
SARS-CoV-2 ORF3a Protein as a Therapeutic Target against COVID-19 and Long-Term Post-Infection Effects

 , , , , , and
, , , , , and 
{kind=link}
Abstract
:1. Introduction
2. SARS-CoV-2 ORF3a Protein as a Promising Antiviral Target
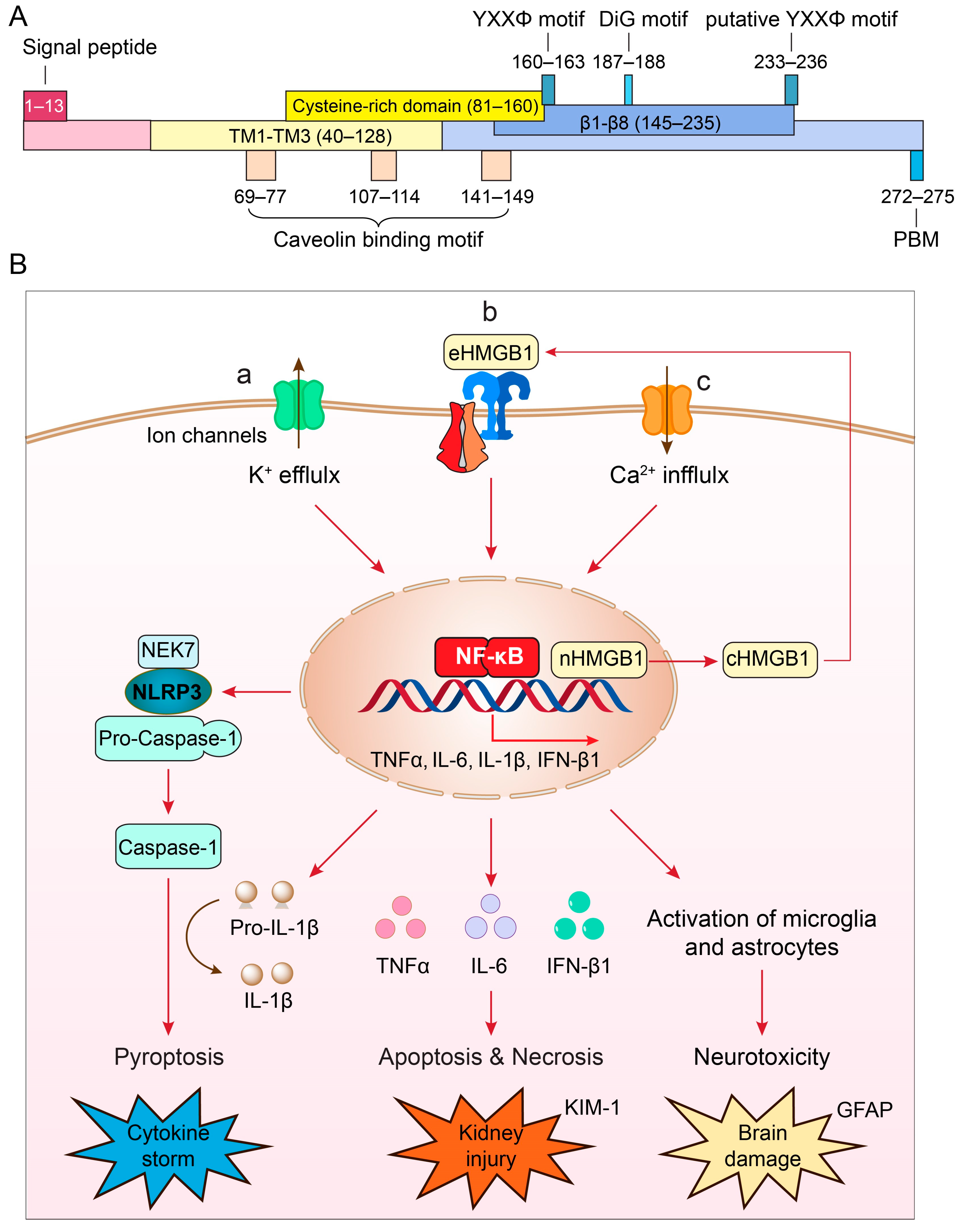
3. Structure and Unique Features of ORF3a Protein

4. Role of ORF3a in Viral Production
5. Role of ORF3a in Viral Pathogenesis: Induction of Cytokine Storm and COVID-19 Severity
6. Roles of ORF3a in Kidney Injury, Neuroinflammation, and Long-COVID Complications
7. Discovery and Development of Inhibitor Drugs Targeting ORF3a Protein
7.1. Testing Well-Established Druggable Cellular Targets of Key ORF3a Regulators with FDA-Approved Drugs and Small Molecule Inhibitors (SMIs)
7.2. Structure-Based Design of ORF3a Inhibitors
7.3. Cell-Based High-Throughput Drug Screening
8. Challenges and Future Prospectives
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Conflicts of Interest
References
- Alcendor, D.J.; Matthews-Juarez, P.; Smoot, D.; Hildreth, J.E.K.; Lamar, K.; Tabatabai, M.; Wilus, D.; Juarez, P.D. Breakthrough COVID-19 Infections in the US: Implications for Prolonging the Pandemic. Vaccines 2022, 10, 755. [Google Scholar] [CrossRef]
- Rubin, R. From Positive to Negative to Positive Again-The Mystery of Why COVID-19 Rebounds in Some Patients Who Take Paxlovid. JAMA 2022, 327, 2380–2382. [Google Scholar] [CrossRef] [PubMed]
- Service, R.F. Bad news for Paxlovid? Resistance may be coming. Science 2022, 377, 138–139. [Google Scholar] [CrossRef]
- Beigel, J.H.; Tomashek, K.M.; Dodd, L.E.; Mehta, A.K.; Zingman, B.S.; Kalil, A.C.; Hohmann, E.; Chu, H.Y.; Luetkemeyer, A.; Kline, S.; et al. Remdesivir for the Treatment of COVID-19—Final Report. N. Engl. J. Med. 2020, 383, 1813–1826. [Google Scholar] [CrossRef] [PubMed]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Kruger, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; Herrler, G.; Wu, N.H.; Nitsche, A.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280.e278. [Google Scholar] [CrossRef] [PubMed]
- Jin, Z.; Du, X.; Xu, Y.; Deng, Y.; Liu, M.; Zhao, Y.; Zhang, B.; Li, X.; Zhang, L.; Peng, C.; et al. Structure of M(pro) from SARS-CoV-2 and discovery of its inhibitors. Nature 2020, 582, 289–293. [Google Scholar] [CrossRef]
- Wang, Y.; Zhang, D.; Du, G.; Du, R.; Zhao, J.; Jin, Y.; Fu, S.; Gao, L.; Cheng, Z.; Lu, Q.; et al. Remdesivir in adults with severe COVID-19: A randomised, double-blind, placebo-controlled, multicentre trial. Lancet 2020, 395, 1569–1578. [Google Scholar] [CrossRef] [PubMed]
- Group, R.C.; Horby, P.; Lim, W.S.; Emberson, J.R.; Mafham, M.; Bell, J.L.; Linsell, L.; Staplin, N.; Brightling, C.; Ustianowski, A.; et al. Dexamethasone in Hospitalized Patients with COVID-19. N. Engl. J. Med. 2021, 384, 693–704. [Google Scholar] [CrossRef]
- Weinreich, D.M.; Sivapalasingam, S.; Norton, T.; Ali, S.; Gao, H.; Bhore, R.; Musser, B.J.; Soo, Y.; Rofail, D.; Im, J.; et al. REGN-COV2, a Neutralizing Antibody Cocktail, in Outpatients with COVID-19. N. Engl. J. Med. 2021, 384, 238–251. [Google Scholar] [CrossRef]
- Zhao, R.Y. Yeast for virus research. Microb. Cell 2017, 4, 311–330. [Google Scholar] [CrossRef]
- Li, G.; Zhao, R.Y. Molecular Cloning and Characterization of Small Viral Genome in Fission Yeast. Methods Mol. Biol. 2018, 1721, 47–61. [Google Scholar] [CrossRef] [PubMed]
- Gordon, D.E.; Jang, G.M.; Bouhaddou, M.; Xu, J.; Obernier, K.; White, K.M.; O’Meara, M.J.; Rezelj, V.V.; Guo, J.Z.; Swaney, D.L.; et al. A SARS-CoV-2 protein interaction map reveals targets for drug repurposing. Nature 2020, 583, 459–468. [Google Scholar] [CrossRef] [PubMed]
- Harcourt, J.; Tamin, A.; Lu, X.; Kamili, S.; Sakthivel, S.K.; Murray, J.; Queen, K.; Tao, Y.; Paden, C.R.; Zhang, J.; et al. Severe Acute Respiratory Syndrome Coronavirus 2 from Patient with Coronavirus Disease, United States. Emerg. Infect. Dis. 2020, 26, 1266–1273. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Elder, R.T.; Chen, M.; Cao, J. Fission yeast expression vectors adapted for positive identification of gene insertion and green fluorescent protein fusion. Biotechniques 1998, 25, 438–440, 442, 444. [Google Scholar] [CrossRef] [PubMed]
- Nkeze, J.; Li, L.; Benko, Z.; Li, G.; Zhao, R.Y. Molecular characterization of HIV-1 genome in fission yeast Schizosaccharomyces pombe. Cell Biosci. 2015, 5, 47. [Google Scholar] [CrossRef]
- Li, G.; Poulsen, M.; Fenyvuesvolgyi, C.; Yashiroda, Y.; Yoshida, M.; Simard, J.M.; Gallo, R.C.; Zhao, R.Y. Characterization of cytopathic factors through genome-wide analysis of the Zika viral proteins in fission yeast. Proc. Natl. Acad. Sci. USA 2017, 114, E376–E385. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Li, Q.; Cruz Cosme, R.S.; Gerzanich, V.; Tang, Q.; Simard, J.M.; Zhao, R.Y. Genome-Wide Characterization of SARS-CoV-2 Cytopathogenic Proteins in the Search of Antiviral Targets. mBio 2021, 13, e0016922. [Google Scholar] [CrossRef] [PubMed]
- Zhang, J.; Ejikemeuwa, A.; Gerzanich, V.; Nasr, M.; Tang, Q.; Simard, J.M.; Zhao, R.Y. Understanding the Role of SARS-CoV-2 ORF3a in Viral Pathogenesis and COVID-19. Front. Microbiol. 2022, 13, 854567. [Google Scholar] [CrossRef]
