Immunol. 2014, 32, 513–545. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Durbin, R.K.; Kotenko, S.V.; Durbin, J.E. Interferon induction and function at the mucosal surface. Immunol. Rev. 2013, 255, 25–39. [Google Scholar] [CrossRef] [PubMed]
- Kaur, S.; Uddin, S.; Platanias, L.C. The PI3’ kinase pathway in interferon signaling. J. Interf. Cytokine Res. 2005, 25, 780–787. [Google Scholar] [CrossRef]
- Zhou, Z.; Hamming, O.J.; Ank, N.; Paludan, S.R.; Nielsen, A.L.; Hartmann, R. Type III interferon (IFN) induces a type I IFN-like response in a restricted subset of cells through signaling pathways involving both the Jak-STAT pathway and the mitogen-activated protein kinases. J. Virol. 2007, 81, 7749–7758. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stanifer, M.L.; Pervolaraki, K.; Boulant, S. Differential Regulation of Type I and Type III Interferon Signaling. Int. J. Mol. Sci. 2019, 20, 1445. [Google Scholar] [CrossRef] [Green Version]
- Lekmine, F.; Uddin, S.; Sassano, A.; Parmar, S.; Brachmann, S.M.; Majchrzak, B.; Sonenberg, N.; Hay, N.; Fish, E.N.; Platanias, L.C. Activation of the p70 S6 Kinase and Phosphorylation of the 4E-BP1 Repressor of mRNA Translation by Type I Interferons*. J. Biol. Chem. 2003, 278, 27772–27780. [Google Scholar] [CrossRef] [Green Version]
- Jordan, W.J.; Eskdale, J.; Srinivas, S.; Pekarek, V.; Kelner, D.; Rodia, M.; Gallagher, G. Human interferon lambda-1 (IFN-lambda1/IL-29) modulates the Th1/Th2 response. Genes Immun 2007, 8, 254–261. [Google Scholar] [CrossRef]
- Wormald, S.; Hilton, D.J. Inhibitors of cytokine signal transduction. J. Biol. Chem. 2004, 279, 821–824. [Google Scholar] [CrossRef] [Green Version]
- Schoggins, J.W. Recent advances in antiviral interferon-stimulated gene biology. F1000Research 2018, 7, 309. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Espenshade, P.J.; Hughes, A.L. Regulation of sterol synthesis in eukaryotes. Annu. Rev. Genet. 2007, 41, 401–427. [Google Scholar] [CrossRef]
- Bordier, B.B.; Marion, P.L.; Ohashi, K.; Kay, M.A.; Greenberg, H.B.; Casey, J.L.; Glenn, J.S. A prenylation inhibitor prevents production of infectious hepatitis delta virus particles. J. Virol. 2002, 76, 10465–10472. [Google Scholar] [CrossRef] [Green Version]
- Bordier, B.B.; Ohkanda, J.; Liu, P.; Lee, S.Y.; Salazar, F.H.; Marion, P.L.; Ohashi, K.; Meuse, L.; Kay, M.A.; Casey, J.L.; et al. In vivo antiviral efficacy of prenylation inhibitors against hepatitis delta virus. J. Clin. Investig. 2003, 112, 407–414. [Google Scholar] [CrossRef] [Green Version]
- Blanc, M.; Hsieh, W.Y.; Robertson, K.A.; Kropp, K.A.; Forster, T.; Shui, G.; Lacaze, P.; Watterson, S.; Griffiths, S.J.; Spann, N.J.; et al. The transcription factor STAT-1 couples macrophage synthesis of 25-hydroxycholesterol to the interferon antiviral response. Immunity 2013, 38, 106–118. [Google Scholar] [CrossRef] [Green Version]
- Cagno, V.; Civra, A.; Rossin, D.; Calfapietra, S.; Caccia, C.; Leoni, V.; Dorma, N.; Biasi, F.; Poli, G.; Lembo, D. Inhibition of herpes simplex-1 virus replication by 25-hydroxycholesterol and 27-hydroxycholesterol. Redox Biol. 2017, 12, 522–527. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.E.; Busse, D.C.; Binter, S.; Weston, S.; Diaz Soria, C.; Laksono, B.M.; Clare, S.; Van Nieuwkoop, S.; Van den Hoogen, B.G.; Clement, M.; et al. Interferon-Induced Transmembrane Protein 1 Restricts Replication of Viruses That Enter Cells via the Plasma Membrane. J. Virol. 2019, 93, e02003-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Reichelt, M.; Stertz, S.; Krijnse-Locker, J.; Haller, O.; Kochs, G. Missorting of LaCrosse virus nucleocapsid protein by the interferon-induced MxA GTPase involves smooth ER membranes. Traffic 2004, 5, 772–784. [Google Scholar] [CrossRef]
- Hefti, H.P.; Frese, M.; Landis, H.; Di Paolo, C.; Aguzzi, A.; Haller, O.; Pavlovic, J. Human MxA protein protects mice lacking a functional alpha/beta interferon system against La crosse virus and other lethal viral infections. J. Virol. 1999, 73, 6984–6991. [Google Scholar] [CrossRef] [Green Version]
- Gao, S.; von der Malsburg, A.; Paeschke, S.; Behlke, J.; Haller, O.; Kochs, G.; Daumke, O. Structural basis of oligomerization in the stalk region of dynamin-like MxA. Nature 2010, 465, 502–506. [Google Scholar] [CrossRef] [Green Version]
- Donaghy, H.; Bosnjak, L.; Harman, A.N.; Marsden, V.; Tyring, S.K.; Meng, T.C.; Cunningham, A.L. Role for Plasmacytoid Dendritic Cells in the Immune Control of Recurrent Human Herpes Simplex Virus Infection. J. Virol. 2009, 83, 1952–1961. [Google Scholar] [CrossRef] [Green Version]
- Ku, C.C.; Che, X.B.; Reichelt, M.; Rajamani, J.; Schaap-Nutt, A.; Huang, K.J.; Sommer, M.H.; Chen, Y.S.; Chen, Y.Y.; Arvin, A.M. Herpes simplex virus-1 induces expression of a novel MxA isoform that enhances viral replication. Immunol. Cell Biol. 2011, 89, 173–182. [Google Scholar] [CrossRef] [Green Version]
- Crameri, M.; Bauer, M.; Caduff, N.; Walker, R.; Steiner, F.; Franzoso, F.D.; Gujer, C.; Boucke, K.; Kucera, T.; Zbinden, A.; et al. MxB is an interferon-induced restriction factor of human herpesviruses. Nat. Commun. 2018, 9, 1980. [Google Scholar] [CrossRef] [Green Version]
- Schilling, M.; Bulli, L.; Weigang, S.; Graf, L.; Naumann, S.; Patzina, C.; Wagner, V.; Bauersfeld, L.; Goujon, C.; Hengel, H.; et al. Human MxB Protein Is a Pan-herpesvirus Restriction Factor. J. Virol. 2018, 92, e01056-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Munir, M.; Berg, M. The multiple faces of proteinkinase R in antiviral defense. Virulence 2013, 4, 85–89. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-khatib, K.; Williams, B.R.; Silverman, R.H.; Halford, W.; Carr, D.J. The murine double-stranded RNA-dependent protein kinase PKR and the murine 2’,5’-oligoadenylate synthetase-dependent RNase L are required for IFN-beta-mediated resistance against herpes simplex virus type 1 in primary trigeminal ganglion culture. Virology 2003, 313, 126–135. [Google Scholar] [CrossRef] [Green Version]
- Low-Calle, A.M.; Prada-Arismendy, J.; Castellanos, J.E. Study of interferon-beta antiviral activity against Herpes simplex virus type 1 in neuron-enriched trigeminal ganglia cultures. Virus Res. 2014, 180, 49–58. [Google Scholar] [CrossRef] [PubMed]
- Floyd-Smith, G.; Slattery, E.; Lengyel, P. Interferon action: RNA cleavage pattern of a (2’-5’)oligoadenylate--dependent endonuclease. Science 1981, 212, 1030–1032. [Google Scholar] [CrossRef]
