
Host and HBV Interactions and Their Potential Impact on Clinical Outcomes


Abstract
:1. Introduction
2. HBV Genotypes and Human Populations
3. SNPs and HBV Clinical Outcomes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Clinical Outcome and Associated Genes | Population | SNP Database | OR | 95% CI | Reference |
|---|---|---|---|---|---|
| Intercellular adhesion molecule-1 (ICAM-1) | China | rs1799969 (A) | 4.197 | 2.550–7.074 | [41] |
| Interferon gamma (IFN-γ) | China | rs2430561 (A) + rs1861494 (A) | 1.485 | 1.065–2.070 | [42] |
| Transforming growth factor (TGF)-beta1 | Korea | rs1982073 (LL) | 3.408 | 1.279–9.085 | [43] |
| Signal transducer and activator of transcription 3 (STAT3) | China | rs4796793 (GG) | 2.17 | 1.11–4.23 | [44] |
| Signal transducer and activator of transcription 4 (STAT4) | China | rs7574865 (TG) | 1.17 | 1.03–1.34 | [45] |
| Complement component 2 (C2) | China | rs9267673 (TC) | 1.37 | 1.15–1.63 | [45] |
| Human leukocyte antigen (HLA)-DRB1 | China | rs2647073 (CA) | 1.63 | 1.29–2.06 | [45] |
| Human leukocyte antigen (HLA)-DRB1 | China | rs3997872 (AT) | 1.86 | 1.32–2.62 | [45] |
| Human leukocyte antigen (HLA)-DQ | China | rs9275319 (GA) | 1.32 | 1.06–1.64 | [45] |
| Hepatocellular carcinoma | |||||