- Hachim, A.; Kavian, N.; Cohen, C.A.; Chin, A.W.H.; Chu, D.K.W.; Mok, C.K.P.; Tsang, O.T.Y.; Yeung, Y.C.; Perera, R.; Poon, L.L.M.; et al. ORF8 and ORF3b antibodies are accurate serological markers of early and late SARS-CoV-2 infection. Nat. Immunol. 2020, 21, 1293–1301. [Google Scholar] [CrossRef]
- Camerini, D.; Randall, A.Z.; Trappl-Kimmons, K.; Oberai, A.; Hung, C.; Edgar, J.; Shandling, A.; Huynh, V.; Teng, A.A.; Hermanson, G.; et al. Mapping SARS-CoV-2 Antibody Epitopes in COVID-19 Patients with a Multi-Coronavirus Protein Microarray. Microbiol. Spectr. 2021, 9, e0141621. [Google Scholar] [CrossRef]
- Liu, I.J.; Hsueh, P.R.; Lin, C.T.; Chiu, C.Y.; Kao, C.L.; Liao, M.Y.; Wu, H.C. Disease-specific B Cell epitopes for serum antibodies from patients with severe acute respiratory syndrome (SARS) and serologic detection of SARS antibodies by epitope-based peptide antigens. J. Infect. Dis. 2004, 190, 797–809. [Google Scholar] [CrossRef] [PubMed]
- Zhong, X.; Guo, Z.; Yang, H.; Peng, L.; Xie, Y.; Wong, T.Y.; Lai, S.T.; Guo, Z. Amino terminus of the SARS coronavirus protein 3a elicits strong, potentially protective humoral responses in infected patients. J. Gen. Virol. 2006, 87, 369–373. [Google Scholar] [CrossRef] [PubMed]
- Zhu, A.; Liu, M.; Li, Y.; Lei, Q.; Wu, Q.; Lin, M.; Lai, D.; Lu, L.; Yu, S.; Guo, S.; et al. Age- and Severity-Associated Humoral Immunity Response in COVID-19 Patients: A Cohort Study from Wuhan, China. J. Clin. Med. 2022, 11, 5974. [Google Scholar] [CrossRef] [PubMed]
- Grifoni, A.; Weiskopf, D.; Ramirez, S.I.; Mateus, J.; Dan, J.M.; Moderbacher, C.R.; Rawlings, S.A.; Sutherland, A.; Premkumar, L.; Jadi, R.S.; et al. Targets of T Cell Responses to SARS-CoV-2 Coronavirus in Humans with COVID-19 Disease and Unexposed Individuals. Cell 2020, 181, 1489–1501.e1415. [Google Scholar] [CrossRef] [PubMed]
- Tan, A.T.; Linster, M.; Tan, C.W.; Le Bert, N.; Chia, W.N.; Kunasegaran, K.; Zhuang, Y.; Tham, C.Y.L.; Chia, A.; Smith, G.J.D.; et al. Early induction of functional SARS-CoV-2-specific T cells associates with rapid viral clearance and mild disease in COVID-19 patients. Cell Rep. 2021, 34, 108728. [Google Scholar] [CrossRef] [PubMed]
- Samandari, T.; Ongalo, J.B.; McCarthy, K.D.; Biegon, R.K.; Madiega, P.A.; Mithika, A.; Orinda, J.; Mboya, G.M.; Mwaura, P.; Anzala, O.; et al. Prevalence and functional profile of SARS-CoV-2 T cells in asymptomatic Kenyan adults. J. Clin. Investig. 2023, 133, 13. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wang, K.; Ping, X.; Yu, W.; Qian, Z.; Xiong, S.; Sun, B. The ns12.9 Accessory Protein of Human Coronavirus OC43 Is a Viroporin Involved in Virion Morphogenesis and Pathogenesis. J. Virol. 2015, 89, 11383–11395. [Google Scholar] [CrossRef]
- Kern, D.M.; Sorum, B.; Mali, S.S.; Hoel, C.M.; Sridharan, S.; Remis, J.P.; Toso, D.B.; Kotecha, A.; Bautista, D.M.; Brohawn, S.G. Cryo-EM structure of SARS-CoV-2 ORF3a in lipid nanodiscs. Nat. Struct. Mol. Biol. 2021, 28, 573–582. [Google Scholar] [CrossRef]
- McClenaghan, C.; Hanson, A.; Lee, S.J.; Nichols, C.G. Coronavirus Proteins as Ion Channels: Current and Potential Research. Front. Immunol. 2020, 11, 573339. [Google Scholar] [CrossRef]
- Issa, E.; Merhi, G.; Panossian, B.; Salloum, T.; Tokajian, S. SARS-CoV-2 and ORF3a: Nonsynonymous Mutations, Functional Domains, and Viral Pathogenesis. mSystems 2020, 5, e00266-20. [Google Scholar] [CrossRef]
- Cruz-Cosme, R.; Zhang, J.; Liu, D.; Mahase, V.; Sallapalli, B.T.; Chang, P.; Zhang, Y.; Teng, S.; Zhao, R.Y.; Tang, Q. A novel diG motif in ORF3a protein of SARS-CoV-2 for intracellular transport. Front. Cell Dev. Biol. 2022, 10, 1011221. [Google Scholar] [CrossRef] [PubMed]
- Padhan, K.; Tanwar, C.; Hussain, A.; Hui, P.Y.; Lee, M.Y.; Cheung, C.Y.; Peiris, J.S.M.; Jameel, S. Severe acute respiratory syndrome coronavirus Orf3a protein interacts with caveolin. J. Gen. Virol. 2007, 88, 3067–3077. [Google Scholar] [CrossRef] [PubMed]
- Tan, Y.J.; Teng, E.; Shen, S.; Tan, T.H.; Goh, P.Y.; Fielding, B.C.; Ooi, E.E.; Tan, H.C.; Lim, S.G.; Hong, W. A novel severe acute respiratory syndrome coronavirus protein, U274, is transported to the cell surface and undergoes endocytosis. J. Virol. 2004, 78, 6723–6734. [Google Scholar] [CrossRef] [PubMed]
- Minakshi, R.; Padhan, K. The YXXPhi motif within the severe acute respiratory syndrome coronavirus (SARS-CoV) 3a protein is crucial for its intracellular transport. Virol. J. 2014, 11, 75. [Google Scholar] [CrossRef] [PubMed]
- Bianchi, M.; Borsetti, A.; Ciccozzi, M.; Pascarella, S. SARS-CoV-2 ORF3a: Mutability and function. Int. J. Biol. Macromol. 2021, 170, 820–826. [Google Scholar] [CrossRef]
- Kakkanas, A.; Karamichali, E.; Koufogeorgou, E.I.; Kotsakis, S.D.; Georgopoulou, U.; Foka, P. Targeting the YXXPhi Motifs of the SARS Coronaviruses 1 and 2 ORF3a Peptides by In Silico Analysis to Predict Novel Virus-Host Interactions. Biomolecules 2022, 12, 1052. [Google Scholar] [CrossRef]
- Siu, K.L.; Yuen, K.S.; Castano-Rodriguez, C.; Ye, Z.W.; Yeung, M.L.; Fung, S.Y.; Yuan, S.; Chan, C.P.; Yuen, K.Y.; Enjuanes, L.; et al. Severe acute respiratory syndrome coronavirus ORF3a protein activates the NLRP3 inflammasome by promoting TRAF3-dependent ubiquitination of ASC. FASEB J. 2019, 33, 8865–8877. [Google Scholar] [CrossRef]
- Jin, D.Y.; Zheng, B.J.; Tang, H.M.V. Mechanism of inflammasome activation by SARS coronavirus 3a protein: Abridged secondary publication. Hong Kong Med. J. 2021, 27 (Suppl. S2), 33–35. [Google Scholar]