- Anderson, B.R.; Muramatsu, H.; Jha, B.K.; Silverman, R.H.; Weissman, D.; Kariko, K. Nucleoside modifications in RNA limit activation of 2’-5’-oligoadenylate synthetase and increase resistance to cleavage by RNase L. Nucleic Acids Res. 2011, 39, 9329–9338. [Google Scholar] [CrossRef] [Green Version]
- Yang, E.; Li, M.M.H. All About the RNA: Interferon-Stimulated Genes That Interfere With Viral RNA Processes. Front. Immunol. 2020, 11, 605024. [Google Scholar] [CrossRef]
- Gusho, E.; Baskar, D.; Banerjee, S. New advances in our understanding of the “unique” RNase L in host pathogen interaction and immune signaling. Cytokine 2020, 133, 153847. [Google Scholar] [CrossRef]
- Carr, D.J.; Al-khatib, K.; James, C.M.; Silverman, R. Interferon-beta suppresses herpes simplex virus type 1 replication in trigeminal ganglion cells through an RNase L-dependent pathway. J. Neuroimmunol. 2003, 141, 40–46. [Google Scholar] [CrossRef]
- Sanchez, R.; Mohr, I. Inhibition of cellular 2’-5’ oligoadenylate synthetase by the herpes simplex virus type 1 Us11 protein. J. Virol. 2007, 81, 3455–3464. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, C.; Collins, M.N.; Hsiang, T.Y.; Krug, R.M. Interferon-induced ISG15 pathway: An ongoing virus-host battle. Trends Microbiol. 2013, 21, 181–186. [Google Scholar] [CrossRef]
- Shi, H.X.; Yang, K.; Liu, X.; Liu, X.Y.; Wei, B.; Shan, Y.F.; Zhu, L.H.; Wang, C. Positive regulation of interferon regulatory factor 3 activation by Herc5 via ISG15 modification. Mol. Cell Biol. 2010, 30, 2424–2436. [Google Scholar] [CrossRef] [Green Version]
- Lenschow, D.J.; Lai, C.; Frias-Staheli, N.; Giannakopoulos, N.V.; Lutz, A.; Wolff, T.; Osiak, A.; Levine, B.; Schmidt, R.E.; Garcia-Sastre, A.; et al. IFN-stimulated gene 15 functions as a critical antiviral molecule against influenza, herpes, and Sindbis viruses. Proc. Natl. Acad. Sci. USA 2007, 104, 1371–1376. [Google Scholar] [CrossRef] [Green Version]
- Chin, K.C.; Cresswell, P. Viperin (cig5), an IFN-inducible antiviral protein directly induced by human cytomegalovirus. Proc. Natl. Acad. Sci. USA 2001, 98, 15125–15130. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shen, G.; Wang, K.; Wang, S.; Cai, M.; Li, M.L.; Zheng, C. Herpes simplex virus 1 counteracts viperin via its virion host shutoff protein UL41. J. Virol. 2014, 88, 12163–12166. [Google Scholar] [CrossRef] [Green Version]
- Tseng, Y.Y.; Gowripalan, A.; Croft, S.N.; Smith, S.A.; Helbig, K.J.; Man, S.M.; Tscharke, D.C. Viperin has species-specific roles in response to herpes simplex virus infection. J. Gen. Virol. 2021, 102, 001638. [Google Scholar] [CrossRef] [PubMed]
- Li, M.L.; Liao, Z.M.; Xu, Z.; Zou, X.M.; Wang, Y.F.; Peng, H.; Li, Y.W.; Ou, X.W.; Deng, Y.X.; Guo, Y.J.; et al. The Interaction Mechanism Between Herpes Simplex Virus 1 Glycoprotein D and Host Antiviral Protein Viperin. Front. Immunol. 2019, 10, 2810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Swiecki, M.; Omattage, N.S.; Brett, T.J. BST-2/tetherin: Structural biology, viral antagonism, and immunobiology of a potent host antiviral factor. Mol. Immunol. 2013, 54, 132–139. [Google Scholar] [CrossRef] [PubMed]
- Blondeau, C.; Pelchen-Matthews, A.; Mlcochova, P.; Marsh, M.; Milne, R.S.B.; Towers, G.J. Tetherin Restricts Herpes Simplex Virus 1 and Is Antagonized by Glycoprotein M. J. Virol. 2013, 87, 13124–13133. [Google Scholar] [CrossRef] [Green Version]
- Zenner, H.L.; Mauricio, R.; Banting, G.; Crump, C.M. Herpes Simplex Virus 1 Counteracts Tetherin Restriction via Its Virion Host Shutoff Activity. J. Virol. 2013, 87, 13115–13123. [Google Scholar] [CrossRef] [Green Version]
- Cohen, J.I. Herpesvirus latency. J. Clin. Investig. 2020, 130, 3361–3369. [Google Scholar] [CrossRef]
- Toma, H.S.; Murina, A.T.; Areaux, R.G., Jr.; Neumann, D.M.; Bhattacharjee, P.S.; Foster, T.P.; Kaufman, H.E.; Hill, J.M. Ocular HSV-1 latency, reactivation and recurrent disease. Semin. Ophthalmol. 2008, 23, 249–273. [Google Scholar] [CrossRef]
- Denes, C.E.; Miranda-Saksena, M.; Cunningham, A.L.; Diefenbach, R.J. Cytoskeletons in the ClosetSubversion in Alphaherpesvirus Infections. Viruses 2018, 10, 79. [Google Scholar] [CrossRef] [Green Version]
- Miranda-Saksena, M.; Denes, C.E.; Diefenbach, R.J.; Cunningham, A.L. Infection and Transport of Herpes Simplex Virus Type 1 in Neurons: Role of the Cytoskeleton. Viruses 2018, 10, 92. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bearer, E.L.; Breakefield, X.O.; Schuback, D.; Reese, T.S.; LaVail, J.H. Retrograde axonal transport of herpes simplex virus: Evidence for a single mechanism and a role for tegument. Proc. Natl. Acad. Sci. USA 2000, 97, 8146–8150. [Google Scholar] [CrossRef] [Green Version]
- LaVail, J.H.; Tauscher, A.N.; Aghaian, E.; Harrabi, O.; Sidhu, S.S. Axonal transport and sorting of herpes simplex virus components in a mature mouse visual system. J. Virol. 2003, 77, 6117–6126. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Antinone, S.E.; Zaichick, S.V.; Smith, G.A. Resolving the assembly state of herpes simplex virus during axon transport by live-cell imaging. J. Virol. 2010, 84, 13019–13030. [Google Scholar] [CrossRef] [Green Version]
- Antinone, S.E.; Smith, G.A. Retrograde axon transport of herpes simplex virus and pseudorabies virus: A live-cell comparative analysis. J. Virol. 2010, 84, 1504–1512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bloom, D.C. Alphaherpesvirus Latency: A Dynamic State of Transcription and Reactivation. Adv. Virus Res. 2016, 94, 53–80. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Tscharke, D.C. Herpes Simplex Virus Latency Is Noisier the Closer We Look. J. Virol. 2020, 94, e01701-19. [Google Scholar] [CrossRef] [Green Version]
- Sharma, N.; Wang, C.; Kessler, P.; Sen, G.C. Herpes simplex virus 1 evades cellular antiviral response by inducing microRNA-24, which attenuates STING synthesis. PLoS Pathog. 2021, 17, e1009950. [Google Scholar] [CrossRef]
- Lin, Y.; Zheng, C. A Tug of War: DNA-Sensing Antiviral Innate Immunity and Herpes Simplex Virus Type I Infection. Front. Microbiol. 2019, 10, 2627. [Google Scholar] [CrossRef] [Green Version]
- Lanfranca, M.P.; Mostafa, H.H.; Davido, D.J. HSV-1 ICP0: An E3 Ubiquitin Ligase That Counteracts Host Intrinsic and Innate Immunity. Cells 2014, 3, 438–454. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagglund, R.; Roizman, B. Characterization of the novel E3 ubiquitin ligase encoded in exon 3 of herpes simplex virus-1-infected cell protein 0. Proc. Natl. Acad. Sci. USA 2002, 99, 7889–7894. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hagglund, R.; Van Sant, C.; Lopez, P.; Roizman, B. Herpes simplex virus 1-infected cell protein 0 contains two E3 ubiquitin ligase sites specific for different E2 ubiquitin-conjugating enzymes. Proc. Natl. Acad. Sci. USA 2002, 99, 631–636. [Google Scholar] [CrossRef] [Green Version]