| Protein Phosphatase 1 Catalytic Subunit Beta (PPP1CB) | China | rs13025377 (AA) | 1.54 | 1.22–1.95 | [47] |
| Mouse double minute 2 homolog (MDM2) | Korea | rs2279744 (G) | 4.27 | 2.23–8.20 | [51] |
| Tumor protein 53 (p53) | Korea | rs1042522 (Pro/Pro) | 3.59 | 1.77–7.31 | [51] |
| MDM2 + p53 | Korea | rs2279744 (G/G) + rs1042522 (Pro/Pro) | 20.78 | 5.25–82.36 | [51] |
| DEP Domain Containing 5 (DEPDC5) | China | rs1012068 (CC) | 2.397 | 1.251–4.595 | [46] |
| X-chromosome long arm band 22.1 (Xq22.1) | China | rs5945919 (AG) | 2.22 | 1.15–4.30 | [49] |
| CD33 molecule (SIGLEC3 or CD33) | Taiwan | rs12459419 (C) | 1.256 | 1.027–1.535 | [50] |
| Occult hepatitis B | |||||
| Human leukocyte antigen (HLA)-DPA1 | Indonesia | rs3077 (CC) | 6.12 | 1.30–28.85 | [52] |
| Human leukocyte antigen (HLA)-DPA1 | Indonesia | rs3077 (T) + rs3135021(G) + rs9277535 (A) | 4.9 | 1.12–21.52 | [52] |
| Human leukocyte antigen (HLA)-DQB1 | China | NA | 2.15 | 1.118–4.161 | [53] |
| Human leukocyte antigen (HLA)-HLA-C*07:01 | China | NA | 2.146 | 1.070–4.306 | [53] |
| Human leukocyte antigen (HLA)-B*44:03 | China | NA | 4.693 | 1.822–12.086 | [53] |
| Human leukocyte antigen (HLA)-DRB1*07:01 | China | NA | 1.919 | 1.188–3.101 | [53] |
| Human leukocyte antigen (HLA)-DQB1*02:02 | China | NA | 2.012 | 1.303–3.107 | [53] |