- Guarnieri, J.W.; Angelin, A.; Murdock, D.G.; Schaefer, P.; Portluri, P.; Lie, T.; Huang, J.; Wallace, D.C. SARS-CoV-2 viroporins activate the NLRP3-inflammasome by the mitochondrial permeability transition pore. Front. Immunol. 2023, 14, 1064293. [Google Scholar] [CrossRef]
- Cai, H.; Chen, Y.; Feng, Y.; Asadi, M.; Kaufman, L.; Lee, K.; Kehrer, T.; Miorin, L.; Garcia-Sastre, A.; Gusella, G.L.; et al. SARS-CoV-2 viral protein ORF3A injures renal tubules by interacting with TRIM59 to induce STAT3 activation. Mol. Ther. 2023, 31, 774–787. [Google Scholar] [CrossRef]
- Zhu, H.; Byrnes, C.; Lee, Y.T.; Tuymetova, G.; Duffy, H.B.D.; Bakir, J.Y.; Pettit, S.N.; Angina, J.; Springer, D.A.; Allende, M.L.; et al. SARS-CoV-2 ORF3a expression in brain disrupts the autophagy-lysosomal pathway, impairs sphingolipid homeostasis, and drives neuropathogenesis. FASEB J. 2023, 37, e22919. [Google Scholar] [CrossRef] [PubMed]
- Han, W.K.; Bailly, V.; Abichandani, R.; Thadhani, R.; Bonventre, J.V. Kidney Injury Molecule-1 (KIM-1): A novel biomarker for human renal proximal tubule injury. Kidney Int. 2002, 62, 237–244. [Google Scholar] [CrossRef] [PubMed]
- Liddelow, S.A.; Guttenplan, K.A.; Clarke, L.E.; Bennett, F.C.; Bohlen, C.J.; Schirmer, L.; Bennett, M.L.; Munch, A.E.; Chung, W.S.; Peterson, T.C.; et al. Neurotoxic reactive astrocytes are induced by activated microglia. Nature 2017, 541, 481–487. [Google Scholar] [CrossRef] [PubMed]
- Schiff, L.; Hadker, N.; Weiser, S.; Rausch, C. A literature review of the feasibility of glial fibrillary acidic protein as a biomarker for stroke and traumatic brain injury. Mol. Diagn. Ther. 2012, 16, 79–92. [Google Scholar] [CrossRef]
- Brant, A.C.; Tian, W.; Majerciak, V.; Yang, W.; Zheng, Z.M. SARS-CoV-2: From its discovery to genome structure, transcription, and replication. Cell Biosci. 2021, 11, 136. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.B.; Jung, M.; Kim, J.; Charles, A.; Christ, W.; Kang, J.; Kang, M.G.; Kwak, C.; Klingstrom, J.; Smed-Sorensen, A.; et al. Super-resolution proximity labeling reveals anti-viral protein network and its structural changes against SARS-CoV-2 viral proteins. Cell Rep. 2023, 42, 112835. [Google Scholar] [CrossRef]
- Zhang, J.; Cruz-Cosme, R.; Zhang, C.; Liu, D.; Tang, Q.; Zhao, R.Y. Endoplasmic reticulum-associated SARS-CoV-2 ORF3a elicits heightened cytopathic effects despite robust ER-associated degradation. mBio 2023, e0303023. [Google Scholar] [CrossRef]
- Nishimura, N.; Balch, W.E. A di-acidic signal required for selective export from the endoplasmic reticulum. Science 1997, 277, 556–558. [Google Scholar] [CrossRef]
- Oostra, M.; de Haan, C.A.; de Groot, R.J.; Rottier, P.J. Glycosylation of the severe acute respiratory syndrome coronavirus triple-spanning membrane proteins 3a and M. J. Virol. 2006, 80, 2326–2336. [Google Scholar] [CrossRef]
- Breitinger, U.; Farag, N.S.; Sticht, H.; Breitinger, H.G. Viroporins: Structure, function, and their role in the life cycle of SARS-CoV-2. Int. J. Biochem. Cell Biol. 2022, 145, 106185. [Google Scholar] [CrossRef]
- Xu, H.; Akinyemi, I.A.; Chitre, S.A.; Loeb, J.C.; Lednicky, J.A.; McIntosh, M.T.; Bhaduri-McIntosh, S. SARS-CoV-2 viroporin encoded by ORF3a triggers the NLRP3 inflammatory pathway. Virology 2022, 568, 13–22. [Google Scholar] [CrossRef] [PubMed]
- Fam, M.S.; Sedky, C.A.; Turky, N.O.; Breitinger, H.G.; Breitinger, U. Channel activity of SARS-CoV-2 viroporin ORF3a inhibited by adamantanes and phenolic plant metabolites. Sci. Rep. 2023, 13, 5328. [Google Scholar] [CrossRef]
- Nieva, J.L.; Madan, V.; Carrasco, L. Viroporins: Structure and biological functions. Nat. Rev. Microbiol. 2012, 10, 563–574. [Google Scholar] [CrossRef] [PubMed]
- Miller, A.N.; Houlihan, P.R.; Matamala, E.; Cabezas-Bratesco, D.; Lee, G.Y.; Cristofori-Armstrong, B.; Dilan, T.L.; Sanchez-Martinez, S.; Matthies, D.; Yan, R.; et al. The SARS-CoV-2 accessory protein Orf3a is not an ion channel, but does interact with trafficking proteins. eLife 2023, 12, e84477. [Google Scholar] [CrossRef] [PubMed]
- Busscher, B.M.; Befekadu, H.B.; Liu, Z.; Xiao, T.S. SARS-CoV-2 ORF3a-Mediated NF-kappaB Activation Is Not Dependent on TRAF-Binding Sequence. Viruses 2023, 15, 2229. [Google Scholar] [CrossRef] [PubMed]
- Caillet-Saguy, C.; Durbesson, F.; Rezelj, V.V.; Gogl, G.; Tran, Q.D.; Twizere, J.C.; Vignuzzi, M.; Vincentelli, R.; Wolff, N. Host PDZ-containing proteins targeted by SARS-CoV-2. FEBS J. 2021, 288, 5148–5162. [Google Scholar] [CrossRef] [PubMed]
- Castano-Rodriguez, C.; Honrubia, J.M.; Gutierrez-Alvarez, J.; DeDiego, M.L.; Nieto-Torres, J.L.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Verdia-Baguena, C.; Queralt-Martin, M.; et al. Role of Severe Acute Respiratory Syndrome Coronavirus Viroporins E, 3a, and 8a in Replication and Pathogenesis. mBio 2018, 9, 10–1128. [Google Scholar] [CrossRef]
- Rice, A.P.; Kimata, J.T. SARS-CoV-2 likely targets cellular PDZ proteins: A common tactic of pathogenic viruses. Future Virol. 2021, 16, 375–377. [Google Scholar] [CrossRef]
- Miura, K.; Suzuki, Y.; Ishida, K.; Arakawa, M.; Wu, H.; Fujioka, Y.; Emi, A.; Maeda, K.; Hamajima, R.; Nakano, T.; et al. Distinct motifs in the E protein are required for SARS-CoV-2 virus particle formation and lysosomal deacidification in host cells. J. Virol. 2023, 97, e0042623. [Google Scholar] [CrossRef]