- van Lint, A.L.; Murawski, M.R.; Goodbody, R.E.; Severa, M.; Fitzgerald, K.A.; Finberg, R.W.; Knipe, D.M.; Kurt-Jones, E.A. Herpes simplex virus immediate-early ICP0 protein inhibits Toll-like receptor 2-dependent inflammatory responses and NF-kappaB signaling. J. Virol. 2010, 84, 10802–10811. [Google Scholar] [CrossRef] [Green Version]
- Daubeuf, S.; Singh, D.; Tan, Y.; Liu, H.; Federoff, H.J.; Bowers, W.J.; Tolba, K. HSV ICP0 recruits USP7 to modulate TLR-mediated innate response. Blood 2009, 113, 3264–3275. [Google Scholar] [CrossRef] [Green Version]
- Sen, J.; Liu, X.; Roller, R.; Knipe, D.M. Herpes simplex virus US3 tegument protein inhibits Toll-like receptor 2 signaling at or before TRAF6 ubiquitination. Virology 2013, 439, 65–73. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peri, P.; Mattila, R.K.; Kantola, H.; Broberg, E.; Karttunen, H.S.; Waris, M.; Vuorinen, T.; Hukkanen, V. Herpes simplex virus type 1 Us3 gene deletion influences toll-like receptor responses in cultured monocytic cells. Virol. J. 2008, 5, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, S.; Wang, K.Z.; Lin, R.T.; Zheng, C.F. Herpes Simplex Virus 1 Serine/Threonine Kinase US3 Hyperphosphorylates IRF3 and Inhibits Beta Interferon Production. J. Virol. 2013, 87, 12814–12827. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Xing, J.; Ni, L.; Wang, S.; Wang, K.; Lin, R.; Zheng, C. Herpes simplex virus 1-encoded tegument protein VP16 abrogates the production of beta interferon (IFN) by inhibiting NF-kappaB activation and blocking IFN regulatory factor 3 to recruit its coactivator CBP. J. Virol. 2013, 87, 9788–9801. [Google Scholar] [CrossRef] [Green Version]
- Xu, H.; Su, C.; Pearson, A.; Mody, C.H.; Zheng, C. Herpes Simplex Virus 1 UL24 Abrogates the DNA Sensing Signal Pathway by Inhibiting NF-kappaB Activation. J. Virol. 2017, 91, e00025-17. [Google Scholar] [CrossRef] [Green Version]
- Wang, S.; Wang, K.; Li, J.; Zheng, C. Herpes simplex virus 1 ubiquitin-specific protease UL36 inhibits beta interferon production by deubiquitinating TRAF3. J. Virol. 2013, 87, 11851–11860. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chapon, M.; Parvatiyar, K.; Aliyari, S.R.; Zhao, J.S.; Cheng, G. Comprehensive Mutagenesis of Herpes Simplex Virus 1 Genome Identifies UL42 as an Inhibitor of Type I Interferon Induction. J. Virol. 2019, 93, e01446-19. [Google Scholar] [CrossRef]
- Su, C.; Zheng, C. Herpes Simplex Virus 1 Abrogates the cGAS/STING-Mediated Cytosolic DNA-Sensing Pathway via Its Virion Host Shutoff Protein, UL41. J. Virol. 2017, 91, e02414-16. [Google Scholar] [CrossRef] [Green Version]
- Zheng, Z.-Q.; Fu, Y.-Z.; Wang, S.-Y.; Xu, Z.-S.; Zou, H.-M.; Wang, Y.-Y. Herpes simplex virus protein UL56 inhibits cGAS-Mediated DNA sensing to evade antiviral immunity. Cell Insight 2022, 1, 100014. [Google Scholar] [CrossRef]
- Huang, J.; You, H.; Su, C.; Li, Y.; Chen, S.; Zheng, C. Herpes Simplex Virus 1 Tegument Protein VP22 Abrogates cGAS/STING-Mediated Antiviral Innate Immunity. J. Virol. 2018, 92, e00841-18. [Google Scholar] [CrossRef] [Green Version]
- Xu, G.; Liu, C.; Zhou, S.; Li, Q.; Feng, Y.; Sun, P.; Feng, H.; Gao, Y.; Zhu, J.; Luo, X.; et al. Viral tegument proteins restrict cGAS-DNA phase separation to mediate immune evasion. Mol. Cell 2021, 81, 2823–2837. [Google Scholar] [CrossRef]
- Zhang, J.J.; Zhao, J.; Xu, S.M.; Li, J.H.; He, S.P.; Zeng, Y.; Xie, L.S.; Xie, N.; Liu, T.; Lee, K.; et al. Species-Specific Deamidation of cGAS by Herpes Simplex Virus UL37 Protein Facilitates Viral Replication. Cell Host Microbe 2018, 24, 234–248. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Barzilai, A.; Zivony-Elbom, I.; Sarid, R.; Noah, E.; Frenkel, N. The herpes simplex virus type 1 vhs-UL41 gene secures viral replication by temporarily evading apoptotic cellular response to infection: Vhs-UL41 activity might require interactions with elements of cellular mRNA degradation machinery. J. Virol. 2006, 80, 505–513. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ye, R.; Su, C.; Xu, H.; Zheng, C. Herpes Simplex Virus 1 Ubiquitin-Specific Protease UL36 Abrogates NF-kappaB Activation in DNA Sensing Signal Pathway. J. Virol. 2017, 91, e02417-16. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bodda, C.; Reinert, L.S.; Fruhwurth, S.; Richardo, T.; Sun, C.L.; Zhang, B.C.; Kalamvoki, M.; Pohlmann, A.; Mogensen, T.H.; Bergstrom, P.; et al. HSV1 VP1-2 deubiquitinates STING to block type I interferon expression and promote brain infection. J. Exp. Med. 2020, 217, e20191422. [Google Scholar] [CrossRef]
- Deschamps, T.; Kalamvoki, M. Evasion of the STING DNA-Sensing Pathway by VP11/12 of Herpes Simplex Virus 1. J. Virol. 2017, 91, e00535-17. [Google Scholar] [CrossRef] [Green Version]
- Pan, S.; Liu, X.; Ma, Y.J.; Cao, Y.J.; He, B. Herpes Simplex Virus 1 gamma(1)34.5 Protein Inhibits STING Activation That Restricts Viral Replication. J. Virol. 2018, 92, e01015-18. [Google Scholar] [CrossRef] [Green Version]
- You, H.J.; Zheng, S.; Huang, Z.M.; Lin, Y.Y.; Shen, Q.T.; Zheng, C.F. Herpes Simplex Virus 1 Tegument Protein UL46 Inhibits TANK-Binding Kinase 1-Mediated Signaling. Mbio 2019, 10, e00919-19. [Google Scholar] [CrossRef] [Green Version]
- Christensen, M.H.; Jensen, S.B.; Miettinen, J.J.; Luecke, S.; Prabakaran, T.; Reinert, L.S.; Mettenleiter, T.; Chen, Z.J.J.; Knipe, D.M.; Sandri-Goldin, R.M.; et al. HSV-1 ICP27 targets the TBK1-activated STING signalsome to inhibit virus-induced type I IFN expression. EMBO J. 2016, 35, 1385–1399. [Google Scholar] [CrossRef] [Green Version]
- Orzalli, M.H.; DeLuca, N.A.; Knipe, D.M. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. USA 2012, 109, E3008–E3017. [Google Scholar] [CrossRef] [Green Version]
- Diner, B.A.; Lum, K.K.; Javitt, A.; Cristea, I.M. Interactions of the Antiviral Factor Interferon Gamma-Inducible Protein 16 (IFI16) Mediate Immune Signaling and Herpes Simplex Virus-1 Immunosuppression. Mol. Cell Proteom. 2015, 14, 2341–2356. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orzalli, M.H.; Broekema, N.M.; Knipe, D.M. Relative Contributions of Herpes Simplex Virus 1 ICP0 and vhs to Loss of Cellular IFI16 Vary in Different Human Cell Types. J. Virol. 2016, 90, 8351–8359. [Google Scholar] [CrossRef] [Green Version]
