Epstein–Barr Virus—Oral Bacterial Link in the Development of Oral Squamous Cell Carcinoma


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Epstein–Barr Virus in Oral Cancer
2.1. Epstein–Barr Virus as a Risk Factor
2.2. Epstein–Barr Virus Replication Cycle in Epithelial Cells
2.3. Epstein–Barr Virus and the Activation of Mechanisms Associated with Oral Cancer
3. Oral Bacterial in the Development of Cancer
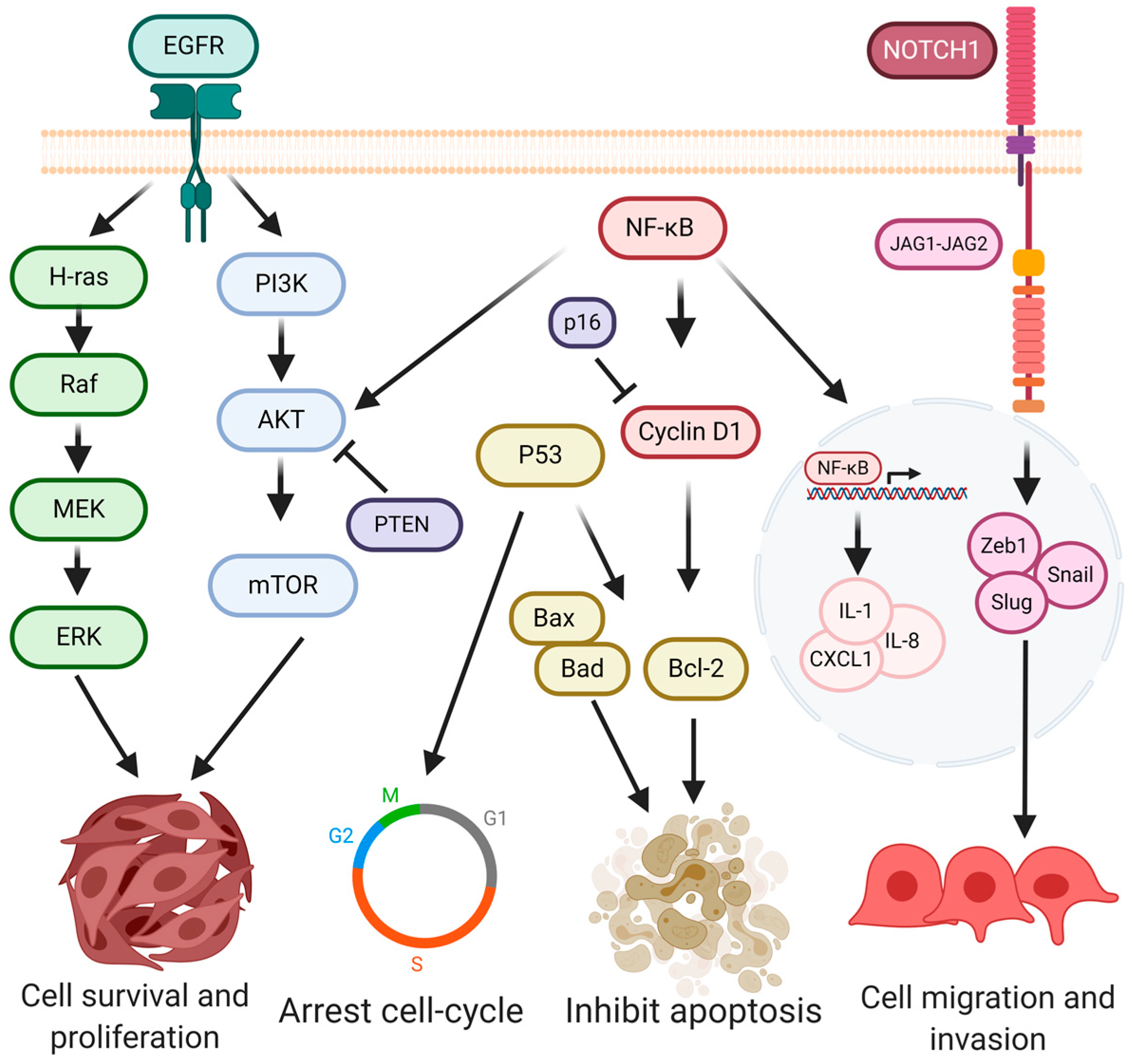
4. Signaling Pathways in Oral Cancer
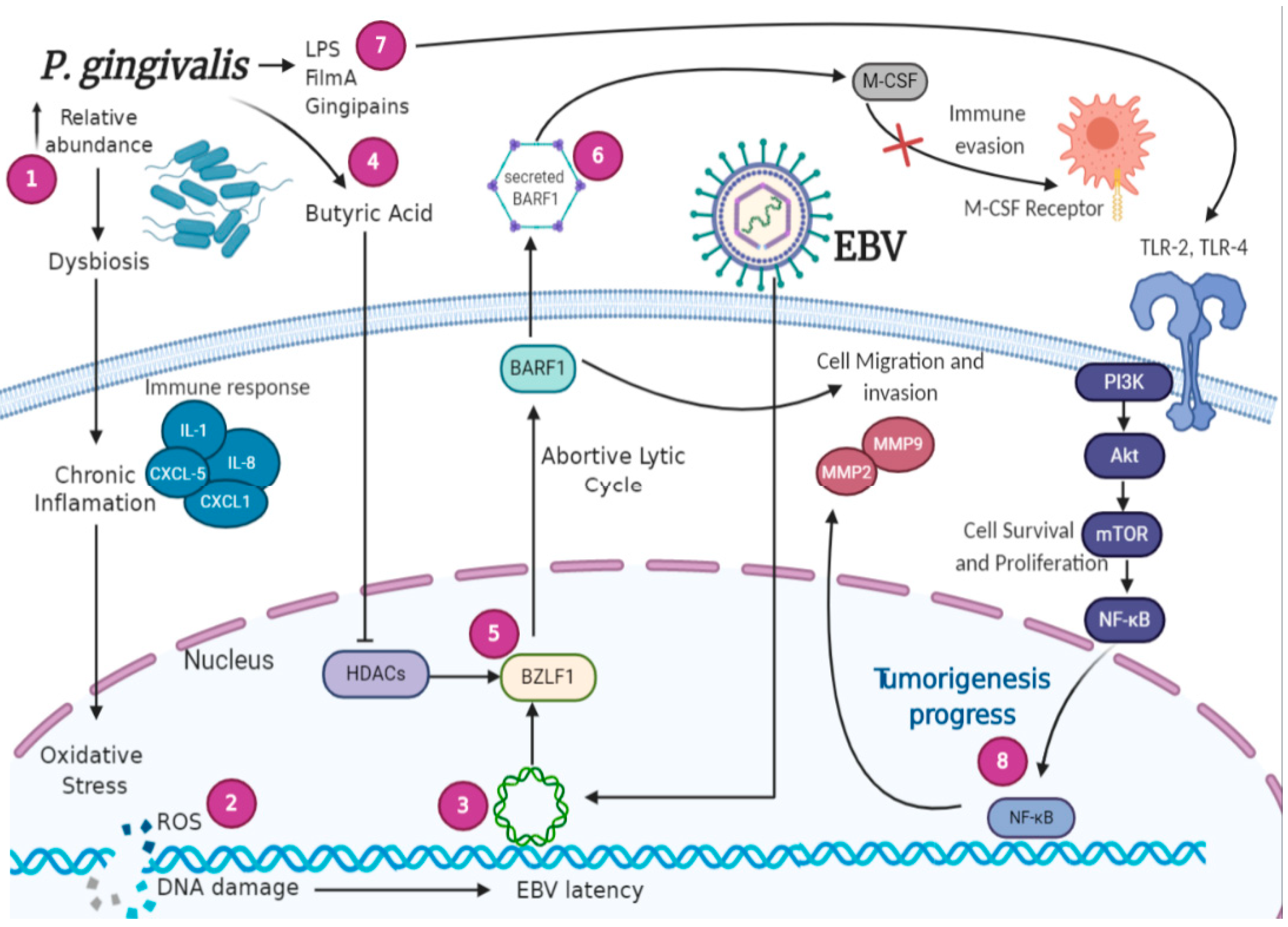
5. Signaling Pathways Involved in EBV-Bacteria Interactions in the Oral Cavity
6. Conclusions and Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Wong, T.; Wiesenfeld, D. Oral Cancer. Aust. Dent. J. 2018, 63, S91–S99. [Google Scholar] [CrossRef] [PubMed]
- Dhanuthai, K.; Rojanawatsirivej, S.; Thosaporn, W.; Kintarak, S.; Subarnbhesaj, A.; Darling, M.; Kryshtalskyj, E.; Chiang, C.P.; Shin, H.I.; Choi, S.Y.; et al. Oral cancer: A multicenter study. Med. Oral Patol. Oral Cir. Bucal 2018, 23, e23–e29. [Google Scholar] [CrossRef] [PubMed]
- Ferlay, J.; Colombet, M.; Soerjomataram, I.; Mathers, C.; Parkin, D.M.; Piñeros, M.; Znaor, A.; Bray, F. Estimating the global cancer incidence and mortality in 2018: GLOBOCAN sources and methods. Int. J. Cancer 2019, 144, 1941–1953. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bray, F.; Ferlay, J.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- de Camargo, C.M.; Voti, L.; Guerra-Yi, M.; Chapuis, F.; Mazuir, M.; Curado, M.P. Oral cavity cancer in developed and in developing countries: Population-based incidence. Head Neck 2010, 32, 357–367. [Google Scholar] [CrossRef] [PubMed]
- Shield, K.D.; Ferlay, J.; Jemal, A.; Sankaranarayanan, R.; Chaturvedi, A.K.; Bray, F.; Soerjomataram, I. The global incidence of lip, oral cavity, and pharyngeal cancers by subsite in 2012. CA Cancer J. Clin. 2017, 67, 51–64. [Google Scholar] [CrossRef]
- Herrera-Serna, B.Y.; Lara-Carrillo, E.; Toral-Rizo, V.H.; do Amaral, R.C.; Aguilera-Eguía, R.A. Relationship between the Human Development Index and its Components with Oral Cancer in Latin America. J. Epidemiol. Glob. Health 2019, 9, 223–232. [Google Scholar] [CrossRef]
- Perdomo, S.; Roa, G.M.; Brennan, P.; Forman, D.; Sierra, M.S. Head and neck cancer burden and preventive measures in Central and South America. Cancer Epidemiol. 2016, 44, S43–S52. [Google Scholar] [CrossRef] [Green Version]