- Honrubia, J.M.; Gutierrez-Alvarez, J.; Sanz-Bravo, A.; Gonzalez-Miranda, E.; Munoz-Santos, D.; Castano-Rodriguez, C.; Wang, L.; Villarejo-Torres, M.; Ripoll-Gomez, J.; Esteban, A.; et al. SARS-CoV-2-Mediated Lung Edema and Replication Are Diminished by Cystic Fibrosis Transmembrane Conductance Regulator Modulators. mBio 2023, 14, e0313622. [Google Scholar] [CrossRef]
- Zhang, X.; Liu, Y.; Liu, J.; Bailey, A.L.; Plante, K.S.; Plante, J.A.; Zou, J.; Xia, H.; Bopp, N.E.; Aguilar, P.V.; et al. A trans-complementation system for SARS-CoV-2 recapitulates authentic viral replication without virulence. Cell 2021, 184, 2229–2238.e2213. [Google Scholar] [CrossRef] [PubMed]
- Chen, D.; Zheng, Q.; Sun, L.; Ji, M.; Li, Y.; Deng, H.; Zhang, H. ORF3a of SARS-CoV-2 promotes lysosomal exocytosis-mediated viral egress. Dev. Cell 2021, 56, 3250–3263.e5. [Google Scholar] [CrossRef] [PubMed]
- Lu, W.; Zheng, B.J.; Xu, K.; Schwarz, W.; Du, L.; Wong, C.K.; Chen, J.; Duan, S.; Deubel, V.; Sun, B. Severe acute respiratory syndrome-associated coronavirus 3a protein forms an ion channel and modulates virus release. Proc. Natl. Acad. Sci. USA 2006, 103, 12540–12545. [Google Scholar] [CrossRef] [PubMed]
- Ghosh, S.; Dellibovi-Ragheb, T.A.; Kerviel, A.; Pak, E.; Qiu, Q.; Fisher, M.; Takvorian, P.M.; Bleck, C.; Hsu, V.W.; Fehr, A.R.; et al. beta-Coronaviruses Use Lysosomes for Egress Instead of the Biosynthetic Secretory Pathway. Cell 2020, 183, 1520–1535.e1514. [Google Scholar] [CrossRef]
- Stewart, H.; Palmulli, R.; Johansen, K.H.; McGovern, N.; Shehata, O.M.; Carnell, G.W.; Jackson, H.K.; Lee, J.S.; Brown, J.C.; Burgoyne, T.; et al. Tetherin antagonism by SARS-CoV-2 ORF3a and spike protein enhances virus release. EMBO Rep. 2023, 24, e57224. [Google Scholar] [CrossRef]
- Martin-Sancho, L.; Lewinski, M.K.; Pache, L.; Stoneham, C.A.; Yin, X.; Becker, M.E.; Pratt, D.; Churas, C.; Rosenthal, S.B.; Liu, S.; et al. Functional landscape of SARS-CoV-2 cellular restriction. Mol. Cell 2021, 81, 2656–2668.e2658. [Google Scholar] [CrossRef]
- Yount, B.; Roberts, R.S.; Sims, A.C.; Deming, D.; Frieman, M.B.; Sparks, J.; Denison, M.R.; Davis, N.; Baric, R.S. Severe acute respiratory syndrome coronavirus group-specific open reading frames encode nonessential functions for replication in cell cultures and mice. J. Virol. 2005, 79, 14909–14922. [Google Scholar] [CrossRef]
- Akerstrom, S.; Mirazimi, A.; Tan, Y.J. Inhibition of SARS-CoV replication cycle by small interference RNAs silencing specific SARS proteins, 7a/7b, 3a/3b and S. Antivir. Res. 2007, 73, 219–227. [Google Scholar] [CrossRef]
- Silvas, J.A.; Vasquez, D.M.; Park, J.G.; Chiem, K.; Allue-Guardia, A.; Garcia-Vilanova, A.; Platt, R.N.; Miorin, L.; Kehrer, T.; Cupic, A.; et al. Contribution of SARS-CoV-2 Accessory Proteins to Viral Pathogenicity in K18 Human ACE2 Transgenic Mice. J. Virol. 2021, 95, e0040221. [Google Scholar] [CrossRef]
- Liu, Y.; Zhang, X.; Liu, J.; Xia, H.; Zou, J.; Muruato, A.E.; Periasamy, S.; Kurhade, C.; Plante, J.A.; Bopp, N.E.; et al. A live-attenuated SARS-CoV-2 vaccine candidate with accessory protein deletions. Nat. Commun. 2022, 13, 4337. [Google Scholar] [CrossRef]
- Duarte, L.F.; Vazquez, Y.; Diethelm-Varela, B.; Pavez, V.; Berrios-Rojas, R.; Mendez, C.; Riedel, C.A.; White, J.A.; Kalergis, A.M.; Bueno, S.M.; et al. Differential Severe Acute Respiratory Syndrome Coronavirus 2-Specific Humoral Response in Inactivated Virus-Vaccinated, Convalescent, and Breakthrough-Infected Subjects. J. Infect. Dis. 2023, 228, 857–867. [Google Scholar] [CrossRef]
- McGrath, M.E.; Xue, Y.; Dillen, C.; Oldfield, L.; Assad-Garcia, N.; Zaveri, J.; Singh, N.; Baracco, L.; Taylor, L.J.; Vashee, S.; et al. SARS-CoV-2 variant spike and accessory gene mutations alter pathogenesis. Proc. Natl. Acad. Sci. USA 2022, 119, e2204717119. [Google Scholar] [CrossRef] [PubMed]
- Ye, C.; Park, J.G.; Chiem, K.; Dravid, P.; Allue-Guardia, A.; Garcia-Vilanova, A.; Pino Tamayo, P.; Shivanna, V.; Kapoor, A.; Walter, M.R.; et al. Immunization with Recombinant Accessory Protein-Deficient SARS-CoV-2 Protects against Lethal Challenge and Viral Transmission. Microbiol. Spectr. 2023, 11, e0065323. [Google Scholar] [CrossRef]
- Zhang, J.; Wu, H.; Yao, X.; Zhang, D.; Zhou, Y.; Fu, B.; Wang, W.; Li, H.; Wang, Z.; Hu, Z.; et al. Pyroptotic macrophages stimulate the SARS-CoV-2-associated cytokine storm. Cell Mol. Immunol. 2021, 18, 1305–1307. [Google Scholar] [CrossRef] [PubMed]
- Ratajczak, M.Z.; Kucia, M. SARS-CoV-2 infection and overactivation of Nlrp3 inflammasome as a trigger of cytokine “storm” and risk factor for damage of hematopoietic stem cells. Leukemia 2020, 34, 1726–1729. [Google Scholar] [CrossRef] [PubMed]
- Hu, B.; Huang, S.; Yin, L. The cytokine storm and COVID-19. J. Med. Virol. 2021, 93, 250–256. [Google Scholar] [CrossRef]
- Freeman, T.L.; Swartz, T.H. Targeting the NLRP3 Inflammasome in Severe COVID-19. Front. Immunol. 2020, 11, 1518. [Google Scholar] [CrossRef]
- Van den Berg, D.F.; Te Velde, A.A. Severe COVID-19: NLRP3 Inflammasome Dysregulated. Front. Immunol. 2020, 11, 1580. [Google Scholar] [CrossRef]