- Xing, J.J.; Wang, S.; Lin, R.T.; Mossman, K.L.; Zheng, C.F. Herpes Simplex Virus 1 Tegument Protein US11 Downmodulates the RLR Signaling Pathway via Direct Interaction with RIG-I and MDA-5. J. Virol. 2012, 86, 3528–3540. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, J.; Zeng, Y.; Xu, S.; Chen, J.; Shen, G.; Yu, C.; Knipe, D.; Yuan, W.; Peng, J.; Xu, W.; et al. A Viral Deamidase Targets the Helicase Domain of RIG-I to Block RNA-Induced Activation. Cell Host Microbe 2016, 20, 770–784. [Google Scholar] [CrossRef] [Green Version]
- Chee, A.V.; Roizman, B. Herpes simplex virus 1 gene products occlude the interferon signaling pathway at multiple sites. J. Virol. 2004, 78, 4185–4196. [Google Scholar] [CrossRef] [Green Version]
- Yuan, H.; You, J.; You, H.; Zheng, C.F. Herpes Simplex Virus 1 UL36USP Antagonizes Type I Interferon-Mediated Antiviral Innate Immunity. J. Virol. 2018, 92, e01161-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Halford, W.P.; Weisend, C.; Grace, J.; Soboleski, M.; Carr, D.J.J.; Balliet, J.W.; Imai, Y.; Margolis, T.P.; Gebhardt, B.M. ICP0 antagonizes Stat I-dependent repression of herpes simplex virus: Implications for the regulation of viral latency. Virol. J. 2006, 3, 44. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.E.; Song, B.; Knipe, D.M. Role for herpes simplex virus 1 ICP27 in the inhibition of type I interferon signaling. Virology 2008, 374, 487–494. [Google Scholar] [CrossRef] [Green Version]
- Johnson, K.E.; Knipe, D.M. Herpes simplex virus-1 infection causes the secretion of a type I interferon-antagonizing protein and inhibits signaling at or before Jak-1 activation. Virology 2010, 396, 21–29. [Google Scholar] [CrossRef] [Green Version]
- Lee, D.H.; Ghiasi, H. Roles of M1 and M2 Macrophages in Herpes Simplex Virus 1 Infectivity. J. Virol. 2017, 91, e00578-17. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, A.J.; Chen, B.; Chew, M.V.; Barra, N.G.; Shenouda, M.M.; Nham, T.; van Rooijen, N.; Jordana, M.; Mossman, K.L.; Schreiber, R.D.; et al. Inflammatory monocytes require type I interferon receptor signaling to activate NK cells via IL-18 during a mucosal viral infection. J. Exp. Med. 2017, 214, 1153–1167. [Google Scholar] [CrossRef] [Green Version]
- Mack, E.A.; Kallal, L.E.; Demers, D.A.; Biron, C.A. Type 1 interferon induction of natural killer cell gamma interferon production for defense during lymphocytic choriomeningitis virus infection. MBio 2011, 2, e00169-11. [Google Scholar] [CrossRef] [Green Version]
- Miner, J.J.; Platt, D.J.; Ghaznavi, C.M.; Chandra, P.; Santeford, A.; Menos, A.M.; Dong, Z.; Wang, E.R.; Qian, W.; Karozichian, E.S.; et al. HSV-1 and Zika Virus but Not SARS-CoV-2 Replicate in the Human Cornea and Are Restricted by Corneal Type III Interferon. Cell Rep. 2020, 33, 108339. [Google Scholar] [CrossRef] [PubMed]
- Jaggi, U.; Bhela, S.; Rouse, B. Role of interferon lambda (IL-28A) in herpes stromal keratitis. J. Immunol. Res. Ther. 2018, 3, 135–144. [Google Scholar]
- Ellermann-Eriksen, S. Macrophages and cytokines in the early defence against herpes simplex virus. Virol. J. 2005, 2, 59. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bryant-Hudson, K.; Conrady, C.D.; Carr, D.J. Type I interferon and lymphangiogenesis in the HSV-1 infected cornea—Are they beneficial to the host? Prog. Retin. Eye Res. 2013, 36, 281–291. [Google Scholar] [CrossRef] [Green Version]
- Hayashi, K.; Hooper, L.C.; Chin, M.S.; Nagineni, C.N.; Detrick, B.; Hooks, J.J. Herpes simplex virus 1 (HSV-1) DNA and immune complex (HSV-1-human IgG) elicit vigorous interleukin 6 release from infected corneal cells via Toll-like receptors. J. Gen. Virol. 2006, 87, 2161–2169. [Google Scholar] [CrossRef] [PubMed]
- Latif, M.B.; Raja, R.; Kessler, P.M.; Sen, G.C. Relative Contributions of the cGAS-STING and TLR3 Signaling Pathways to Attenuation of Herpes Simplex Virus 1 Replication. J. Virol. 2020, 94, e01717-19. [Google Scholar] [CrossRef]
- Almine, J.F.; O’Hare, C.A.J.; Dunphy, G.; Haga, I.R.; Naik, R.J.; Atrih, A.; Connolly, D.J.; Taylor, J.; Kelsall, I.R.; Bowie, A.G.; et al. IFI16 and cGAS cooperate in the activation of STING during DNA sensing in human keratinocytes. Nat. Commun. 2017, 8, 14392. [Google Scholar] [CrossRef] [Green Version]
- Conrady, C.D.; Zheng, M.; Mandal, N.A.; van Rooijen, N.; Carr, D.J. IFN-α-driven CCL2 production recruits inflammatory monocytes to infection site in mice. Mucosal. Immunol. 2013, 6, 45–55. [Google Scholar] [CrossRef] [Green Version]
- Conrady, C.D.; Thapa, M.; Wuest, T.; Carr, D.J. Loss of mandibular lymph node integrity is associated with an increase in sensitivity to HSV-1 infection in CD118-deficient mice. J. Immunol. 2009, 182, 3678–3687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Madera, S.; Rapp, M.; Firth, M.A.; Beilke, J.N.; Lanier, L.L.; Sun, J.C. Type I IFN promotes NK cell expansion during viral infection by protecting NK cells against fratricide. J. Exp. Med. 2016, 213, 225–233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vogel, K.; Thomann, S.; Vogel, B.; Schuster, P.; Schmidt, B. Both plasmacytoid dendritic cells and monocytes stimulate natural killer cells early during human herpes simplex virus type 1 infections. Immunology 2014, 143, 588–600. [Google Scholar] [CrossRef] [Green Version]
- Lee, A.J.; Mian, F.; Poznanski, S.M.; Stackaruk, M.; Chan, T.; Chew, M.V.; Ashkar, A.A. Type I Interferon Receptor on NK Cells Negatively Regulates Interferon-γ Production. Front. Immunol. 2019, 10, 1261. [Google Scholar] [CrossRef] [Green Version]
- Broggi, A.; Granucci, F.; Zanoni, I. Type III interferons: Balancing tissue tolerance and resistance to pathogen invasion. J. Exp. Med. 2020, 217, e20190295. [Google Scholar] [CrossRef] [PubMed]
- Kurt-Jones, E.A.; Orzalli, M.H.; Knipe, D.M. Innate Immune Mechanisms and Herpes Simplex Virus Infection and Disease. Adv. Anat. Embryol. Cell Biol. 2017, 223, 49–75. [Google Scholar] [CrossRef]
- Krishnan, R.; Stuart, P.M. Developments in Vaccination for Herpes Simplex Virus. Front. Microbiol. 2021, 12, 798927. [Google Scholar] [CrossRef]
- Chaloulis, S.K.; Mousteris, G.; Tsaousis, K.T. Incidence and Risk Factors of Bilateral Herpetic Keratitis: 2022 Update. Trop. Med. Infect. Dis. 2022, 7, 92. [Google Scholar] [CrossRef]
- Shukla, S.D.; Valyi-Nagy, T. Host Molecules That Promote Pathophysiology of Ocular Herpes. Front. Microbiol. 2022, 13, 818658. [Google Scholar] [CrossRef]