- Mello, F.W.; Miguel, A.F.P.; Dutra, K.L.; Porporatti, A.L.; Warnakulasuriya, S.; Guerra, E.N.S.; Rivero, E.R.C. Prevalence of oral potentially malignant disorders: A systematic review and meta-analysis. J. Oral Pathol. Med. 2018, 47, 633–640. [Google Scholar] [CrossRef]
- Conway, D.I.; Purkayastha, M.; Chestnutt, I.G. The changing epidemiology of oral cancer: Definitions, trends, and risk factors. Br. Dent. J. 2018, 225, 867–873. [Google Scholar] [CrossRef] [Green Version]
- Jehn, P.; Dittmann, J.; Zimmerer, R.; Stier, R.; Jehn, M.; Gellrich, N.C.; Tavassol, F.; Spalthoff, S. Survival Rates According to Tumour Location in Patients With Surgically Treated Oral and Oropharyngeal Squamous Cell Carcinoma. Anticancer Res. 2019, 39, 2527–2533. [Google Scholar] [CrossRef] [PubMed]
- Listl, S.; Jansen, L.; Stenzinger, A.; Freier, K.; Emrich, K.; Holleczek, B.; Katalinic, A.; Gondos, A.; Brenner, H.; Group, G.C.S.W. Survival of patients with oral cavity cancer in Germany. PLoS ONE 2013, 8, e53415. [Google Scholar] [CrossRef] [PubMed]
- Warnakulasuriya, S.; Johnson, N.W.; van der Waal, I. Nomenclature and classification of potentially malignant disorders of the oral mucosa. J. Oral Pathol. Med. 2007, 36, 575–580. [Google Scholar] [CrossRef] [PubMed]
- Hashibe, M.; Hunt, J.; Wei, M.; Buys, S.; Gren, L.; Lee, Y.C. Tobacco, alcohol, body mass index, physical activity, and the risk of head and neck cancer in the prostate, lung, colorectal, and ovarian (PLCO) cohort. Head Neck 2013, 35, 914–922. [Google Scholar] [CrossRef]
- Castellsagué, X.; Quintana, M.J.; Martínez, M.C.; Nieto, A.; Sánchez, M.J.; Juan, A.; Monner, A.; Carrera, M.; Agudo, A.; Quer, M.; et al. The role of type of tobacco and type of alcoholic beverage in oral carcinogenesis. Int. J. Cancer 2004, 108, 741–749. [Google Scholar] [CrossRef]
- Hashibe, M.; Brennan, P.; Benhamou, S.; Castellsague, X.; Chen, C.; Curado, M.P.; Dal Maso, L.; Daudt, A.W.; Fabianova, E.; Fernandez, L.; et al. Alcohol drinking in never users of tobacco, cigarette smoking in never drinkers, and the risk of head and neck cancer: Pooled analysis in the International Head and Neck Cancer Epidemiology Consortium. J. Natl. Cancer Inst. 2007, 99, 777–789. [Google Scholar] [CrossRef]
- Garrote, L.F.; Herrero, R.; Reyes, R.M.; Vaccarella, S.; Anta, J.L.; Ferbeye, L.; Muñoz, N.; Franceschi, S. Risk factors for cancer of the oral cavity and oro-pharynx in Cuba. Br. J. Cancer 2001, 85, 46–54. [Google Scholar] [CrossRef] [Green Version]