| HBV reactivation | |||||
| Human leukocyte antigen (HLA)-DPB2 | Japan | rs872956 (AA) | 8.277 | 1.540–51.550 | [56] |
| Interleukin 13 (IL-13) | Taiwan | rs1295686 (AA) | 4.683 | 1.030–19.156 | [57] |
| Clearance or protection | |||||
| Sodium taurocholate cotransporting polypeptide (NTCP) | Korea | rs2296651(CT) | 0.455 | 0.220–0.942 | [58] |
| Toll-like receptor (TLR5) | Taiwan | rs5744174 (T) | 1.32 | 1.03–1.69 | [59] |
| Interferon-induced helicase C domain-containing protein 1 (IFIH1) | China | rs2111485 (G) | 0.47 | 0.25–0.87 | [60] |
| DExD/H-box helicase 58 (DDX58) | China | rs3824456 (C) or rs2074160 (A) | 0.69 | 0.49–0.97 | [60] |
| Sodium taurocholate cotransporting polypeptide (NTCP) | Taiwan | rs2296651(AA) | 0.13 | 0.05–0.34 | [63] |
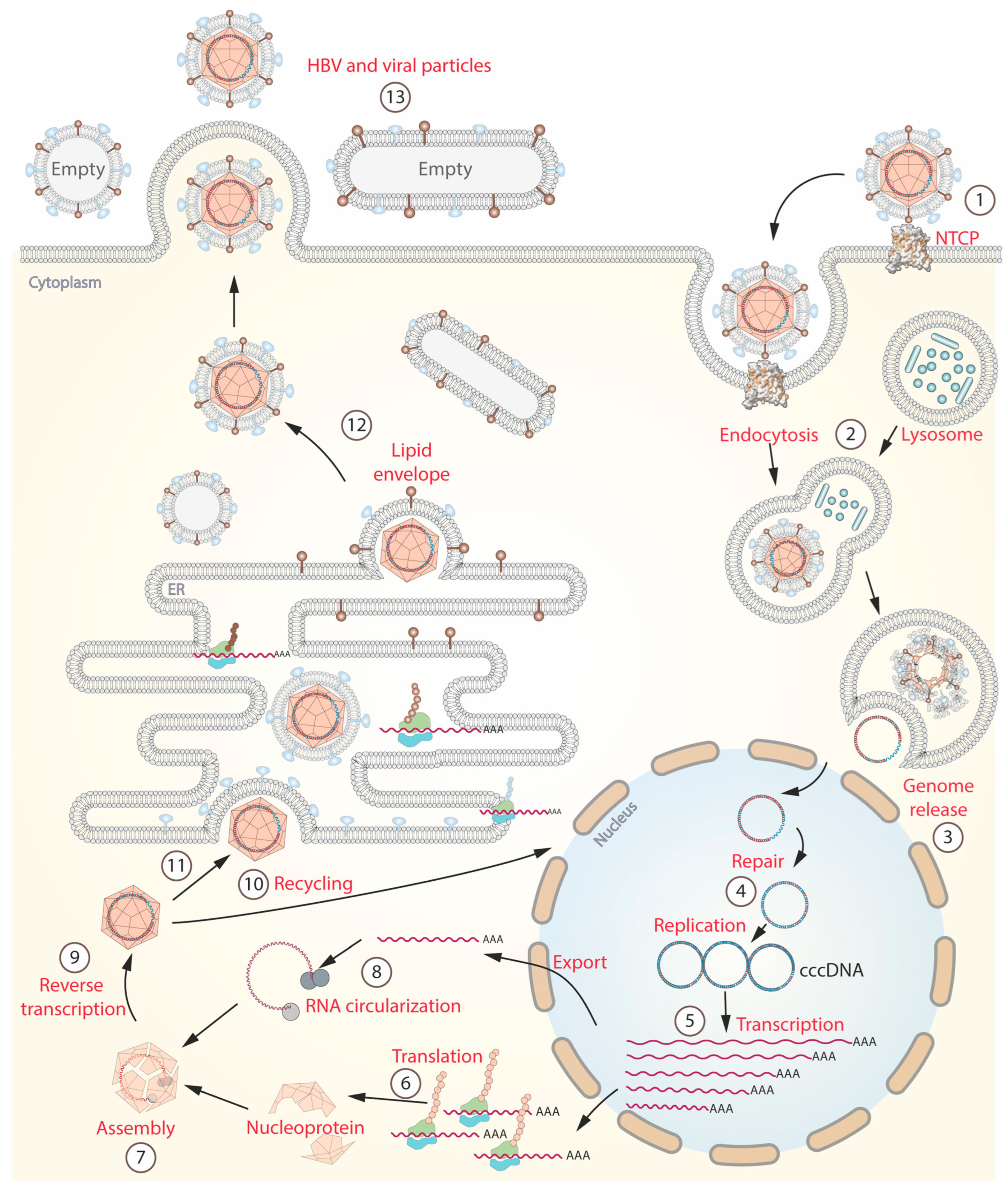
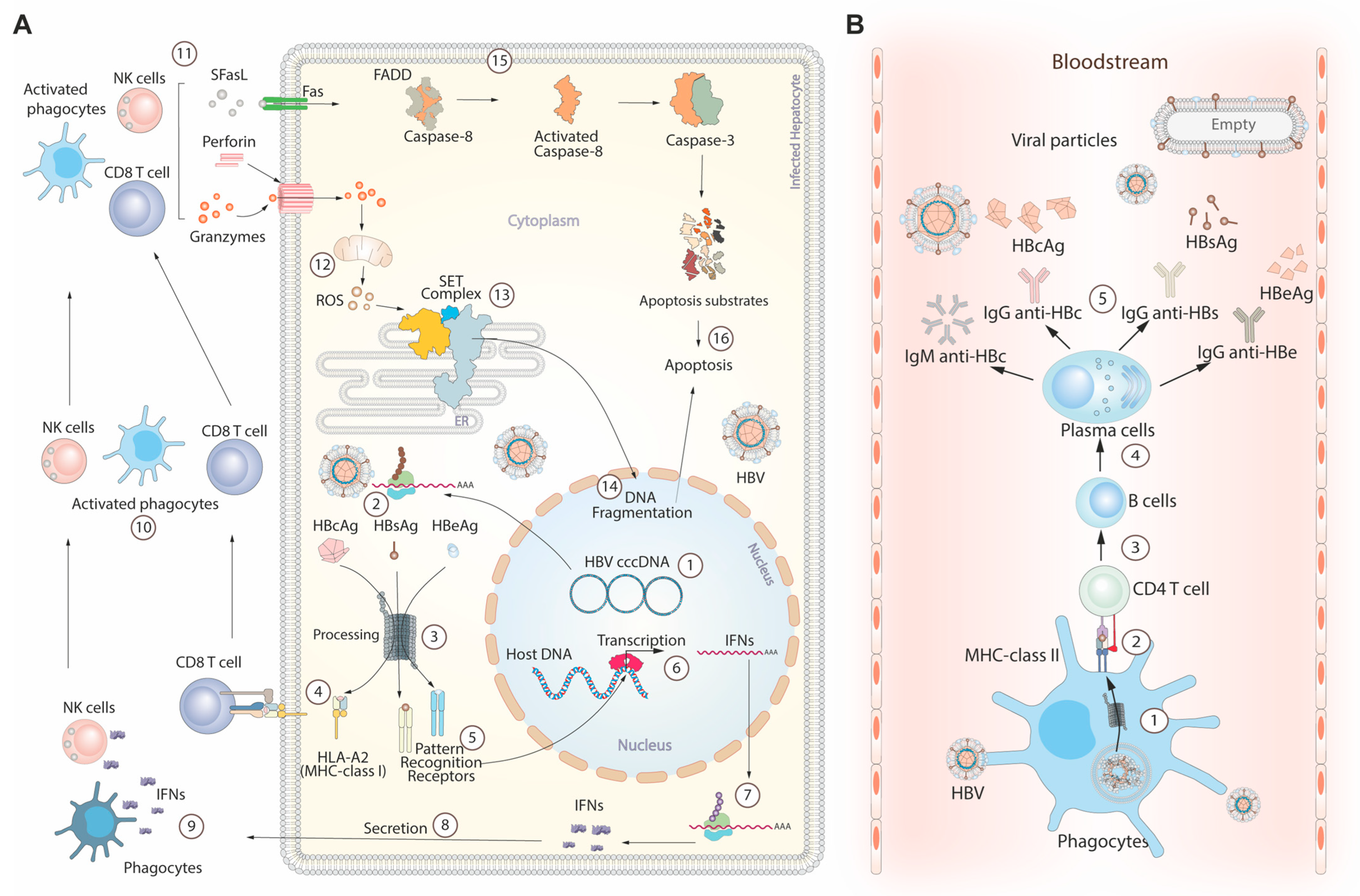
4. HBV Life Cycle and Immune Response
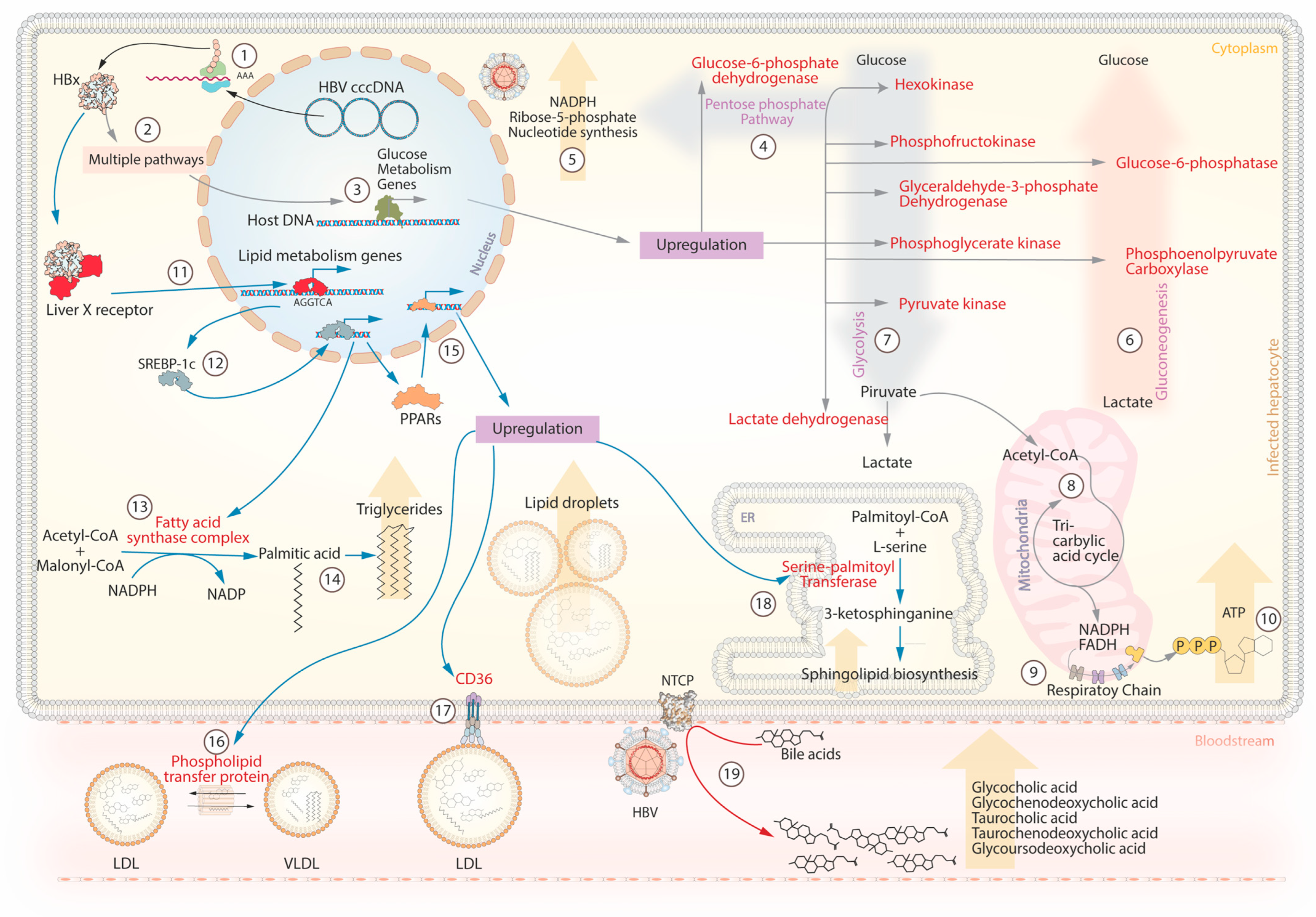
5. HBV and Metabolism
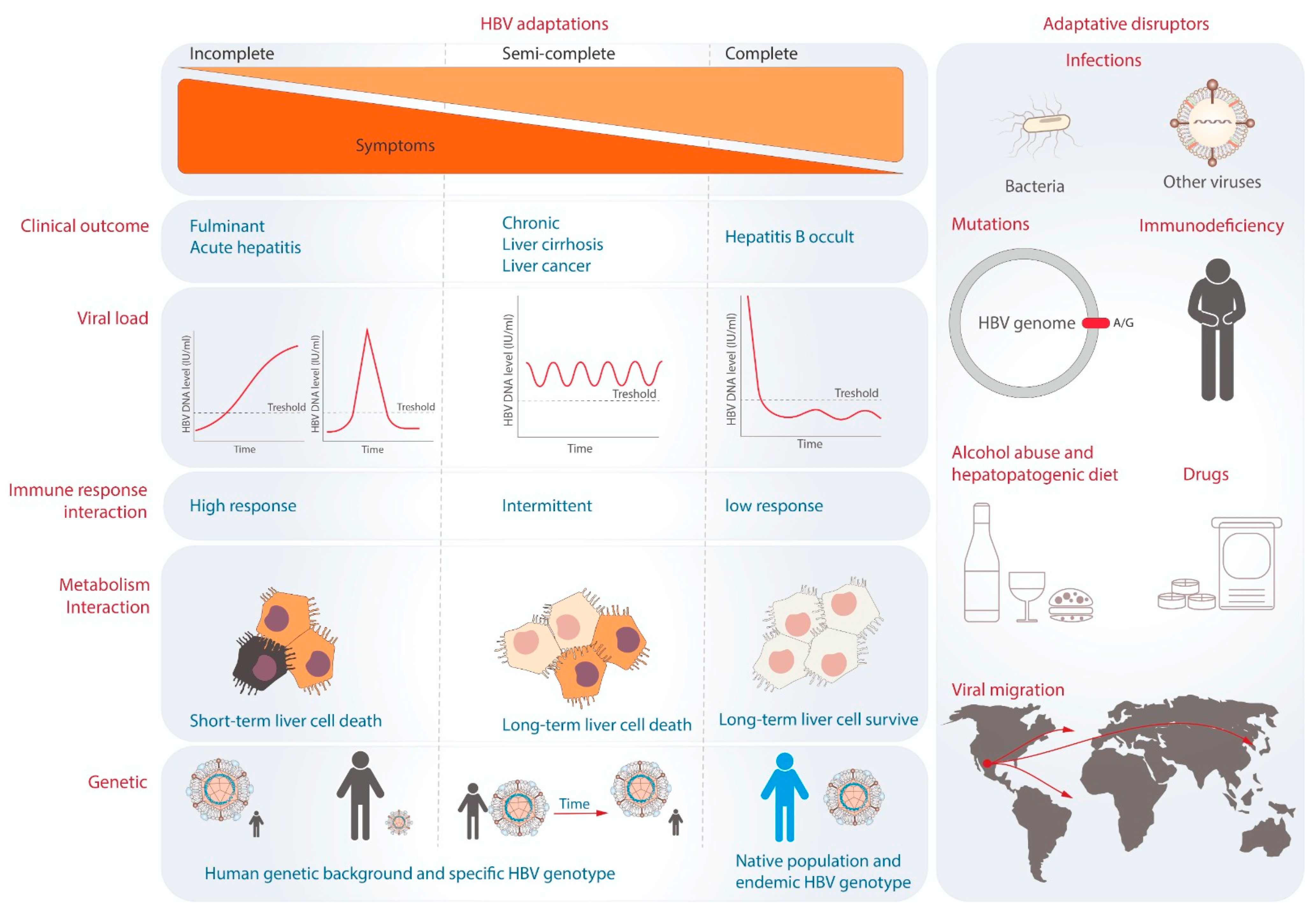
6. The Theory of Viral Adaptation
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- World Health Organization. Hepatitis B; World Health Organization: Geneva, Switzerland, 2022; p. 4. Available online: https://www.who.int/news-room/fact-sheets/detail/hepatitis-b (accessed on 13 June 2023).