- Tsatsos, M.; MacGregor, C.; Athanasiadis, I.; Moschos, M.M.; Hossain, P.; Anderson, D. Herpes simplex virus keratitis: An update of the pathogenesis and current treatment with oral and topical antiviral agents—Response. Clin. Exp. Ophthalmol. 2017, 45, 317. [Google Scholar] [CrossRef] [Green Version]
- Tabery, H.M. Early epithelial changes in recurrent herpes simplex virus keratitis: A non-contact photomicrographic study in vivo in the human cornea. Acta Ophthalmol. Scand. 1998, 76, 349–352. [Google Scholar] [CrossRef] [PubMed]
- Kaye, S.; Choudhary, A. Herpes simplex keratitis. Prog. Retin. Eye Res. 2006, 25, 355–380. [Google Scholar] [CrossRef]
- O’Brien, W.J.; Palmer, M.L.; Guy, J.; Taylor, J.L. Endothelial barrier function and Na+/K(+)-ATPase pump density in herpetic stromal disease. Investig. Ophthalmol. Vis. Sci. 1996, 37, 29–36. [Google Scholar]
- Vemuganti, G.K.; Murthy, S.I.; Das, S. Update on pathologic diagnosis of corneal infections and inflammations. Middle E. Afr. J. Ophthalmol. 2011, 18, 277–284. [Google Scholar] [CrossRef]
- Webre, J.M.; Hill, J.M.; Nolan, N.M.; Clement, C.; McFerrin, H.E.; Bhattacharjee, P.S.; Hsia, V.; Neumann, D.M.; Foster, T.P.; Lukiw, W.J.; et al. Rabbit and mouse models of HSV-1 latency, reactivation, and recurrent eye diseases. J. Biomed. Biotechnol. 2012, 2012, 612316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Azher, T.N.; Yin, X.T.; Stuart, P.M. Understanding the Role of Chemokines and Cytokines in Experimental Models of Herpes Simplex Keratitis. J. Immunol. Res. 2017, 2017, 7261980. [Google Scholar] [CrossRef]
- Niemialtowski, M.G.; Rouse, B.T. Phenotypic and Functional-Studies on Ocular T-Cells during Herpetic Infections of the Eye. J. Immunol. 1992, 148, 1864–1870. [Google Scholar] [CrossRef]
- Niemialtowski, M.G.; Rouse, B.T. Predominance of Th1-Cells in Ocular-Tissues during Herpetic Stromal Keratitis. J. Immunol. 1992, 149, 3035–3039. [Google Scholar] [CrossRef]
- Niemialtowski, M.G.; Rouse, B.T. Herpetic Stromal Keratitis [Hsk]—Phenotypic and Functional-Analysis of Ocular T-Cells. Faseb J. 1992, 6, A1428. [Google Scholar]
- Russell, R.G.; Nasisse, M.P.; Larsen, H.S.; Rouse, B.T. Role of T-lymphocytes in the pathogenesis of herpetic stromal keratitis. Investig. Ophthalmol. Vis. Sci. 1984, 25, 938–944. [Google Scholar]
- Gimenez, F.; Suryawanshi, A.; Rouse, B.T. Pathogenesis of herpes stromal keratitis—A focus on corneal neovascularization. Prog. Retin. Eye Res. 2013, 33, 1–9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajasagi, N.K.; Rouse, B.T. The role of T cells in herpes stromal keratitis. Front. Immunol. 2019, 10, 512. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Conrady, C.D.; Zheng, M.; Stone, D.U.; Carr, D.J. CD8+ T cells suppress viral replication in the cornea but contribute to VEGF-C–induced lymphatic vessel genesis. J. Immunol. 2012, 189, 425–432. [Google Scholar] [CrossRef] [Green Version]
- Rajasagi, N.K.; Rouse, B.T. Application of our understanding of pathogenesis of herpetic stromal keratitis for novel therapy. Microbes Infect. 2018, 20, 526–530. [Google Scholar] [CrossRef]
- Suryawanshi, A.; Veiga-Parga, T.; Rajasagi, N.K.; Reddy, P.B.; Sehrawat, S.; Sharma, S.; Rouse, B.T. Role of IL-17 and Th17 cells in herpes simplex virus-induced corneal immunopathology. J. Immunol. 2011, 187, 1919–1930. [Google Scholar] [CrossRef] [Green Version]
- Veiga-Parga, T.; Suryawanshi, A.; Mulik, S.; Gimenez, F.; Sharma, S.; Sparwasser, T.; Rouse, B.T. On the role of regulatory T cells during viral-induced inflammatory lesions. J. Immunol. 2012, 189, 5924–5933. [Google Scholar] [CrossRef] [Green Version]
- Veiga-Parga, T.; Suryawanshi, A.; Rouse, B.T. Controlling viral immuno-inflammatory lesions by modulating aryl hydrocarbon receptor signaling. PLoS Pathog. 2011, 7, e1002427. [Google Scholar] [CrossRef] [Green Version]
- Smith, J.B.; Herbert, J.J.; Truong, N.R.; Cunningham, A.L. Cytokines and chemokines: The vital role they play in herpes simplex virus mucosal immunology. Front. Immunol. 2022, 13, 936235. [Google Scholar] [CrossRef]
- Ambati, B.K.; Nozaki, M.; Singh, N.; Takeda, A.; Jani, P.D.; Suthar, T.; Albuquerque, R.J.; Richter, E.; Sakurai, E.; Newcomb, M.T.; et al. Corneal avascularity is due to soluble VEGF receptor-1. Nature 2006, 443, 993–997. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zheng, M.; Deshpande, S.; Lee, S.; Ferrara, N.; Rouse, B.T. Contribution of vascular endothelial growth factor in the neovascularization process during the pathogenesis of herpetic stromal keratitis. J. Virol. 2001, 75, 9828–9835. [Google Scholar] [CrossRef] [Green Version]
- Zheng, M.; Schwarz, M.A.; Lee, S.; Kumaraguru, U.; Rouse, B.T. Control of stromal keratitis by inhibition of neovascularization. Am. J. Pathol. 2001, 159, 1021–1029. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Biswas, P.S.; Banerjee, K.; Kinchington, P.R.; Rouse, B.T. Involvement of IL-6 in the paracrine production of VEGF in ocular HSV-1 infection. Exp. Eye Res. 2006, 82, 46–54. [Google Scholar] [CrossRef]
- Wuest, T.R.; Carr, D.J. VEGF-A expression by HSV-1-infected cells drives corneal lymphangiogenesis. J. Exp. Med. 2010, 207, 101–115. [Google Scholar] [CrossRef] [Green Version]
- Suryawanshi, A.; Mulik, S.; Sharma, S.; Reddy, P.B.J.; Sehrawat, S.; Rouse, B.T. Ocular Neovascularization Caused by Herpes Simplex Virus Type 1 Infection Results from Breakdown of Binding between Vascular Endothelial Growth Factor A and Its Soluble Receptor. J. Immunol. 2011, 186, 3653–3665. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Suryawanshi, A.; Veiga-Parga, T.; Reddy, P.B.; Rajasagi, N.K.; Rouse, B.T. IL-17A differentially regulates corneal vascular endothelial growth factor (VEGF)-A and soluble VEGF receptor 1 expression and promotes corneal angiogenesis after herpes simplex virus infection. J. Immunol. 2012, 188, 3434–3446. [Google Scholar] [CrossRef] [Green Version]
- Wilhelmus, K.R.; Gee, L.; Hauck, W.W.; Kurinij, N.; Dawson, C.R.; Jones, D.B.; Barron, B.A.; Kaufman, H.E.; Sugar, J.; Hyndiuk, R.A.; et al. Herpetic Eye Disease Study—A Controlled Trial of Topical Corticosteroids for Herpes-Simplex Stromal Keratitis. Ophthalmology 1994, 101, 1883–1895. [Google Scholar] [CrossRef]
- White, M.; Chodosh, J.; Street, C.; Feder, R.S.; Pavan-Langston, D.; Liesegang, T.J.; Margolis, T.P.; Sokol, H.; Tuli, S.S. Herpes Simplex Virus Keratitis: A Treatment Guideline. Hoskins Center for Quality Eye Care and American Academy of Ophthalmology Website. 2014. Available online: https://www.aao.org/education/clinical-statement/herpes-simplex-virus-keratitis-treatment-guideline (accessed on 8 February 2023).