- Perry, B.J.; Zammit, A.P.; Lewandowski, A.W.; Bashford, J.J.; Dragovic, A.S.; Perry, E.J.; Hayatbakhsh, R.; Perry, C.F. Sites of origin of oral cavity cancer in nonsmokers vs smokers: Possible evidence of dental trauma carcinogenesis and its importance compared with human papillomavirus. JAMA Otolaryngol. Head Neck Surg. 2015, 141, 5–11. [Google Scholar] [CrossRef] [Green Version]
- Bravi, F.; Bosetti, C.; Filomeno, M.; Levi, F.; Garavello, W.; Galimberti, S.; Negri, E.; La Vecchia, C. Foods, nutrients and the risk of oral and pharyngeal cancer. Br. J. Cancer 2013, 109, 2904–2910. [Google Scholar] [CrossRef]
- Gupta, B.; Bray, F.; Kumar, N.; Johnson, N.W. Associations between oral hygiene habits, diet, tobacco and alcohol and risk of oral cancer: A case-control study from India. Cancer Epidemiol. 2017, 51, 7–14. [Google Scholar] [CrossRef]
- Kruse, A.L.; Bredell, M.; Grätz, K.W. Oral squamous cell carcinoma in non-smoking and non-drinking patients. Head Neck Oncol. 2010, 2, 24. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dahlstrom, K.R.; Little, J.A.; Zafereo, M.E.; Lung, M.; Wei, Q.; Sturgis, E.M. Squamous cell carcinoma of the head and neck in never smoker-never drinkers: A descriptive epidemiologic study. Head Neck 2008, 30, 75–84. [Google Scholar] [CrossRef] [PubMed]
- Foy, J.P.; Bertolus, C.; Boutolleau, D.; Agut, H.; Gessain, A.; Herceg, Z.; Saintigny, P. Arguments to Support a Viral Origin of Oral Squamous Cell Carcinoma in Non-Smoker and Non-Drinker Patients. Front. Oncol. 2020, 10, 822. [Google Scholar] [CrossRef] [PubMed]
- Bebek, G.; Bennett, K.L.; Funchain, P.; Campbell, R.; Seth, R.; Scharpf, J.; Burkey, B.; Eng, C. Microbiomic subprofiles and MDR1 promoter methylation in head and neck squamous cell carcinoma. Hum. Mol. Genet. 2012, 21, 1557–1565. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Termine, N.; Panzarella, V.; Falaschini, S.; Russo, A.; Matranga, D.; Lo Muzio, L.; Campisi, G. HPV in oral squamous cell carcinoma vs head and neck squamous cell carcinoma biopsies: A meta-analysis (1988–2007). Ann. Oncol. 2008, 19, 1681–1690. [Google Scholar] [CrossRef] [PubMed]
- Haeggblom, L.; Ramqvist, T.; Tommasino, M.; Dalianis, T.; Näsman, A. Time to change perspectives on HPV in oropharyngeal cancer. A systematic review of HPV prevalence per oropharyngeal sub-site the last 3 years. Papillomavirus Res. 2017, 4, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Ndiaye, C.; Mena, M.; Alemany, L.; Arbyn, M.; Castellsagué, X.; Laporte, L.; Bosch, F.X.; de Sanjosé, S.; Trottier, H. HPV DNA, E6/E7 mRNA, and p16INK4a detection in head and neck cancers: A systematic review and meta-analysis. Lancet Oncol. 2014, 15, 1319–1331. [Google Scholar] [CrossRef]