- Jeng, W.J.; Papatheodoridis, G.V.; Lok, A.S.F. Hepatitis B. Lancet 2023, 401, 1039–1052. [Google Scholar] [CrossRef] [PubMed]
- Weisberg, I.S.; Brown, R.S., Jr.; Sigal, S.H. Hepatitis B and end-stage liver disease. Clin. Liver Dis. 2007, 11, 893–916. [Google Scholar] [CrossRef] [PubMed]
- Schillie, S.; Vellozzi, C.; Reingold, A.; Harris, A.; Haber, P.; Ward, J.W.; Nelson, N.P. Prevention of Hepatitis B Virus Infection in the United States: Recommendations of the Advisory Committee on Immunization Practices. MMWR Recomm. Rep. 2018, 67, 1–31. [Google Scholar] [CrossRef] [PubMed]
- Lee, W.M. Acute liver failure. N. Engl. J. Med. 1993, 329, 1862–1872. [Google Scholar] [CrossRef] [PubMed]
- Petrosillo, N.; Ippolito, G.; Solforosi, L.; Varaldo, P.E.; Clementi, M.; Manzin, A. Molecular epidemiology of an outbreak of fulminant hepatitis B. J. Clin. Microbiol. 2000, 38, 2975–2981. [Google Scholar] [CrossRef]
- Ichai, P.; Samuel, D. Management of Fulminant Hepatitis B. Curr. Infect. Dis. Rep. 2019, 21, 25. [Google Scholar] [CrossRef]
- Ostapowicz, G.; Fontana, R.J.; Schiodt, F.V.; Larson, A.; Davern, T.J.; Han, S.H.; McCashland, T.M.; Shakil, A.O.; Hay, J.E.; Hynan, L.; et al. Results of a prospective study of acute liver failure at 17 tertiary care centers in the United States. Ann. Intern. Med. 2002, 137, 947–954. [Google Scholar] [CrossRef]
- Pollicino, T.; Raimondo, G. Occult hepatitis B infection. J. Hepatol. 2014, 61, 688–689. [Google Scholar] [CrossRef]
- Panduro, A.; Maldonado-Gonzalez, M.; Fierro, N.A.; Roman, S. Distribution of HBV genotypes F and H in Mexico and Central America. Antivir. Ther. 2013, 18, 475–484. [Google Scholar] [CrossRef]
- Raimondo, G.; Filomia, R.; Maimone, S. Therapy of occult hepatitis B virus infection and prevention of reactivation. Intervirology 2014, 57, 189–195. [Google Scholar] [CrossRef]
- Kafeero, H.M.; Ndagire, D.; Ocama, P.; Kato, C.D.; Wampande, E.; Walusansa, A.; Kajumbula, H.; Kateete, D.; Ssenku, J.E.; Sendagire, H. Mapping hepatitis B virus genotypes on the African continent from 1997 to 2021: A systematic review with meta-analysis. Sci. Rep. 2023, 13, 5723. [Google Scholar] [CrossRef] [PubMed]
- Okamoto, H.; Tsuda, F.; Sakugawa, H.; Sastrosoewignjo, R.I.; Imai, M.; Miyakawa, Y.; Mayumi, M. Typing hepatitis B virus by homology in nucleotide sequence: Comparison of surface antigen subtypes. J. Gen. Virol. 1988, 69 Pt 10, 2575–2583. [Google Scholar] [CrossRef] [PubMed]
- Norder, H.; Hammas, B.; Lofdahl, S.; Courouce, A.M.; Magnius, L.O. Comparison of the amino acid sequences of nine different serotypes of hepatitis B surface antigen and genomic classification of the corresponding hepatitis B virus strains. J. Gen. Virol. 1992, 73 Pt 5, 1201–1208. [Google Scholar] [CrossRef] [PubMed]
- Stuyver, L.; De Gendt, S.; Van Geyt, C.; Zoulim, F.; Fried, M.; Schinazi, R.F.; Rossau, R. A new genotype of hepatitis B virus: Complete genome and phylogenetic relatedness. J. Gen. Virol. 2000, 81, 67–74. [Google Scholar] [CrossRef]
- Arauz-Ruiz, P.; Norder, H.; Robertson, B.H.; Magnius, L.O. Genotype H: A new Amerindian genotype of hepatitis B virus revealed in Central America. J. Gen. Virol. 2002, 83, 2059–2073. [Google Scholar] [CrossRef] [PubMed]
- Hannoun, C.; Norder, H.; Lindh, M. An aberrant genotype revealed in recombinant hepatitis B virus strains from Vietnam. J. Gen. Virol. 2000, 81, 2267–2272. [Google Scholar] [CrossRef]
- Olinger, C.M.; Jutavijittum, P.; Hubschen, J.M.; Yousukh, A.; Samountry, B.; Thammavong, T.; Toriyama, K.; Muller, C.P. Possible new hepatitis B virus genotype, southeast Asia. Emerg. Infect. Dis. 2008, 14, 1777–1780. [Google Scholar] [CrossRef]
- Tatematsu, K.; Tanaka, Y.; Kurbanov, F.; Sugauchi, F.; Mano, S.; Maeshiro, T.; Nakayoshi, T.; Wakuta, M.; Miyakawa, Y.; Mizokami, M. A genetic variant of hepatitis B virus divergent from known human and ape genotypes isolated from a Japanese patient and provisionally assigned to new genotype J. J. Virol. 2009, 83, 10538–10547. [Google Scholar] [CrossRef]
- Velkov, S.; Ott, J.J.; Protzer, U.; Michler, T. The Global Hepatitis B Virus Genotype Distribution Approximated from Available Genotyping Data. Genes 2018, 9, 495. [Google Scholar] [CrossRef]
- Osiowy, C.; Coffin, C.; Andonov, A. Review of Laboratory Tests used in Monitoring Hepatitis B Response to Pegylated Interferon and Nucleos(t)ide Analog Therapy. Curr. Treat. Options Infect. Dis. 2016, 8, 177–193. [Google Scholar] [CrossRef]
- Alvarado-Mora, M.V.; Pinho, J.R. Distribution of HBV genotypes in Latin America. Antivir. Ther. 2013, 18, 459–465. [Google Scholar] [CrossRef] [PubMed]
- Roman, S.; Panduro, A. HBV endemicity in Mexico is associated with HBV genotypes H and G. World J. Gastroenterol. 2013, 19, 5446–5453. [Google Scholar] [CrossRef]
- Shah, A.A.; Bodewes, R.; Reijnen, L.; Boelsums, T.; Weller, C.M.; Fanoy, E.B.; Veldhuijzen, I.K. Outbreaks of mumps genotype G viruses in the Netherlands between October 2019 and March 2020: Clusters associated with multiple introductions. BMC Infect. Dis. 2021, 21, 1035. [Google Scholar] [CrossRef] [PubMed]
- Jose-Abrego, A.; Roman, S.; Laguna-Meraz, S.; Rebello-Pinho, J.R.; Justo Arevalo, S.; Panduro, A. Tracing the evolutionary history of hepatitis B virus genotype H endemic to Mexico. Front. Microbiol. 2023, 14, 1180931. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Zhang, Y.; Xu, M.; Li, X.; Zhang, Z. Distribution of hepatitis B virus genotypes and subgenotypes: A meta-analysis. Medicine 2021, 100, e27941. [Google Scholar] [CrossRef]