- Koganti, R.; Yadavalli, T.; Shukla, D. Current and Emerging Therapies for Ocular Herpes Simplex Virus Type-1 Infections. Microorganisms 2019, 7, 429. [Google Scholar] [CrossRef] [Green Version]
- Wilhelmus, K.R. Antiviral treatment and other therapeutic interventions for herpes simplex virus epithelial keratitis. Cochrane Database Syst. Rev. 2010, 1, CD002898. [Google Scholar] [CrossRef]
- Azher, T.N.; Yin, X.T.; Tajfirouz, D.; Huang, A.J.; Stuart, P.M. Herpes simplex keratitis: Challenges in diagnosis and clinical management. Clin. Ophthalmol. 2017, 11, 185–191. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qi, X.; Wang, M.; Li, X.; Jia, Y.; Li, S.; Shi, W.; Gao, H. Characteristics of New Onset Herpes Simplex Keratitis after Keratoplasty. J. Ophthalmol. 2018, 2018, 4351460. [Google Scholar] [CrossRef] [Green Version]
- Singhal, D.; Nagpal, R.; Maharana, P.K.; Sinha, R.; Agarwal, T.; Sharma, N.; Titiyal, J.S. Surgical alternatives to keratoplasty in microbial keratitis. Surv. Ophthalmol. 2021, 66, 290–307. [Google Scholar] [CrossRef]
- Herpetic Eye Disease Study Group. Acyclovir for the prevention of recurrent herpes simplex virus eye disease. N. Engl. J. Med. 1998, 339, 300–306. [Google Scholar] [CrossRef] [Green Version]
- Tambasco, F.P.; Cohen, E.J.; Nguyen, L.H.; Rapuano, C.J.; Laibson, P.R. Oral acyclovir after penetrating keratoplasty for herpes simplex keratitis. Arch Ophthalmol.-Chic. 1999, 117, 445–449. [Google Scholar] [CrossRef] [Green Version]
- Fleischer, R.; Johnson, M. Acyclovir nephrotoxicity: A case report highlighting the importance of prevention, detection, and treatment of acyclovir-induced nephropathy. Case Rep. Med. 2010, 2010, 602783. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carmine, A.A.; Brogden, R.N.; Heel, R.C.; Speight, T.M.; Avery, G.S. Trifluridine: A review of its antiviral activity and therapeutic use in the topical treatment of viral eye infections. Drugs 1982, 23, 329–353. [Google Scholar] [CrossRef] [PubMed]
- Duan, R.; de Vries, R.D.; Osterhaus, A.D.; Remeijer, L.; Verjans, G.M. Acyclovir-resistant corneal HSV-1 isolates from patients with herpetic keratitis. J. Infect. Dis. 2008, 198, 659–663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Duan, R.; de Vries, R.D.; van Dun, J.M.; van Loenen, F.B.; Osterhaus, A.D.; Remeijer, L.; Verjans, G.M. Acyclovir susceptibility and genetic characteristics of sequential herpes simplex virus type 1 corneal isolates from patients with recurrent herpetic keratitis. J. Infect. Dis. 2009, 200, 1402–1414. [Google Scholar] [CrossRef] [Green Version]
- Toriyama, K.; Inoue, T.; Suzuki, T.; Kobayashi, T.; Ohashi, Y. Necrotizing keratitis caused by acyclovir-resistant herpes simplex virus. Case Rep. Ophthalmol. 2014, 5, 325–328. [Google Scholar] [CrossRef] [Green Version]
- van Velzen, M.; van de Vijver, D.A.; van Loenen, F.B.; Osterhaus, A.D.; Remeijer, L.; Verjans, G.M. Acyclovir prophylaxis predisposes to antiviral-resistant recurrent herpetic keratitis. J. Infect. Dis. 2013, 208, 1359–1365. [Google Scholar] [CrossRef] [Green Version]
- Bacon, T.H.; Levin, M.J.; Leary, J.J.; Sarisky, R.T.; Sutton, D. Herpes simplex virus resistance to acyclovir and penciclovir after two decades of antiviral therapy. Clin. Microbiol. Rev. 2003, 16, 114–128. [Google Scholar] [CrossRef] [Green Version]
- Broekema, F.I.; Dikkers, F.G. Side-effects of cidofovir in the treatment of recurrent respiratory papillomatosis. Eur. Arch. Oto-Rhino-Laryngol. 2008, 265, 871–879. [Google Scholar] [CrossRef] [PubMed]
- Jacobson, M.A. Review of the Toxicities of Foscarnet. J. Acquir. Immune Defic. Syndr. 1992, 5, S11–S17. [Google Scholar]
- Rao, S.N. Treatment of herpes simplex virus stromal keratitis unresponsive to topical prednisolone 1% with topical cyclosporine 0.05%. Am. J. Ophthalmol. 2006, 141, 771–772. [Google Scholar] [CrossRef] [PubMed]
- Gunduz, K.; Ozdemir, O. Topical cyclosporin as an adjunct to topical acyclovir treatment in herpetic stromal keratitis. Ophthalmic Res. 1997, 29, 405–408. [Google Scholar] [CrossRef] [PubMed]
- Heiligenhaus, A.; Steuhl, K.P. Treatment of HSV-1 stromal keratitis with topical cyclosporin A: A pilot study. Graef. Arch. Clin. Exp. 1999, 237, 435–438. [Google Scholar] [CrossRef]
- Knickelbein, J.E.; Khanna, K.M.; Yee, M.B.; Baty, C.J.; Kinchington, P.R.; Hendricks, R.L. Noncytotoxic lytic granule-mediated CD8+ T cell inhibition of HSV-1 reactivation from neuronal latency. Science 2008, 322, 268–271. [Google Scholar] [CrossRef] [Green Version]
- Yu, W.; Geng, S.; Suo, Y.; Wei, X.; Cai, Q.; Wu, B.; Zhou, X.; Shi, Y.; Wang, B. Critical Role of Regulatory T Cells in the Latency and Stress-Induced Reactivation of HSV-1. Cell Rep. 2018, 25, 2379–2389. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Gupta, N.; Vanathi, M.; Tandon, R. Corneal transplantation in the modern era. Indian J. Med. Res. 2019, 150, 7–22. [Google Scholar] [CrossRef]
- Kuffova, L.; Knickelbein, J.E.; Yu, T.; Medina, C.; Amescua, G.; Rowe, A.M.; Hendricks, R.L.; Forrester, J.V. High-Risk Corneal Graft Rejection in the Setting of Previous Corneal Herpes Simplex Virus (HSV)-1 Infection. Investig. Ophthalmol. Vis. Sci. 2016, 57, 1578–1587. [Google Scholar] [CrossRef] [Green Version]
- Hawthorne, K.M.; Dana, R.; Chodosh, J. Delayed type hypersensitivity in the pathogenesis of recurrent herpes stromal keratitis. Semin. Ophthalmol. 2011, 26, 246–250. [Google Scholar] [CrossRef]
- Shtein, R.M.; Garcia, D.D.; Musch, D.C.; Elner, V.M. Herpes simplex virus keratitis: Histopathologic inflammation and corneal allograft rejection. Ophthalmology 2009, 116, 1301–1305. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jaishankar, D.; Yakoub, A.M.; Yadavalli, T.; Agelidis, A.; Thakkar, N.; Hadigal, S.; Ames, J.; Shukla, D. An off-target effect of BX795 blocks herpes simplex virus type 1 infection of the eye. Sci. Transl. Med. 2018, 10, eaan5861. [Google Scholar] [CrossRef] [Green Version]