- She, Y.; Nong, X.; Zhang, M.; Wang, M. Epstein-Barr virus infection and oral squamous cell carcinoma risk: A meta-analysis. PLoS ONE 2017, 12, e0186860. [Google Scholar] [CrossRef] [Green Version]
- de Lima, M.A.P.; Teodoro, I.P.P.; Galiza, L.E.; Filho, P.H.B.M.; Marques, F.M.; Junior, R.F.F.P.; Macedo, G.E.C.; Facundo, H.T.; da Silva, C.G.L.; Lima, M.V.A. Association between Epstein-Barr Virus and Oral Carcinoma: A Systematic Review with Meta-Analysis. Crit. Rev. Oncog. 2019, 24, 349–368. [Google Scholar] [CrossRef]
- Gao, Z.; Lv, J.; Wang, M. Epstein-Barr virus is associated with periodontal diseases: A meta-analysis based on 21 case-control studies. Medicine 2017, 96, e5980. [Google Scholar] [CrossRef]
- Farid, E.; Al-Biltagi, M. Trend and seroprevalence of Epstein-Barr virus in Bahrain: 2001–2015. East Mediterr Health J. 2018, 23, 821–829. [Google Scholar] [CrossRef] [PubMed]
- Dowd, J.B.; Palermo, T.; Brite, J.; McDade, T.W.; Aiello, A. Seroprevalence of Epstein-Barr virus infection in U.S. children ages 6–19, 2003–2010. PLoS ONE 2013, 8, e64921. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kuri, A.; Jacobs, B.M.; Vickaryous, N.; Pakpoor, J.; Middeldorp, J.; Giovannoni, G.; Dobson, R. Epidemiology of Epstein-Barr virus infection and infectious mononucleosis in the United Kingdom. BMC Public Health 2020, 20, 912. [Google Scholar] [CrossRef] [PubMed]
- IARC. Proceedings of the IARC Working Group on the Evaluation of Carcinogenic Risks to Humans. Epstein-Barr Virus and Kaposi’s Sarcoma Herpesvirus/Human Herpesvirus 8, Lyon, France, 17–24 June 1997. IARC Monogr. Eval. Carcinog. Risks Hum. 1997, 70, 1–492. [Google Scholar]
- Weiss, L.M.; Chen, Y.Y.; Liu, X.F.; Shibata, D. Epstein-Barr virus and Hodgkin’s disease. A correlative in situ hybridization and polymerase chain reaction study. Am. J. Pathol. 1991, 139, 1259–1265. [Google Scholar] [PubMed]
- Suzuki, R.; Yamaguchi, M.; Izutsu, K.; Yamamoto, G.; Takada, K.; Harabuchi, Y.; Isobe, Y.; Gomyo, H.; Koike, T.; Okamoto, M.; et al. Prospective measurement of Epstein-Barr virus-DNA in plasma and peripheral blood mononuclear cells of extranodal NK/T-cell lymphoma, nasal type. Blood 2011, 118, 6018–6022. [Google Scholar] [CrossRef]