- Bottecchia, M.; Madejon, A.; Sheldon, J.; Garcia-Samaniego, J.; Barreiro, P.; Soriano, V. Hepatitis B virus genotype A2 harbours an L217R polymorphism which may account for a lower response to adefovir. J. Antimicrob. Chemother. 2008, 62, 626–627. [Google Scholar] [CrossRef]
- Chan, H.L.; Hui, A.Y.; Wong, M.L.; Tse, A.M.; Hung, L.C.; Wong, V.W.; Sung, J.J. Genotype C hepatitis B virus infection is associated with an increased risk of hepatocellular carcinoma. Gut 2004, 53, 1494–1498. [Google Scholar] [CrossRef]
- Gounder, P.P.; Bulkow, L.R.; Snowball, M.; Negus, S.; Spradling, P.R.; McMahon, B.J. Hepatocellular Carcinoma Risk in Alaska Native Children and Young Adults with Hepatitis B Virus: Retrospective Cohort Analysis. J. Pediatr. 2016, 178, 206–213. [Google Scholar] [CrossRef]
- Shi, Y.H. Correlation between hepatitis B virus genotypes and clinical outcomes. Jpn. J. Infect. Dis. 2012, 65, 476–482. [Google Scholar] [CrossRef]
- Jose-Abrego, A.; Roman, S.; Rebello-Pinho, J.R.; Gomes-Gouvea, M.; Panduro, A. High Frequency of Antiviral Resistance Mutations in HBV Genotypes A2 and H: Multidrug Resistance Strains in Mexico. J. Clin. Transl. Hepatol. 2023, 11, 1023–1034. [Google Scholar] [CrossRef]
- Roca, T.P.; Villar, L.M.; Nogueira Lima, F.S.; Vasconcelos, M.P.A.; Borzacov, L.M.P.; Silva, E.C.E.; Lago, B.V.D.; Silva, M.; Botelho Souza, L.F.; Salcedo, J.M.V.; et al. Genomic Variability of Hepatitis B Virus Circulating in Brazilian Western Amazon. Viruses 2022, 14, 2100. [Google Scholar] [CrossRef] [PubMed]
- di Filippo Villa, D.; Cortes-Mancera, F.; Payares, E.; Montes, N.; de la Hoz, F.; Arbelaez, M.P.; Correa, G.; Navas, M.C. Hepatitis D virus and hepatitis B virus infection in Amerindian communities of the Amazonas state, Colombia. Virol. J. 2015, 12, 172. [Google Scholar] [CrossRef] [PubMed]
- Roman, S.; Panduro, A.; Aguilar-Gutierrez, Y.; Maldonado, M.; Vazquez-Vandyck, M.; Martinez-Lopez, E.; Ruiz-Madrigal, B.; Hernandez-Nazara, Z. A low steady HBsAg seroprevalence is associated with a low incidence of HBV-related liver cirrhosis and hepatocellular carcinoma in Mexico: A systematic review. Hepatol. Int. 2009, 3, 343–355. [Google Scholar] [CrossRef]
- Oba, U.; Koga, Y.; Hoshina, T.; Suminoe, A.; Abe, K.; Hayashida, M.; Taguchi, T.; Hara, T. An adolescent female having hepatocellular carcinoma associated with hepatitis B virus genotype H with a deletion mutation in the pre-S2 region. J. Infect. Chemother. 2015, 21, 302–304. [Google Scholar] [CrossRef]
- Simmonds, P.; Midgley, S. Recombination in the genesis and evolution of hepatitis B virus genotypes. J. Virol. 2005, 79, 15467–15476. [Google Scholar] [CrossRef] [PubMed]
- Toan, N.L.; Song, L.H.; Kremsner, P.G.; Duy, D.N.; Binh, V.Q.; Koeberlein, B.; Kaiser, S.; Kandolf, R.; Torresi, J.; Bock, C.T. Impact of the hepatitis B virus genotype and genotype mixtures on the course of liver disease in Vietnam. Hepatology 2006, 43, 1375–1384. [Google Scholar] [CrossRef]
- Ghosh, S.; Banerjee, P.; Deny, P.; Mondal, R.K.; Nandi, M.; Roychoudhury, A.; Das, K.; Banerjee, S.; Santra, A.; Zoulim, F.; et al. New HBV subgenotype D9, a novel D/C recombinant, identified in patients with chronic HBeAg-negative infection in Eastern India. J. Viral Hepat. 2013, 20, 209–218. [Google Scholar] [CrossRef]
- Jose-Abrego, A.; Roman, S.; Rebello Pinho, J.R.; de Castro, V.F.D.; Panduro, A. Hepatitis B Virus (HBV) Genotype Mixtures, Viral Load, and Liver Damage in HBV Patients Co-infected With Human Immunodeficiency Virus. Front. Microbiol. 2021, 12, 640889. [Google Scholar] [CrossRef]
- Sakamoto, T.; Tanaka, Y.; Watanabe, T.; Iijima, S.; Kani, S.; Sugiyama, M.; Murakami, S.; Matsuura, K.; Kusakabe, A.; Shinkai, N.; et al. Mechanism of the dependence of hepatitis B virus genotype G on co-infection with other genotypes for viral replication. J. Viral Hepat. 2013, 20, e27–e36. [Google Scholar] [CrossRef]
- Zhang, X.Q.; Hong, X.J.; Bai, X.J. Susceptibility to active decompensated cirrhosis is associated with polymorphisms of intercellular adhesion molecule-1 (ICAM-1) in chronic HBV carriers. J. Viral. Hepat. 2008, 15, 173–178. [Google Scholar] [CrossRef]
- Sun, Y.; Lu, Y.; Xie, L.; Deng, Y.; Li, S.; Qin, X. Interferon gamma polymorphisms and hepatitis B virus-related liver cirrhosis risk in a Chinese population. Cancer Cell Int. 2015, 15, 35. [Google Scholar] [CrossRef] [PubMed]
- Yu, S.K.; Kwon, O.S.; Jung, H.S.; Bae, K.S.; Kwon, K.A.; Kim, Y.K.; Kim, Y.S.; Kim, J.H. Influence of transforming growth factor-beta1 gene polymorphism at codon 10 on the development of cirrhosis in chronic hepatitis B virus carriers. J. Korean Med. Sci. 2010, 25, 564–569. [Google Scholar] [CrossRef] [PubMed]
- Yan, X.H.; Wu, J.L.; Yu, R.; Ma, X.H.; Li, Q.F.; Xie, R.F. Associations between gene polymorphisms of signal transducer and activator of transcription 3 and the susceptibility to hepatitis B virus related liver cirrhosis. Zhonghua Yu Fang Yi Xue Za Zhi 2022, 56, 185–191. [Google Scholar] [CrossRef]
- Jiang, D.K.; Ma, X.P.; Wu, X.; Peng, L.; Yin, J.; Dan, Y.; Huang, H.X.; Ding, D.L.; Zhang, L.Y.; Shi, Z.; et al. Genetic variations in STAT4, C2, HLA-DRB1 and HLA-DQ associated with risk of hepatitis B virus-related liver cirrhosis. Sci. Rep. 2015, 5, 16278. [Google Scholar] [CrossRef]
- Liu, W.; Ma, N.; Zhao, D.; Gao, X.; Zhang, X.; Yang, L.; Liu, D. Correlation between the DEPDC5 rs1012068 polymorphism and the risk of HBV-related hepatocellular carcinoma. Clin. Res. Hepatol. Gastroenterol. 2019, 43, 446–450. [Google Scholar] [CrossRef]
- Mai, H.; Xie, H.; Hou, J.; Chen, H.; Zhou, B.; Hou, J.; Jiang, D. A Genetic Variant of PPP1CB Influences Risk of Hepatitis B Virus-Related Hepatocellular Carcinoma in Han Chinese: A Pathway Based Analysis. J. Hepatocell. Carcinoma 2021, 8, 1055–1064. [Google Scholar] [CrossRef] [PubMed]