- Yadavalli, T.; Suryawanshi, R.; Koganti, R.; Hopkins, J.; Ames, J.; Koujah, L.; Iqbal, A.; Madavaraju, K.; Agelidis, A.; Shukla, D. Standalone or combinatorial phenylbutyrate therapy shows excellent antiviral activity and mimics CREB3 silencing. Sci. Adv. 2020, 6, eabd9443. [Google Scholar] [CrossRef] [PubMed]
- Tiwari, V.; Liu, J.; Valyi-Nagy, T.; Shukla, D. Anti-heparan sulfate peptides that block herpes simplex virus infection in vivo. J. Biol. Chem. 2011, 286, 25406–25415. [Google Scholar] [CrossRef] [Green Version]
- Yadavalli, T.; Agelidis, A.; Jaishankar, D.; Mangano, K.; Thakkar, N.; Penmetcha, K.; Shukla, D. Targeting Herpes Simplex Virus-1 gD by a DNA Aptamer Can Be an Effective New Strategy to Curb Viral Infection. Mol. Ther. Nucleic Acids 2017, 9, 365–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berdugo, M.; Larsen, I.V.; Abadie, C.; Deloche, C.; Kowalczuk, L.; Touchard, E.; Dubielzig, R.; Brandt, C.R.; Behar-Cohen, F.; Combette, J.M. Ocular distribution, spectrum of activity, and in vivo viral neutralization of a fully humanized anti-herpes simplex virus IgG Fab fragment following topical application. Antimicrob. Agents Chemother. 2012, 56, 1390–1402. [Google Scholar] [CrossRef] [Green Version]
- Dix, R.D.; Pereira, L.; Baringer, J.R. Use of monoclonal antibody directed against herpes simplex virus glycoproteins to protect mice against acute virus-induced neurological disease. Infect. Immun. 1981, 34, 192–199. [Google Scholar] [CrossRef] [Green Version]
- Andersen, J.H.; Jenssen, H.; Sandvik, K.; Gutteberg, T.J. Anti-HSV activity of lactoferrin and lactoferricin is dependent on the presence of heparan sulphate at the cell surface. J. Med. Virol. 2004, 74, 262–271. [Google Scholar] [CrossRef] [PubMed]
- Kennedy, P.G.; Chaudhuri, A. Herpes simplex encephalitis. J. Neurol. Neurosurg. Psychiatry 2002, 73, 237–238. [Google Scholar] [CrossRef] [Green Version]
- Virtanen, J.O.; Jacobson, S. Viruses and multiple sclerosis. CNS Neurol. Disord. Drug Targets 2012, 11, 528–544. [Google Scholar] [CrossRef] [Green Version]
- Leibovitch, E.C.; Caruso, B.; Ha, S.K.; Schindler, M.K.; Lee, N.J.; Luciano, N.J.; Billioux, B.J.; Guy, J.R.; Yen, C.; Sati, P.; et al. Herpesvirus trigger accelerates neuroinflammation in a nonhuman primate model of multiple sclerosis. Proc. Natl. Acad. Sci. USA 2018, 115, 11292–11297. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lovheim, H.; Gilthorpe, J.; Adolfsson, R.; Nilsson, L.G.; Elgh, F. Reactivated herpes simplex infection increases the risk of Alzheimer’s disease. Alzheimers Dement. 2015, 11, 593–599. [Google Scholar] [CrossRef] [PubMed]
- Lovheim, H.; Gilthorpe, J.; Johansson, A.; Eriksson, S.; Hallmans, G.; Elgh, F. Herpes simplex infection and the risk of Alzheimer’s disease: A nested case-control study. Alzheimers Dement. 2015, 11, 587–592. [Google Scholar] [CrossRef]
- Harris, S.A.; Harris, E.A. Herpes Simplex Virus Type 1 and Other Pathogens are Key Causative Factors in Sporadic Alzheimer’s Disease. J. Alzheimers Dis. 2015, 48, 319–353. [Google Scholar] [CrossRef] [Green Version]
- Itzhaki, R.F. Herpes simplex virus type 1 and Alzheimer’s disease: Increasing evidence for a major role of the virus. Front. Aging Neurosci. 2014, 6, 202. [Google Scholar] [CrossRef]
- Hendricks, R.L.; Tumpey, T.M.; Finnegan, A. IFN-gamma and IL-2 are protective in the skin but pathologic in the corneas of HSV-1-infected mice. J. Immunol. 1992, 149, 3023–3028. [Google Scholar] [CrossRef]
- Rajasagi, N.K.; Suryawanshi, A.; Sehrawat, S.; Reddy, P.B.; Mulik, S.; Hirashima, M.; Rouse, B.T. Galectin-1 reduces the severity of herpes simplex virus-induced ocular immunopathological lesions. J. Immunol. 2012, 188, 4631–4643. [Google Scholar] [CrossRef] [Green Version]
- Sehrawat, S.; Reddy, P.B.; Rajasagi, N.; Suryawanshi, A.; Hirashima, M.; Rouse, B.T. Galectin-9/TIM-3 interaction regulates virus-specific primary and memory CD8 T cell response. PLoS Pathog. 2010, 6, e1000882. [Google Scholar] [CrossRef] [PubMed]
- Varanasi, S.K.; Donohoe, D.; Jaggi, U.; Rouse, B.T. Manipulating Glucose Metabolism during Different Stages of Viral Pathogenesis Can Have either Detrimental or Beneficial Effects. J. Immunol. 2017, 199, 1748–1761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berber, E.; Sumbria, D.; Newkirk, K.M.; Rouse, B.T. Inhibiting Glucose Metabolism Results in Herpes Simplex Encephalitis. J. Immunol. 2021, 207, 1824–1835. [Google Scholar] [CrossRef]
- Sumbria, D.; Berber, E.; Miller, L.; Rouse, B.T. Modulating glutamine metabolism to control viral immuno-inflammatory lesions. Cell Immunol. 2021, 370, 104450. [Google Scholar] [CrossRef]
- Chucair-Elliott, A.J.; Jinkins, J.; Carr, M.M.; Carr, D.J. IL-6 Contributes to Corneal Nerve Degeneration after Herpes Simplex Virus Type I Infection. Am. J. Pathol. 2016, 186, 2665–2678. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rajasagi, N.K.; Reddy, P.B.; Mulik, S.; Gjorstrup, P.; Rouse, B.T. Neuroprotectin D1 reduces the severity of herpes simplex virus-induced corneal immunopathology. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6269–6279. [Google Scholar] [CrossRef] [Green Version]
- Rajasagi, N.K.; Reddy, P.B.; Suryawanshi, A.; Mulik, S.; Gjorstrup, P.; Rouse, B.T. Controlling herpes simplex virus-induced ocular inflammatory lesions with the lipid-derived mediator resolvin E1. J. Immunol. 2011, 186, 1735–1746. [Google Scholar] [CrossRef] [Green Version]