- Bertoni, A.; Penco, F.; Mollica, H.; Bocca, P.; Prigione, I.; Corcione, A.; Cangelosi, D.; Schena, F.; Del Zotto, G.; Amaro, A.; et al. Spontaneous NLRP3 inflammasome-driven IL1-beta secretion is induced in severe COVID-19 patients and responds to anakinra treatment. J. Allergy Clin. Immunol. 2022, 150, 796–805. [Google Scholar] [CrossRef]
- Jorgensen, I.; Miao, E.A. Pyroptotic cell death defends against intracellular pathogens. Immunol. Rev. 2015, 265, 130–142. [Google Scholar] [CrossRef]
- Yang, Y.; Wang, H.; Kouadir, M.; Song, H.; Shi, F. Recent advances in the mechanisms of NLRP3 inflammasome activation and its inhibitors. Cell Death Dis. 2019, 10, 128. [Google Scholar] [CrossRef]
- Kayagaki, N.; Stowe, I.B.; Lee, B.L.; O’Rourke, K.; Anderson, K.; Warming, S.; Cuellar, T.; Haley, B.; Roose-Girma, M.; Phung, Q.T.; et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature 2015, 526, 666–671. [Google Scholar] [CrossRef] [PubMed]
- Shi, J.; Zhao, Y.; Wang, K.; Shi, X.; Wang, Y.; Huang, H.; Zhuang, Y.; Cai, T.; Wang, F.; Shao, F. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature 2015, 526, 660–665. [Google Scholar] [CrossRef] [PubMed]
- Chen, I.Y.; Moriyama, M.; Chang, M.F.; Ichinohe, T. Severe Acute Respiratory Syndrome Coronavirus Viroporin 3a Activates the NLRP3 Inflammasome. Front. Microbiol. 2019, 10, 50. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Torres, J.L.; Verdia-Baguena, C.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Castano-Rodriguez, C.; Fernandez-Delgado, R.; Torres, J.; Aguilella, V.M.; Enjuanes, L. Severe acute respiratory syndrome coronavirus E protein transports calcium ions and activates the NLRP3 inflammasome. Virology 2015, 485, 330–339. [Google Scholar] [CrossRef]
- Nieto-Torres, J.L.; DeDiego, M.L.; Verdia-Baguena, C.; Jimenez-Guardeno, J.M.; Regla-Nava, J.A.; Fernandez-Delgado, R.; Castano-Rodriguez, C.; Alcaraz, A.; Torres, J.; Aguilella, V.M.; et al. Severe acute respiratory syndrome coronavirus envelope protein ion channel activity promotes virus fitness and pathogenesis. PLoS Pathog. 2014, 10, e1004077. [Google Scholar] [CrossRef] [PubMed]
- Yalcinkaya, M.; Liu, W.; Islam, M.N.; Kotini, A.G.; Gusarova, G.A.; Fidler, T.P.; Papapetrou, E.P.; Bhattacharya, J.; Wang, N.; Tall, A.R. Modulation of the NLRP3 inflammasome by SARS-CoV-2 Envelope protein. Sci. Rep. 2021, 11, 24432. [Google Scholar] [CrossRef]
- Yang, H.; Wang, H.; Andersson, U. Targeting Inflammation Driven by HMGB1. Front. Immunol. 2020, 11, 484. [Google Scholar] [CrossRef]
- Gowda, P.; Patrick, S.; Joshi, S.D.; Kumawat, R.K.; Sen, E. Glycyrrhizin prevents SARS-CoV-2 S1 and Orf3a induced high mobility group box 1 (HMGB1) release and inhibits viral replication. Cytokine 2021, 142, 155496. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Shaikh, M.F.; Chakraborti, A.; Kumari, Y.; Aledo-Serrano, A.; Aleksovska, K.; Alvim, M.K.M.; Othman, I. HMGB1: A Common Biomarker and Potential Target for TBI, Neuroinflammation, Epilepsy, and Cognitive Dysfunction. Front. Neurosci. 2018, 12, 628. [Google Scholar] [CrossRef]
- Peng, T.; Du, S.Y.; Son, M.; Diamond, B. HIF-1alpha is a negative regulator of interferon regulatory factors: Implications for interferon production by hypoxic monocytes. Proc. Natl. Acad. Sci. USA 2021, 118, e2106017118. [Google Scholar] [CrossRef]
- Bolay, H.; Karadas, O.; Ozturk, B.; Sonkaya, R.; Tasdelen, B.; Bulut, T.D.S.; Gulbahar, O.; Ozge, A.; Baykan, B. HMGB1, NLRP3, IL-6 and ACE2 levels are elevated in COVID-19 with headache: A window to the infection-related headache mechanism. J. Headache Pain 2021, 22, 94. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Long, X.; Xu, Q.; Tan, J.; Wang, G.; Cao, Y.; Wei, J.; Luo, H.; Zhu, H.; Huang, L.; et al. Elevated serum levels of S100A8/A9 and HMGB1 at hospital admission are correlated with inferior clinical outcomes in COVID-19 patients. Cell Mol. Immunol. 2020, 17, 992–994. [Google Scholar] [CrossRef] [PubMed]
- Legrand, M.; Bell, S.; Forni, L.; Joannidis, M.; Koyner, J.L.; Liu, K.; Cantaluppi, V. Pathophysiology of COVID-19-associated acute kidney injury. Nat. Rev. Nephrol. 2021, 17, 751–764. [Google Scholar] [CrossRef] [PubMed]
- May, R.M.; Cassol, C.; Hannoudi, A.; Larsen, C.P.; Lerma, E.V.; Haun, R.S.; Braga, J.R.; Hassen, S.I.; Wilson, J.; VanBeek, C.; et al. A multi-center retrospective cohort study defines the spectrum of kidney pathology in Coronavirus 2019 Disease (COVID-19). Kidney Int. 2021, 100, 1303–1315. [Google Scholar] [CrossRef] [PubMed]
- Bowe, B.; Cai, M.; Xie, Y.; Gibson, A.K.; Maddukuri, G.; Al-Aly, Z. Acute Kidney Injury in a National Cohort of Hospitalized US Veterans with COVID-19. Clin. J. Am. Soc. Nephrol. 2020, 16, 14–25. [Google Scholar] [CrossRef]
- Chan, L.; Chaudhary, K.; Saha, A.; Chauhan, K.; Vaid, A.; Zhao, S.; Paranjpe, I.; Somani, S.; Richter, F.; Miotto, R.; et al. AKI in Hospitalized Patients with COVID-19. J. Am. Soc. Nephrol. 2021, 32, 151–160. [Google Scholar] [CrossRef]
- Geri, G.; Darmon, M.; Zafrani, L.; Fartoukh, M.; Voiriot, G.; Le Marec, J.; Nemlaghi, S.; Vieillard-Baron, A.; Azoulay, E. Acute kidney injury in SARS-CoV2-related pneumonia ICU patients: A retrospective multicenter study. Ann. Intensive Care 2021, 11, 86. [Google Scholar] [CrossRef]