- Zeisel, M.B.; Guerrieri, F.; Levrero, M. Host Epigenetic Alterations and Hepatitis B Virus-Associated Hepatocellular Carcinoma. J. Clin. Med. 2021, 10, 1715. [Google Scholar] [CrossRef]
- Jiang, J.H.; Gao, Q.; Shen, X.Z.; Yu, Y.; Gu, F.M.; Yan, J.; Pan, J.F.; Jin, F.; Fan, J.; Zhou, J.; et al. An X-chromosomal association study identifies a susceptibility locus at Xq22.1 for hepatitis B virus-related hepatocellular carcinoma. Clin. Res. Hepatol. Gastroenterol. 2013, 37, 586–595. [Google Scholar] [CrossRef]
- Tsai, T.Y.; Huang, M.T.; Sung, P.S.; Peng, C.Y.; Tao, M.H.; Yang, H.I.; Chang, W.C.; Yang, A.S.; Yu, C.M.; Lin, Y.P.; et al. SIGLEC-3 (CD33) serves as an immune checkpoint receptor for HBV infection. J. Clin. Investig. 2021, 131, e141965. [Google Scholar] [CrossRef]
- Yoon, Y.J.; Chang, H.Y.; Ahn, S.H.; Kim, J.K.; Park, Y.K.; Kang, D.R.; Park, J.Y.; Myoung, S.M.; Kim, D.Y.; Chon, C.Y.; et al. MDM2 and p53 polymorphisms are associated with the development of hepatocellular carcinoma in patients with chronic hepatitis B virus infection. Carcinogenesis 2008, 29, 1192–1196. [Google Scholar] [CrossRef]
- Mardian, Y.; Yano, Y.; Wasityastuti, W.; Ratnasari, N.; Liang, Y.; Putri, W.A.; Triyono, T.; Hayashi, Y. Genetic polymorphisms of HLA-DP and isolated anti-HBc are important subsets of occult hepatitis B infection in Indonesian blood donors: A case-control study. Virol. J. 2017, 14, 201. [Google Scholar] [CrossRef] [PubMed]
- Wang, T.; Shen, C.; Chen, L.; Liu, S.; Ji, Y. Association of human leukocyte antigen polymorphisms with occult hepatitis B virus infection in a Shaanxi Han population. J. Gene Med. 2017, 19, e2987. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Han, S.B. Hepatitis B Reactivation: A Review of Clinical Guidelines. J. Clin. Gastroenterol. 2021, 55, 393–399. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Y.; Li, H.; Wang, X.; Zheng, X.; Huang, Y.; Chen, J.; Meng, Z.; Gao, Y.; Qian, Z.; Liu, F.; et al. Hepatitis B Virus Reactivation Increased the Risk of Developing Hepatic Failure and Mortality in Cirrhosis with Acute Exacerbation. Front. Microbiol. 2022, 13, 910549. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, H.; Hiramatsu, K.; Akazawa, Y.; Nosaka, T.; Saito, Y.; Ozaki, Y.; Hayama, R.; Takahashi, K.; Naito, T.; Ofuji, K.; et al. Genetic polymorphism and decreased expression of HLA class II DP genes are associated with HBV reactivation in patients treated with immunomodulatory agents. J. Med. Virol. 2018, 90, 712–720. [Google Scholar] [CrossRef]
- Hsiao, L.T.; Wang, H.Y.; Yang, C.F.; Chiou, T.J.; Gau, J.P.; Yu, Y.B.; Liu, H.L.; Chang, W.C.; Chen, P.M.; Tzeng, C.H.; et al. Human Cytokine Genetic Variants Associated with HBsAg Reverse Seroconversion in Rituximab-Treated Non-Hodgkin Lymphoma Patients. Medicine 2016, 95, e3064. [Google Scholar] [CrossRef]
- Lee, H.W.; Park, H.J.; Jin, B.; Dezhbord, M.; Kim, D.Y.; Han, K.H.; Ryu, W.S.; Kim, S.; Ahn, S.H. Effect of S267F variant of NTCP on the patients with chronic hepatitis B. Sci. Rep. 2017, 7, 17634. [Google Scholar] [CrossRef]
- Wu, J.F.; Chen, C.H.; Ni, Y.H.; Lin, Y.T.; Chen, H.L.; Hsu, H.Y.; Chang, M.H. Toll-like receptor and hepatitis B virus clearance in chronic infected patients: A long-term prospective cohort study in Taiwan. J. Infect. Dis. 2012, 206, 662–668. [Google Scholar] [CrossRef]
- Yao, Y.; Shen, Y.; Shao, H.; Liu, Y.; Ji, Y.; Du, G.; Ye, X.; Huang, P.; Chen, H. Polymorphisms of RIG-I-like receptor influence HBV clearance in Chinese Han population. J. Med. Virol. 2021, 93, 4957–4965. [Google Scholar] [CrossRef]
- Shen, Z.; Yang, H.; Yang, S.; Wang, W.; Cui, X.; Zhou, X.; Liu, W.; Pan, S.; Liu, Y.; Zhang, J.; et al. Hepatitis B virus persistence in mice reveals IL-21 and IL-33 as regulators of viral clearance. Nat. Commun. 2017, 8, 2119. [Google Scholar] [CrossRef]
- Wang, L.; Zou, Z.Q.; Wang, K. Clinical Relevance of HLA Gene Variants in HBV Infection. J. Immunol. Res. 2016, 2016, 9069375. [Google Scholar] [CrossRef] [PubMed]
- Hu, H.H.; Liu, J.; Lin, Y.L.; Luo, W.S.; Chu, Y.J.; Chang, C.L.; Jen, C.L.; Lee, M.H.; Lu, S.N.; Wang, L.Y.; et al. The rs2296651 (S267F) variant on NTCP (SLC10A1) is inversely associated with chronic hepatitis B and progression to cirrhosis and hepatocellular carcinoma in patients with chronic hepatitis B. Gut 2016, 65, 1514–1521. [Google Scholar] [CrossRef] [PubMed]
- Hu, P.; Liu, J.; Zhang, D. Association of NTCP Gene Polymorphisms and Spontaneous Clearance of Hepatitis B Virus in Asia: A Meta-Analysis. Hepat. Mon. 2019, 19, 2–8. [Google Scholar] [CrossRef]
- Yan, H.; Zhong, G.; Xu, G.; He, W.; Jing, Z.; Gao, Z.; Huang, Y.; Qi, Y.; Peng, B.; Wang, H.; et al. Sodium taurocholate cotransporting polypeptide is a functional receptor for human hepatitis B and D virus. eLife 2012, 1, e00049. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Summers, J. Integration of hepadnavirus DNA in infected liver: Evidence for a linear precursor. J. Virol. 1999, 73, 9710–9717. [Google Scholar] [CrossRef]
- Xia, Y.; Guo, H. Hepatitis B virus cccDNA: Formation, regulation and therapeutic potential. Antivir. Res. 2020, 180, 104824. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.H.; Kang, S.; Kim, J.; Ahn, B.Y. Hepatitis B virus core protein stimulates the proteasome-mediated degradation of viral X protein. J. Virol. 2003, 77, 7166–7173. [Google Scholar] [CrossRef]
- Mogensen, T.H. Pathogen recognition and inflammatory signaling in innate immune defenses. Clin. Microbiol. Rev. 2009, 22, 240–273. [Google Scholar] [CrossRef]