- Gordon, S.; Martinez, F.O. Alternative activation of macrophages: Mechanism and functions. Immunity 2010, 32, 593–604. [Google Scholar] [CrossRef] [Green Version]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative activation of macrophages: An immunologic functional perspective. Annu. Rev. Immunol. 2009, 27, 451–483. [Google Scholar] [CrossRef] [Green Version]
- Jaggi, U.; Yang, M.; Matundan, H.H.; Hirose, S.; Shah, P.K.; Sharifi, B.G.; Ghiasi, H. Increased phagocytosis in the presence of enhanced M2-like macrophage responses correlates with increased primary and latent HSV-1 infection. PLoS Pathog. 2020, 16, e1008971. [Google Scholar] [CrossRef]
- Jaggi, U.; Matundan, H.H.; Yu, J.; Hirose, S.; Mueller, M.; Wormley, F.L., Jr.; Ghiasi, H. Essential role of M1 macrophages in blocking cytokine storm and pathology associated with murine HSV-1 infection. PLoS Pathog. 2021, 17, e1009999. [Google Scholar] [CrossRef]
- Lee, S.; Zheng, M.; Kim, B.; Rouse, B.T. Role of matrix metalltoproteinase-9 in angiogenesis caused by ocular infection with herpes simplex virus. J. Clin. Investig. 2002, 110, 1105–1111. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Mulik, S.; Kumar, N.; Suryawanshi, A.; Rouse, B.T. An Anti-Inflammatory Role of VEGFR2/Src Kinase Inhibitor in Herpes Simplex Virus 1-Induced Immunopathology. J. Virol. 2011, 85, 5995–6007. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mulik, S.; Sharma, S.; Suryawanshi, A.; Veiga-Parga, T.; Reddy, P.B.J.; Rajasagi, N.K.; Rouse, B.T. Activation of Endothelial Roundabout Receptor 4 Reduces the Severity of Virus-Induced Keratitis. J. Immunol. 2011, 186, 7195–7204. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arimoto, K.I.; Löchte, S.; Stoner, S.A.; Burkart, C.; Zhang, Y.; Miyauchi, S.; Wilmes, S.; Fan, J.B.; Heinisch, J.J.; Li, Z.; et al. STAT2 is an essential adaptor in USP18-mediated suppression of type I interferon signaling. Nat. Struct. Mol. Biol. 2017, 24, 279–289. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Blazek, K.; Eames, H.L.; Weiss, M.; Byrne, A.J.; Perocheau, D.; Pease, J.E.; Doyle, S.; McCann, F.; Williams, R.O.; Udalova, I.A. IFN-λ resolves inflammation via suppression of neutrophil infiltration and IL-1β production. J. Exp. Med. 2015, 212, 845–853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nice, T.J.; Baldridge, M.T.; McCune, B.T.; Norman, J.M.; Lazear, H.M.; Artyomov, M.; Diamond, M.S.; Virgin, H.W. Interferon-lambda cures persistent murine norovirus infection in the absence of adaptive immunity. Science 2015, 347, 269–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hemann, E.A.; Green, R.; Turnbull, J.B.; Langlois, R.A.; Savan, R.; Gale, M., Jr. Interferon-lambda modulates dendritic cells to facilitate T cell immunity during infection with influenza A virus. Nat. Immunol. 2019, 20, 1035–1045. [Google Scholar] [CrossRef]
- Sommereyns, C.; Paul, S.; Staeheli, P.; Michiels, T. IFN-lambda (IFN-lambda) is expressed in a tissue-dependent fashion and primarily acts on epithelial cells in vivo. PLoS Pathog. 2008, 4, e1000017. [Google Scholar] [CrossRef]





| ISGs | Mechanism of Action | Roles in HSV-1 Life Cycle | References |
|---|---|---|---|
| CH25H | Blocks viral fusion with the host cell membrane by altering cell membrane; inhibits viral proteins prenylation by converting cholesterol into 25-hydroxycholesterol (25HC) | Inhibits HSV-1 envelope fusion with the host cell membrane | [193,194,195,196] |
| IFITM1 | IFITM1 proteins block viral fusion with the host cell membrane and also trap viral proteins in the endolysosomal compartment to inhibit viral replication | Blocks HSV-1 entry into host cell and inhibits its replication | [183,184,198] |
| MxA (Mx1) and MxB (Mx2) | Affects early viral replication by inhibiting capsid transport within the cells | MxA and MxB inhibits viral replication | [21,183,184,199,200,201,203,204,205] |
| Forms MxA-oligomer ring for nucleocapsid degradation | |||
| PKR | Binds to dsRNA and phosphorylates eukaryotic translation initiation factor-2 α (eIF-2α) to inhibit viral protein synthesis | Inhibits HSV-1 replication | [206,207,208] |
| OAS/RNase L | Binds to viral dsRNA to promote the synthesis of 2’,5’-oligoadenylate and activate latent RNase L to inhibit viral replication | Inhibits HSV-1 replication in neurons | [207,209,210,211,212,213] |
| ISG15 | ISGylation of IRF3 prevents ubiquitin-mediated degradation, promotes stability and transcriptional activation | Inhibits viral replication | [21,215,216,217] |
| Viperin | Interacts with HSV-1 gD to promote IRF7 mediated IFN-β signaling | Inhibits HSV-1 replication | [183,184,219,220,221] |
| Tetherin | Restrains the release of virus progeny particles budding from infected host cells during the late stage of viral replication | Inhibits HSV-1 spread | [136,222,223,224] |
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ren, J.; Antony, F.; Rouse, B.T.; Suryawanshi, A. Role of Innate Interferon Responses at the Ocular Surface in Herpes Simplex Virus-1-Induced Herpetic Stromal Keratitis. Pathogens 2023, 12, 437. https://doi.org/10.3390/pathogens12030437
Ren J, Antony F, Rouse BT, Suryawanshi A. Role of Innate Interferon Responses at the Ocular Surface in Herpes Simplex Virus-1-Induced Herpetic Stromal Keratitis. Pathogens. 2023; 12(3):437. https://doi.org/10.3390/pathogens12030437
Chicago/Turabian StyleRen, Jiayi, Ferrin Antony, Barry T. Rouse, and Amol Suryawanshi. 2023. "Role of Innate Interferon Responses at the Ocular Surface in Herpes Simplex Virus-1-Induced Herpetic Stromal Keratitis" Pathogens 12, no. 3: 437. https://doi.org/10.3390/pathogens12030437
APA StyleRen, J., Antony, F., Rouse, B. T., & Suryawanshi, A. (2023). Role of Innate Interferon Responses at the Ocular Surface in Herpes Simplex Virus-1-Induced Herpetic Stromal Keratitis. Pathogens, 12(3), 437. https://doi.org/10.3390/pathogens12030437