- Kim, W.Y.; Montes-Mojarro, I.A.; Fend, F.; Quintanilla-Martinez, L. Epstein-Barr Virus-Associated T and NK-Cell Lymphoproliferative Diseases. Front. Pediatr. 2019, 7, 71. [Google Scholar] [CrossRef] [Green Version]
- Taylor, G.S.; Long, H.M.; Brooks, J.M.; Rickinson, A.B.; Hislop, A.D. The immunology of Epstein-Barr virus-induced disease. Annu. Rev. Immunol. 2015, 33, 787–821. [Google Scholar] [CrossRef]
- Qiao, Y.W.; Zhao, X.Q.; Liu, J.; Yang, W.J. Clinicopathological features of Epstein-Barr virus-associated gastric carcinoma: A systematic review and meta-analysis. J. BUON 2019, 24, 1092–1099. [Google Scholar] [PubMed]
- Heawchaiyaphum, C.; Iizasa, H.; Ekalaksananan, T.; Burassakarn, A.; Kiyono, T.; Kanehiro, Y.; Yoshiyama, H.; Pientong, C. Epstein-Barr Virus Infection of Oral Squamous Cells. Microorganisms 2020, 8, 419. [Google Scholar] [CrossRef] [Green Version]
- Sand, L.P.; Jalouli, J.; Larsson, P.A.; Hirsch, J.M. Prevalence of Epstein-Barr virus in oral squamous cell carcinoma, oral lichen planus, and normal oral mucosa. Oral Surg. Oral Med. Oral Pathol. Oral Radiol. Endod. 2002, 93, 586–592. [Google Scholar] [CrossRef] [PubMed]
- Acharya, S.; Ekalaksananan, T.; Vatanasapt, P.; Loyha, K.; Phusingha, P.; Promthet, S.; Kongyingyoes, B.; Pientong, C. Association of Epstein-Barr virus infection with oral squamous cell carcinoma in a case-control study. J. Oral Pathol. Med. 2015, 44, 252–257. [Google Scholar] [CrossRef] [PubMed]
- Yen, C.Y.; Lu, M.C.; Tzeng, C.C.; Huang, J.Y.; Chang, H.W.; Chen, R.S.; Liu, S.Y.; Liu, S.T.; Shieh, B.; Li, C. Detection of EBV infection and gene expression in oral cancer from patients in Taiwan by microarray analysis. J. Biomed. Biotechnol. 2009, 2009, 904589. [Google Scholar] [CrossRef] [PubMed]
- Guidry, J.T.; Birdwell, C.E.; Scott, R.S. Epstein-Barr virus in the pathogenesis of oral cancers. Oral Dis. 2018, 24, 497–508. [Google Scholar] [CrossRef] [PubMed]
- Chesnokova, L.S.; Hutt-Fletcher, L.M. Fusion of Epstein-Barr virus with epithelial cells can be triggered by αvβ5 in addition to αvβ6 and αvβ8, and integrin binding triggers a conformational change in glycoproteins gHgL. J. Virol. 2011, 85, 13214–13223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, H.; Li, Y.; Wang, H.B.; Zhang, A.; Chen, M.L.; Fang, Z.X.; Dong, X.D.; Li, S.B.; Du, Y.; Xiong, D.; et al. Ephrin receptor A2 is an epithelial cell receptor for Epstein-Barr virus entry. Nat. Microbiol. 