- Puelles, V.G.; Lutgehetmann, M.; Lindenmeyer, M.T.; Sperhake, J.P.; Wong, M.N.; Allweiss, L.; Chilla, S.; Heinemann, A.; Wanner, N.; Liu, S.; et al. Multiorgan and Renal Tropism of SARS-CoV-2. N. Engl. J. Med. 2020, 383, 590–592. [Google Scholar] [CrossRef]
- Copur, S.; Berkkan, M.; Basile, C.; Tuttle, K.; Kanbay, M. Post-acute COVID-19 syndrome and kidney diseases: What do we know? J. Nephrol. 2022, 35, 795–805. [Google Scholar] [CrossRef]
- Amraei, R.; Yin, W.; Napoleon, M.A.; Suder, E.L.; Berrigan, J.; Zhao, Q.; Olejnik, J.; Chandler, K.B.; Xia, C.; Feldman, J.; et al. CD209L/L-SIGN and CD209/DC-SIGN Act as Receptors for SARS-CoV-2. ACS Cent. Sci. 2021, 7, 1156–1165. [Google Scholar] [CrossRef] [PubMed]
- Kalejaiye, T.D.; Bhattacharya, R.; Burt, M.A.; Travieso, T.; Okafor, A.E.; Mou, X.; Blasi, M.; Musah, S. SARS-CoV-2 Employ BSG/CD147 and ACE2 Receptors to Directly Infect Human Induced Pluripotent Stem Cell-Derived Kidney Podocytes. Front. Cell Dev. Biol. 2022, 10, 855340. [Google Scholar] [CrossRef] [PubMed]
- Khan, S.; Chen, L.; Yang, C.R.; Raghuram, V.; Khundmiri, S.J.; Knepper, M.A. Does SARS-CoV-2 Infect the Kidney? J. Am. Soc. Nephrol. 2020, 31, 2746–2748. [Google Scholar] [CrossRef] [PubMed]
- Armaly, Z.; Kinaneh, S.; Skorecki, K. Renal Manifestations of COVID-19: Physiology and Pathophysiology. J. Clin. Med. 2021, 10, 1216. [Google Scholar] [CrossRef] [PubMed]
- Su, H.; Yang, M.; Wan, C.; Yi, L.X.; Tang, F.; Zhu, H.Y.; Yi, F.; Yang, H.C.; Fogo, A.B.; Nie, X.; et al. Renal histopathological analysis of 26 postmortem findings of patients with COVID-19 in China. Kidney Int. 2020, 98, 219–227. [Google Scholar] [CrossRef] [PubMed]
- Braun, F.; Lutgehetmann, M.; Pfefferle, S.; Wong, M.N.; Carsten, A.; Lindenmeyer, M.T.; Norz, D.; Heinrich, F.; Meissner, K.; Wichmann, D.; et al. SARS-CoV-2 renal tropism associates with acute kidney injury. Lancet 2020, 396, 597–598. [Google Scholar] [CrossRef]
- Papadimitriou, J.C.; Drachenberg, C.B.; Kleiner, D.; Choudhri, N.; Haririan, A.; Cebotaru, V. Tubular Epithelial and Peritubular Capillary Endothelial Injury in COVID-19 AKI. Kidney Int. Rep. 2021, 6, 518–525. [Google Scholar] [CrossRef]
- Alexander, M.P.; Mangalaparthi, K.K.; Madugundu, A.K.; Moyer, A.M.; Adam, B.A.; Mengel, M.; Singh, S.; Herrmann, S.M.; Rule, A.D.; Cheek, E.H.; et al. Acute Kidney Injury in Severe COVID-19 Has Similarities to Sepsis-Associated Kidney Injury: A Multi-Omics Study. Mayo Clin. Proc. 2021, 96, 2561–2575. [Google Scholar] [CrossRef]
- Diao, B.; Wang, C.; Wang, R.; Feng, Z.; Zhang, J.; Yang, H.; Tan, Y.; Wang, H.; Wang, C.; Liu, L.; et al. Human kidney is a target for novel severe acute respiratory syndrome coronavirus 2 infection. Nat. Commun. 2021, 12, 2506. [Google Scholar] [CrossRef]
- Sharfuddin, A.A.; Molitoris, B.A. Pathophysiology of ischemic acute kidney injury. Nat. Rev. Nephrol. 2011, 7, 189–200. [Google Scholar] [CrossRef]
- Aranda-Rivera, A.K.; Cruz-Gregorio, A.; Aparicio-Trejo, O.E.; Pedraza-Chaverri, J. Mitochondrial Redox Signaling and Oxidative Stress in Kidney Diseases. Biomolecules 2021, 11, 1144. [Google Scholar] [CrossRef] [PubMed]
- Guo, L.P.; Liu, S.X.; Yang, Q.; Liu, H.Y.; Xu, L.L.; Hao, Y.H.; Zhang, X.Q. Effect of Thymoquinone on Acute Kidney Injury Induced by Sepsis in BALB/c Mice. Biomed. Res. Int. 2020, 2020, 1594726. [Google Scholar] [CrossRef]
- Dong, W.; Mead, H.; Tian, L.; Park, J.G.; Garcia, J.I.; Jaramillo, S.; Barr, T.; Kollath, D.S.; Coyne, V.K.; Stone, N.E.; et al. The K18-Human ACE2 Transgenic Mouse Model Recapitulates Non-severe and Severe COVID-19 in Response to an Infectious Dose of the SARS-CoV-2 Virus. J. Virol. 2022, 96, e0096421. [Google Scholar] [CrossRef] [PubMed]
- Meinhardt, J.; Radke, J.; Dittmayer, C.; Franz, J.; Thomas, C.; Mothes, R.; Laue, M.; Schneider, J.; Brunink, S.; Greuel, S.; et al. Olfactory transmucosal SARS-CoV-2 invasion as a port of central nervous system entry in individuals with COVID-19. Nat. Neurosci. 2021, 24, 168–175. [Google Scholar] [CrossRef] [PubMed]
- Spudich, S.; Nath, A. Nervous system consequences of COVID-19. Science 2022, 375, 267–269. [Google Scholar] [CrossRef]
- Crunfli, F.; Carregari, V.C.; Veras, F.P.; Silva, L.S.; Nogueira, M.H.; Antunes, A.; Vendramini, P.H.; Valenca, A.G.F.; Brandao-Teles, C.; Zuccoli, G.D.S.; et al. Morphological, cellular, and molecular basis of brain infection in COVID-19 patients. Proc. Natl. Acad. Sci. USA 2022, 119, e2200960119. [Google Scholar] [CrossRef]
- Dedoni, S.; Avdoshina, V.; Camoglio, C.; Siddi, C.; Fratta, W.; Scherma, M.; Fadda, P. K18- and CAG-hACE2 Transgenic Mouse Models and SARS-CoV-2: Implications for Neurodegeneration Research. Molecules 2022, 27, 4142. [Google Scholar] [CrossRef]
- Xu, E.; Xie, Y.; Al-Aly, Z. Long-term neurologic outcomes of COVID-19. Nat. Med. 2022, 28, 2406–2415. [Google Scholar] [CrossRef]
- Marshall, M. How COVID-19 can damage the brain. Nature 2020, 585, 342–343. [Google Scholar] [CrossRef]
- Shariq, M.; Malik, A.A.; Sheikh, J.A.; Hasnain, S.E.; Ehtesham, N.Z. Regulation of autophagy by SARS-CoV-2: The multifunctional contributions of ORF3a. J. Med. Virol. 2023, 95, e28959. [Google Scholar] [CrossRef]
- Treeza, M.M.; Augustine, S.; Mathew, A.A.; Kanthlal, S.K.; Panonummal, R. Targeting Viral ORF3a Protein: A New Approach to Mitigate COVID-19 Induced Immune Cell Apoptosis and Associated Respiratory Complications. Adv. Pharm. Bull. 2023, 13, 678–687. [Google Scholar] [CrossRef] [PubMed]