- Li, Q.; Sun, B.; Zhuo, Y.; Jiang, Z.; Li, R.; Lin, C.; Jin, Y.; Gao, Y.; Wang, D. Interferon and interferon-stimulated genes in HBV treatment. Front. Immunol. 2022, 13, 1034968. [Google Scholar] [CrossRef]
- Liu, Q.; Zheng, Y.; Yu, Y.; Tan, Q.; Huang, X. Identification of HLA-A*0201-restricted CD8+ T-cell epitope C64–72 from hepatitis B virus core protein. Int. Immunopharmacol. 2012, 13, 141–147. [Google Scholar] [CrossRef]
- Lee, J.Y.; Chae, D.W.; Kim, S.M.; Nam, E.S.; Jang, M.K.; Lee, J.H.; Kim, H.Y.; Yoo, J.Y. Expression of FasL and perforin/granzyme B mRNA in chronic hepatitis B virus infection. J. Viral. Hepat. 2004, 11, 130–135. [Google Scholar] [CrossRef]
- Schreiber, S.; Honz, M.; Mamozai, W.; Kurktschiev, P.; Schiemann, M.; Witter, K.; Moore, E.; Zielinski, C.; Sette, A.; Protzer, U.; et al. Characterization of a library of 20 HBV-specific MHC class II-restricted T cell receptors. Mol. Ther. Methods Clin. Dev. 2021, 23, 476–489. [Google Scholar] [CrossRef] [PubMed]
- Asao, H. Interleukin-21 in Viral Infections. Int. J. Mol. Sci. 2021, 22, 9521. [Google Scholar] [CrossRef]
- Wang, S.; Wang, J.; Kumar, V.; Karnell, J.L.; Naiman, B.; Gross, P.S.; Rahman, S.; Zerrouki, K.; Hanna, R.; Morehouse, C.; et al. IL-21 drives expansion and plasma cell differentiation of autoreactive CD11chiT-bet+ B cells in SLE. Nat. Commun. 2018, 9, 1758. [Google Scholar] [CrossRef] [PubMed]
- Schillie, S.; Murphy, T.V.; Sawyer, M.; Ly, K.; Hughes, E.; Jiles, R.; de Perio, M.A.; Reilly, M.; Byrd, K.; Ward, J.W.; et al. CDC guidance for evaluating healthcare personnel for hepatitis B virus protection and for administering postexposure management. MMWR Recomm. Rep. 2013, 62, 1–19. [Google Scholar] [PubMed]
- Conners, E.E.; Panagiotakopoulos, L.; Hofmeister, M.G.; Spradling, P.R.; Hagan, L.M.; Harris, A.M.; Rogers-Brown, J.S.; Wester, C.; Nelson, N.P.; Rapposelli, K.; et al. Screening and Testing for Hepatitis B Virus Infection: CDC Recommendations—United States, 2023. MMWR Recomm. Rep. 2023, 72, 1–25. [Google Scholar] [CrossRef]
- Lin, S.; Zhang, Y.J. Interference of Apoptosis by Hepatitis B Virus. Viruses 2017, 9, 230. [Google Scholar] [CrossRef]
- Ogden, S.K.; Lee, K.C.; Barton, M.C. Hepatitis B viral transactivator HBx alleviates p53-mediated repression of alpha-fetoprotein gene expression. J. Biol. Chem. 2000, 275, 27806–27814. [Google Scholar] [CrossRef]
- Shi, Y.X.; Huang, C.J.; Yang, Z.G. Impact of hepatitis B virus infection on hepatic metabolic signaling pathway. World J. Gastroenterol. 2016, 22, 8161–8167. [Google Scholar] [CrossRef]
- Hu, J.J.; Song, W.; Zhang, S.D.; Shen, X.H.; Qiu, X.M.; Wu, H.Z.; Gong, P.H.; Lu, S.; Zhao, Z.J.; He, M.L.; et al. HBx-upregulated lncRNA UCA1 promotes cell growth and tumorigenesis by recruiting EZH2 and repressing p27Kip1/CDK2 signaling. Sci. Rep. 2016, 6, 23521. [Google Scholar] [CrossRef]
- Li, Z.; Li, X.; Wu, S.; Xue, M.; Chen, W. Long non-coding RNA UCA1 promotes glycolysis by up-regulating hexokinase 2 through the mTOR-STAT3/microRNA143 pathway. Cancer Sci. 2014, 105, 951–955. [Google Scholar] [CrossRef] [PubMed]
- Lamontagne, R.J.; Casciano, J.C.; Bouchard, M.J. A broad investigation of the HBV-mediated changes to primary hepatocyte physiology reveals HBV significantly alters metabolic pathways. Metabolism 2018, 83, 50–59. [Google Scholar] [CrossRef]
- Sadrolodabaee, L.; Low, T.K.; Feng, H.; Chen, W.N. Role of HBV Replication in Host Cell Metabolism: A Proteomics Analysis. Curr. Proteom. 2013, 10, 29–37. [Google Scholar] [CrossRef]
- Shin, H.J.; Park, Y.H.; Kim, S.U.; Moon, H.B.; Park, D.S.; Han, Y.H.; Lee, C.H.; Lee, D.S.; Song, I.S.; Lee, D.H.; et al. Hepatitis B virus X protein regulates hepatic glucose homeostasis via activation of inducible nitric oxide synthase. J. Biol. Chem. 2011, 286, 29872–29881. [Google Scholar] [CrossRef] [PubMed]
- Borchani-Chabchoub, I.; Mokdad-Gargouri, R.; Gargouri, A. Glucose dependent [correction of dependant] negative translational control of the heterologous expression of the preS2 HBV antigen in yeast. Gene 2003, 311, 165–170. [Google Scholar] [CrossRef] [PubMed]
- Liu, B.; Fang, M.; He, Z.; Cui, D.; Jia, S.; Lin, X.; Xu, X.; Zhou, T.; Liu, W. Hepatitis B virus stimulates G6PD expression through HBx-mediated Nrf2 activation. Cell Death Dis. 2015, 6, e1980. [Google Scholar] [CrossRef]
- Chen, Y.; Wang, B.; Ou, X.; Wu, Y.; He, Y.; Lin, X.; Lin, X. Small Hepatitis B Virus Surface Antigen Promotes Hepatic Gluconeogenesis via Enhancing Glucagon/cAMP/Protein Kinase A/CREB Signaling. J. Virol. 2022, 96, e0102022. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.; Kim, K.H.; Kim, H.H.; Cheong, J. Hepatitis B virus X protein induces lipogenic transcription factor SREBP1 and fatty acid synthase through the activation of nuclear receptor LXRalpha. Biochem. J. 2008, 416, 219–230. [Google Scholar] [CrossRef] [PubMed]
- Willy, P.J.; Umesono, K.; Ong, E.S.; Evans, R.M.; Heyman, R.A.; Mangelsdorf, D.J. LXR, a nuclear receptor that defines a distinct retinoid response pathway. Genes. Dev. 1995, 9, 1033–1045. [Google Scholar] [CrossRef]
- DeBose-Boyd, R.A.; Ye, J. SREBPs in Lipid Metabolism, Insulin Signaling, and Beyond. Trends Biochem. Sci. 2018, 43, 358–368. [Google Scholar] [CrossRef]
- Wang, Y.; Nakajima, T.; Gonzalez, F.J.; Tanaka, N. PPARs as Metabolic Regulators in the Liver: Lessons from Liver-Specific PPAR-Null Mice. Int. J. Mol. Sci. 2020, 21, 2061. [Google Scholar] [CrossRef] [PubMed]
- Wakil, S.J.; Abu-Elheiga, L.A. Fatty acid metabolism: Target for metabolic syndrome. J. Lipid Res. 2009, 50, 138–143. [Google Scholar] [CrossRef] [PubMed]