2018, 3, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Tsai, K.; Thikmyanova, N.; Wojcechowskyj, J.A.; Delecluse, H.J.; Lieberman, P.M. EBV tegument protein BNRF1 disrupts DAXX-ATRX to activate viral early gene transcription. PLoS Pathog. 2011, 7, e1002376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feederle, R.; Kost, M.; Baumann, M.; Janz, A.; Drouet, E.; Hammerschmidt, W.; Delecluse, H.J. The Epstein-Barr virus lytic program is controlled by the co-operative functions of two transactivators. EMBO J. 2000, 19, 3080–3089. [Google Scholar] [CrossRef] [Green Version]
- Murata, T. Encyclopedia of EBV-Encoded Lytic Genes: An Update. Adv. Exp. Med. Biol. 2018, 1045, 395–412. [Google Scholar] [CrossRef]
- Tonoyan, L.; Vincent-Bugnas, S.; Olivieri, C.V.; Doglio, A. New Viral Facets in Oral Diseases: The EBV Paradox. Int. J. Mol. Sci. 2019, 20, 5861. [Google Scholar] [CrossRef] [Green Version]
- Hatton, O.L.; Harris-Arnold, A.; Schaffert, S.; Krams, S.M.; Martinez, O.M. The interplay between Epstein-Barr virus and B lymphocytes: Implications for infection, immunity, and disease. Immunol. Res. 2014, 58, 268–276. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Eichelberg, M.R.; Welch, R.; Guidry, J.T.; Ali, A.; Ohashi, M.; Makielski, K.R.; McChesney, K.; Van Sciver, N.; Lambert, P.F.; Keleș, S.; et al. Epstein-Barr Virus Infection Promotes Epithelial Cell Growth by Attenuating Differentiation-Dependent Exit from the Cell Cycle. MBio 2019, 10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tsang, C.M.; Deng, W.; Yip, Y.L.; Zeng, M.S.; Lo, K.W.; Tsao, S.W. Epstein-Barr virus infection and persistence in nasopharyngeal epithelial cells. Chin. J. Cancer 2014, 33, 549–555. [Google Scholar] [CrossRef] [PubMed]
- Tsang, C.M.; Yip, Y.L.; Lo, K.W.; Deng, W.; To, K.F.; Hau, P.M.; Lau, V.M.; Takada, K.; Lui, V.W.; Lung, M.L.; et al. Cyclin D1 overexpression supports stable EBV infection in nasopharyngeal epithelial cells. Proc. Natl. Acad. Sci. USA 2012, 109, E3473–E3482. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pathmanathan, R.; Prasad, U.; Sadler, R.; Flynn, K.; Raab-Traub, N. Clonal proliferations of cells infected with Epstein-Barr virus in preinvasive lesions related to nasopharyngeal carcinoma. N. Engl. J. Med. 1995, 333, 693–698. [Google Scholar] [CrossRef] [PubMed]
- Tarbouriech, N.; Ruggiero, F.; de Turenne-Tessier, M.; Ooka, T.; Burmeister, W.P. Structure of the Epstein-Barr virus oncogene BARF1. J. Mol. Biol. 2006, 359, 667–678. [Google Scholar] [CrossRef] [PubMed]