- Bakhshi, S.; Shamsi, S. MCC950 in the treatment of NLRP3-mediated inflammatory diseases: Latest evidence and therapeutic outcomes. Int. Immunopharmacol. 2022, 106, 108595. [Google Scholar] [CrossRef] [PubMed]
- Ma, J.; Chen, X.; Xue, R.; Wang, F.; Dong, J.; Tao, N.; Qin, Z. Cinnamaldehyde inhibits cytokine storms induced by the ORF3a protein of SARS-CoV-2 via ROS-elimination in activated T cells. Phytother. Res. 2023, 37, 6006–6020. [Google Scholar] [CrossRef] [PubMed]
- He, Y.; Chang, Y.; Peng, Y.; Zhu, J.; Liu, K.; Chen, J.; Wu, Y.; Ji, Z.; Lin, Z.; Wang, S.; et al. Glibenclamide Directly Prevents Neuroinflammation by Targeting SUR1-TRPM4-Mediated NLRP3 Inflammasome Activation in Microglia. Mol. Neurobiol. 2022, 59, 6590–6607. [Google Scholar] [CrossRef]
- Laplantine, E.; Chable-Bessia, C.; Oudin, A.; Swain, J.; Soria, A.; Merida, P.; Gourdelier, M.; Mestiri, S.; Besseghe, I.; Bremaud, E.; et al. The FDA-approved drug Auranofin has a dual inhibitory effect on SARS-CoV-2 entry and NF-kappaB signaling. iScience 2022, 25, 105066. [Google Scholar] [CrossRef]
- Su, C.M.; Wang, L.; Yoo, D. Activation of NF-kappaB and induction of proinflammatory cytokine expressions mediated by ORF7a protein of SARS-CoV-2. Sci. Rep. 2021, 11, 13464. [Google Scholar] [CrossRef]
- Sayah, W.; Berkane, I.; Guermache, I.; Sabri, M.; Lakhal, F.Z.; Yasmine Rahali, S.; Djidjeli, A.; Lamara Mahammed, L.; Merah, F.; Belaid, B.; et al. Interleukin-6, procalcitonin and neutrophil-to-lymphocyte ratio: Potential immune-inflammatory parameters to identify severe and fatal forms of COVID-19. Cytokine 2021, 141, 155428. [Google Scholar] [CrossRef]
- Del Valle, D.M.; Kim-Schulze, S.; Huang, H.H.; Beckmann, N.D.; Nirenberg, S.; Wang, B.; Lavin, Y.; Swartz, T.H.; Madduri, D.; Stock, A.; et al. An inflammatory cytokine signature predicts COVID-19 severity and survival. Nat. Med. 2020, 26, 1636–1643. [Google Scholar] [CrossRef]
- Izadi, Z.; Brenner, E.J.; Mahil, S.K.; Dand, N.; Yiu, Z.Z.N.; Yates, M.; Ungaro, R.C.; Zhang, X.; Agrawal, M.; Colombel, J.F.; et al. Association Between Tumor Necrosis Factor Inhibitors and the Risk of Hospitalization or Death Among Patients with Immune-Mediated Inflammatory Disease and COVID-19. JAMA Netw. Open 2021, 4, e2129639. [Google Scholar] [CrossRef]
- Lonze, B.E.; Spiegler, P.; Wesson, R.N.; Alachkar, N.; Petkova, E.; Weldon, E.P.; Dieter, R.A.; Li, Y.; Quinn, M.; Mattoo, A.; et al. A Randomized Double-Blinded Placebo Controlled Trial of Clazakizumab for the Treatment of COVID-19 Pneumonia with Hyperinflammation. Crit. Care Med. 2022, 50, 1348–1359. [Google Scholar] [CrossRef]
- Zhang, T.; Wu, K.Y.; Ma, N.; Wei, L.L.; Garstka, M.; Zhou, W.; Li, K. The C5a/C5aR2 axis promotes renal inflammation and tissue damage. JCI Insight 2020, 5, e134081. [Google Scholar] [CrossRef] [PubMed]
- Tan, S.W.; Zhao, Y.; Li, P.; Ning, Y.L.; Huang, Z.Z.; Yang, N.; Liu, D.; Zhou, Y.G. HMGB1 mediates cognitive impairment caused by the NLRP3 inflammasome in the late stage of traumatic brain injury. J. Neuroinflamm. 2021, 18, 241. [Google Scholar] [CrossRef] [PubMed]
- Lebedeva, N.S.; Gubarev, Y.A.; Mamardashvili, G.M.; Zaitceva, S.V.; Zdanovich, S.A.; Malyasova, A.S.; Romanenko, J.V.; Koifman, M.O.; Koifman, O.I. Theoretical and experimental study of interaction of macroheterocyclic compounds with ORF3a of SARS-CoV-2. Sci. Rep. 2021, 11, 19481. [Google Scholar] [CrossRef] [PubMed]
- Arora, A.; Maiti, S. Differential biophysical behavior of human telomeric RNA and DNA quadruplex. J. Phys. Chem. B 2009, 113, 10515–10520. [Google Scholar] [CrossRef] [PubMed]
- Qin, G.; Zhao, C.; Liu, Y.; Zhang, C.; Yang, G.; Yang, J.; Wang, Z.; Wang, C.; Tu, C.; Guo, Z.; et al. RNA G-quadruplex formed in SARS-CoV-2 used for COVID-19 treatment in animal models. Cell Discov. 2022, 8, 86. [Google Scholar] [CrossRef]
- Zhang, J.T.; Nasr, M.; Zhao, R.Y. Unpublished data. 2024. [Google Scholar]
- Benko, Z.; Elder, R.T.; Liang, D.; Zhao, R.R. Fission yeast as a HTS platform for molecular probes of HIV-1 Vpr-induced cell death. Int. J. High Throughput Screen. 2010, 1, 151–162. [Google Scholar]
- Benko, Z.; Zhang, J.; Zhao, R.Y. Development of A Fission Yeast Cell-Based Platform for High Throughput Screening of HIV-1 Protease Inhibitors. Curr. HIV Res. 2019, 17, 429–440. [Google Scholar] [CrossRef]
- Benko, Z.; Elder, R.T.; Li, G.; Liang, D.; Zhao, R.Y. HIV-1 Protease in the Fission Yeast Schizosaccharomyces pombe. PLoS ONE 2016, 11, e0151286. [Google Scholar] [CrossRef]
- Benko, Z.; Liang, D.; Li, G.; Elder, R.T.; Sarkar, A.; Takayama, J.; Ghosh, A.K.; Zhao, R.Y. A fission yeast cell-based system for multidrug resistant HIV-1 proteases. Cell Biosci. 2017, 7, 5. [Google Scholar] [CrossRef]
- Yang, H.; Nkeze, J.; Zhao, R.Y. Effects of HIV-1 protease on cellular functions and their potential applications in antiretroviral therapy. Cell Biosci. 2012, 2, 32. [Google Scholar] [CrossRef]
- Zhang, J.; Vernon, K.; Li, Q.; Benko, Z.; Amoroso, A.; Nasr, M.; Zhao, R.Y. Single-Agent and Fixed-Dose Combination HIV-1 Protease Inhibitor Drugs in Fission Yeast (Schizosaccharomyces pombe). Pathogens 2021, 10, 804. [Google Scholar] [CrossRef]