- Marechal, L.; Laviolette, M.; Rodrigue-Way, A.; Sow, B.; Brochu, M.; Caron, V.; Tremblay, A. The CD36-PPARgamma Pathway in Metabolic Disorders. Int. J. Mol. Sci. 2018, 19, 1529. [Google Scholar] [CrossRef]
- Zakrzewicz, D.; Geyer, J. Interactions of Na(+)/taurocholate cotransporting polypeptide with host cellular proteins upon hepatitis B and D virus infection: Novel potential targets for antiviral therapy. Biol. Chem. 2023, 404, 673–690. [Google Scholar] [CrossRef]
- Park, J.H.; Iwamoto, M.; Yun, J.H.; Uchikubo-Kamo, T.; Son, D.; Jin, Z.; Yoshida, H.; Ohki, M.; Ishimoto, N.; Mizutani, K.; et al. Structural insights into the HBV receptor and bile acid transporter NTCP. Nature 2022, 606, 1027–1031. [Google Scholar] [CrossRef]
- Wang, X.; Xie, G.; Zhao, A.; Zheng, X.; Huang, F.; Wang, Y.; Yao, C.; Jia, W.; Liu, P. Serum Bile Acids Are Associated with Pathological Progression of Hepatitis B-Induced Cirrhosis. J. Proteome Res. 2016, 15, 1126–1134. [Google Scholar] [CrossRef] [PubMed]
- Xun, Z.; Yao, X.; Ou, Q. Emerging roles of bile acids in chronic hepatitis, liver cirrhosis, and hepatocellular carcinoma. Cell Mol. Immunol. 2023, 20, 1087–1089. [Google Scholar] [CrossRef]
- Lauber, C.; Seitz, S.; Mattei, S.; Suh, A.; Beck, J.; Herstein, J.; Borold, J.; Salzburger, W.; Kaderali, L.; Briggs, J.A.G.; et al. Deciphering the Origin and Evolution of Hepatitis B Viruses by Means of a Family of Non-enveloped Fish Viruses. Cell Host Microbe 2017, 22, 387–399.e6. [Google Scholar] [CrossRef]
- Kocher, A.; Papac, L.; Barquera, R.; Key, F.M.; Spyrou, M.A.; Hubler, R.; Rohrlach, A.B.; Aron, F.; Stahl, R.; Wissgott, A.; et al. Ten millennia of hepatitis B virus evolution. Science 2021, 374, 182–188. [Google Scholar] [CrossRef]
- Larsen, C.S. The past 12,000 years of behavior, adaptation, population, and evolution shaped who we are today. Proc. Natl. Acad. Sci. USA 2023, 120, e2209613120. [Google Scholar] [CrossRef]
- Roman, S.; Jose-Abrego, A.; Fierro, N.A.; Escobedo-Melendez, G.; Ojeda-Granados, C.; Martinez-Lopez, E.; Panduro, A. Hepatitis B virus infection in Latin America: A genomic medicine approach. World J. Gastroenterol. 2014, 20, 7181–7196. [Google Scholar] [CrossRef] [PubMed]
- Peacock, T.P.; Penrice-Randal, R.; Hiscox, J.A.; Barclay, W.S. SARS-CoV-2 one year on: Evidence for ongoing viral adaptation. J. Gen. Virol. 2021, 102, 1584. [Google Scholar] [CrossRef]
- Liang, T.J. Hepatitis B: The virus and disease. Hepatology 2009, 49, 13–21. [Google Scholar] [CrossRef] [PubMed]
- Tu, T.; Budzinska, M.A.; Shackel, N.A.; Urban, S. HBV DNA Integration: Molecular Mechanisms and Clinical Implications. Viruses 2017, 9, 75. [Google Scholar] [CrossRef]
- Roman, S.; Tanaka, Y.; Khan, A.; Kurbanov, F.; Kato, H.; Mizokami, M.; Panduro, A. Occult hepatitis B in the genotype H-infected Nahuas and Huichol native Mexican population. J. Med. Virol. 2010, 82, 1527–1536. [Google Scholar] [CrossRef] [PubMed]
- Jaramillo, C.M.; de La Hoz, F.; Porras, A.; di Filippo, D.; Choconta-Piraquive, L.A.; Payares, E.; Montes, N.; Navas, M.C. Characterization of hepatitis B virus in Amerindian children and mothers from Amazonas State, Colombia. PLoS ONE 2017, 12, e0181643. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Wang, K.; Zou, Z.Q. Crosstalk between innate and adaptive immunity in hepatitis B virus infection. World J. Hepatol. 2015, 7, 2980–2991. [Google Scholar] [CrossRef]
- Yamada, N.; Shigefuku, R.; Sugiyama, R.; Kobayashi, M.; Ikeda, H.; Takahashi, H.; Okuse, C.; Suzuki, M.; Itoh, F.; Yotsuyanagi, H.; et al. Acute hepatitis B of genotype H resulting in persistent infection. World J. Gastroenterol. 2014, 20, 3044–3049. [Google Scholar] [CrossRef]
- Centers for Disease Control and Prevention. The Age-Adjusted Death Rate for Hepatitis C and Hepatitis B during 2020. 2020. Available online: https://www.cdc.gov/hepatitis/policy/npr/2022/reduce-reported-hepatitis-b-deaths.htm#:~:text=The%20age%2Dadjusted%20hepatitis%20B,the%20target%20rate%20of%200.42 (accessed on 4 July 2023).
- Centers for Disease Control and Prevention. Viral Hepatitis and Liver Cancer. 2012. Available online: https://www.cdc.gov/nchhstp/newsroom/docs/factsheets/viral-hep-liver-cancer.pdf (accessed on 4 July 2023).
- World Health Organization. Guidelines on Hepatitis B and C Testing. 2017. Available online: https://www.who.int/publications/i/item/9789241549981 (accessed on 4 July 2023).
- Forni, D.; Cagliani, R.; Pontremoli, C.; Pozzoli, U.; Vertemara, J.; De Gioia, L.; Clerici, M.; Sironi, M. Evolutionary Analysis Provides Insight into the Origin and Adaptation of HCV. Front. Microbiol. 2018, 9, 854. [Google Scholar] [CrossRef]




Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2023 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Jose-Abrego, A.; Roman, S.; Laguna-Meraz, S.; Panduro, A. Host and HBV Interactions and Their Potential Impact on Clinical Outcomes. Pathogens 2023, 12, 1146. https://doi.org/10.3390/pathogens12091146
Jose-Abrego A, Roman S, Laguna-Meraz S, Panduro A. Host and HBV Interactions and Their Potential Impact on Clinical Outcomes. Pathogens. 2023; 12(9):1146. https://doi.org/10.3390/pathogens12091146
Chicago/Turabian StyleJose-Abrego, Alexis, Sonia Roman, Saul Laguna-Meraz, and Arturo Panduro. 2023. "Host and HBV Interactions and Their Potential Impact on Clinical Outcomes" Pathogens 12, no. 9: 1146. https://doi.org/10.3390/pathogens12091146
APA StyleJose-Abrego, A., Roman, S., Laguna-Meraz, S., & Panduro, A. (2023). Host and HBV Interactions and Their Potential Impact on Clinical Outcomes. Pathogens, 12(9), 1146. https://doi.org/10.3390/pathogens12091146