- Zhang, J.; Li, Q.; Kawashima, S.A.; Nasr, M.; Xue, F.; Zhao, R.Y. Improving Drug Sensitivity of HIV-1 Protease Inhibitors by Restriction of Cellular Efflux System in a Fission Yeast Model. Pathogens 2022, 11, 804. [Google Scholar] [CrossRef] [PubMed]
- Zhao, Y.; Elder, R.T. Yeast perspectives on HIV-1 Vpr. Front. Biosci. 2000, 5, 905–916. [Google Scholar] [CrossRef] [PubMed]
- Andreola, M.L.; Litvak, S. Yeast and the AIDS virus: The odd couple. J. Biomed. Biotechnol. 2012, 2012, 549020. [Google Scholar] [CrossRef] [PubMed]
- Ren, Y.; Shu, T.; Wu, D.; Mu, J.; Wang, C.; Huang, M.; Han, Y.; Zhang, X.Y.; Zhou, W.; Qiu, Y.; et al. The ORF3a protein of SARS-CoV-2 induces apoptosis in cells. Cell Mol. Immunol. 2020, 17, 881–883. [Google Scholar] [CrossRef]
- Donia, A.; Bokhari, H. Apoptosis induced by SARS-CoV-2: Can we target it? Apoptosis 2021, 26, 7–8. [Google Scholar] [CrossRef]
- Zhang, Y.; Sun, H.; Pei, R.; Mao, B.; Zhao, Z.; Li, H.; Lin, Y.; Lu, K. The SARS-CoV-2 protein ORF3a inhibits fusion of autophagosomes with lysosomes. Cell Discov. 2021, 7, 31. [Google Scholar] [CrossRef]
- Chu, H.; Chan, J.F.; Yuen, T.T.; Shuai, H.; Yuan, S.; Wang, Y.; Hu, B.; Yip, C.C.; Tsang, J.O.; Huang, X.; et al. Comparative tropism, replication kinetics, and cell damage profiling of SARS-CoV-2 and SARS-CoV with implications for clinical manifestations, transmissibility, and laboratory studies of COVID-19: An observational study. Lancet Microbe 2020, 1, e14–e23. [Google Scholar] [CrossRef]
- Majumdar, P.; Niyogi, S. ORF3a mutation associated with higher mortality rate in SARS-CoV-2 infection. Epidemiol. Infect. 2020, 148, e262. [Google Scholar] [CrossRef]
- Nagy, A.; Pongor, S.; Gyorffy, B. Different mutations in SARS-CoV-2 associate with severe and mild outcome. Int. J. Antimicrob. Agents 2021, 57, 106272. [Google Scholar] [CrossRef]
- Tzou, P.L.; Tao, K.; Nouhin, J.; Rhee, S.Y.; Hu, B.D.; Pai, S.; Parkin, N.; Shafer, R.W. Coronavirus Antiviral Research Database (CoV-RDB): An Online Database Designed to Facilitate Comparisons between Candidate Anti-Coronavirus Compounds. Viruses 2020, 12, 1006. [Google Scholar] [CrossRef] [PubMed]
- Tzou, P.L.; Tao, K.; Pond, S.L.K.; Shafer, R.W. Coronavirus Resistance Database (CoV-RDB): SARS-CoV-2 susceptibility to monoclonal antibodies, convalescent plasma, and plasma from vaccinated persons. PLoS ONE 2022, 17, e0261045. [Google Scholar] [CrossRef] [PubMed]
- Lednicky, J.A.; Cherabuddi, K.; Tagliamonte, M.S.; Elbadry, M.A.; Subramaniam, K.; Waltzek, T.B.; Morris, J.G., Jr. In-Frame 12-Nucleotide Deletion within Open Reading Frame 3a in a SARS-CoV-2 Strain Isolated from a Patient Hospitalized with COVID-19. Microbiol. Resour. Announc. 2021, 10, 10–1128. [Google Scholar] [CrossRef] [PubMed]
- Cheng, L.; Zhang, X.; Chen, Y.; Wang, D.; Zhang, D.; Yan, S.; Wang, H.; Xiao, M.; Liang, T.; Li, H.; et al. Dynamic landscape mapping of humoral immunity to SARS-CoV-2 identifies non-structural protein antibodies associated with the survival of critical COVID-19 patients. Signal Transduct. Target. Ther. 2021, 6, 304. [Google Scholar] [CrossRef] [PubMed]
- de Silva, T.I.; Liu, G.; Lindsey, B.B.; Dong, D.; Moore, S.C.; Hsu, N.S.; Shah, D.; Wellington, D.; Mentzer, A.J.; Angyal, A.; et al. The impact of viral mutations on recognition by SARS-CoV-2 specific T-cells. iScience 2021, 24, 103353. [Google Scholar] [CrossRef]
- COVID is here to stay: Countries must decide how to adapt. Nature 2022, 601, 165. [CrossRef]
- Xie, R.; Edwards, K.M.; Adam, D.C.; Leung, K.S.M.; Tsang, T.K.; Gurung, S.; Xiong, W.; Wei, X.; Ng, D.Y.M.; Liu, G.Y.Z.; et al. Resurgence of Omicron BA.2 in SARS-CoV-2 infection-naive Hong Kong. Nat. Commun. 2023, 14, 2422. [Google Scholar] [CrossRef]
Disclaimer/Publisher’s Note: The statements, opinions and data contained in all publications are solely those of the individual author(s) and contributor(s) and not of MDPI and/or the editor(s). MDPI and/or the editor(s) disclaim responsibility for any injury to people or property resulting from any ideas, methods, instructions or products referred to in the content. |
© 2024 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, J.; Hom, K.; Zhang, C.; Nasr, M.; Gerzanich, V.; Zhang, Y.; Tang, Q.; Xue, F.; Simard, J.M.; Zhao, R.Y. SARS-CoV-2 ORF3a Protein as a Therapeutic Target against COVID-19 and Long-Term Post-Infection Effects. Pathogens 2024, 13, 75. https://doi.org/10.3390/pathogens13010075
Zhang J, Hom K, Zhang C, Nasr M, Gerzanich V, Zhang Y, Tang Q, Xue F, Simard JM, Zhao RY. SARS-CoV-2 ORF3a Protein as a Therapeutic Target against COVID-19 and Long-Term Post-Infection Effects. Pathogens. 2024; 13(1):75. https://doi.org/10.3390/pathogens13010075
Chicago/Turabian StyleZhang, Jiantao, Kellie Hom, Chenyu Zhang, Mohamed Nasr, Volodymyr Gerzanich, Yanjin Zhang, Qiyi Tang, Fengtian Xue, J. Marc Simard, and Richard Y. Zhao. 2024. "SARS-CoV-2 ORF3a Protein as a Therapeutic Target against COVID-19 and Long-Term Post-Infection Effects" Pathogens 13, no. 1: 75. https://doi.org/10.3390/pathogens13010075
APA StyleZhang, J., Hom, K., Zhang, C., Nasr, M., Gerzanich, V., Zhang, Y., Tang, Q., Xue, F., Simard, J. M., & Zhao, R. Y. (2024). SARS-CoV-2 ORF3a Protein as a Therapeutic Target against COVID-19 and Long-Term Post-Infection Effects. Pathogens, 13(1), 75. https://doi.org/10.3390/pathogens13010075