- Seto, E.; Yang, L.; Middeldorp, J.; Sheen, T.S.; Chen, J.Y.; Fukayama, M.; Eizuru, Y.; Ooka, T.; Takada, K. Epstein-Barr virus (EBV)-encoded BARF1 gene is expressed in nasopharyngeal carcinoma and EBV-associated gastric carcinoma tissues in the absence of lytic gene expression. J. Med. Virol. 2005, 76, 82–88. [Google Scholar] [CrossRef]
- Decaussin, G.; Sbih-Lammali, F.; de Turenne-Tessier, M.; Bouguermouh, A.; Ooka, T. Expression of BARF1 gene encoded by Epstein-Barr virus in nasopharyngeal carcinoma biopsies. Cancer Res. 2000, 60, 5584–5588. [Google Scholar]
- zur Hausen, A.; Brink, A.A.; Craanen, M.E.; Middeldorp, J.M.; Meijer, C.J.; van den Brule, A.J. Unique transcription pattern of Epstein-Barr virus (EBV) in EBV-carrying gastric adenocarcinomas: Expression of the transforming BARF1 gene. Cancer Res. 2000, 60, 2745–2748. [Google Scholar]
- Hoebe, E.; Wille, C.; Hagemeier, S.; Kenney, S.; Greijer, A.; Middeldorp, J. Epstein-Barr Virus Gene BARF1 Expression is Regulated by the Epithelial Differentiation Factor ΔNp63α in Undifferentiated Nasopharyngeal Carcinoma. Cancers 2018, 10, 76. [Google Scholar] [CrossRef] [Green Version]
- Sheng, W.; Decaussin, G.; Sumner, S.; Ooka, T. N-terminal domain of BARF1 gene encoded by Epstein-Barr virus is essential for malignant transformation of rodent fibroblasts and activation of BCL-2. Oncogene 2001, 20, 1176–1185. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hoebe, E.K.; Le Large, T.Y.; Tarbouriech, N.; Oosterhoff, D.; De Gruijl, T.D.; Middeldorp, J.M.; Greijer, A.E. Epstein-Barr virus-encoded BARF1 protein is a decoy receptor for macrophage colony stimulating factor and interferes with macrophage differentiation and activation. Viral Immunol. 2012, 25, 461–470. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jiang, R.; Cabras, G.; Sheng, W.; Zeng, Y.; Ooka, T. Synergism of BARF1 with Ras induces malignant transformation in primary primate epithelial cells and human nasopharyngeal epithelial cells. Neoplasia 2009, 11, 964–973. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, Y.; Xu, M.; Zhang, X.; Chu, F.; Zhou, T. MAPK/c-Jun signaling pathway contributes to the upregulation of the anti-apoptotic proteins Bcl-2 and Bcl-xL induced by Epstein-Barr virus-encoded. Oncol. Lett. 2018, 15, 7537–7544. [Google Scholar] [CrossRef] [Green Version]
- Kim, D.H.; Chang, M.S.; Yoon, C.J.; Middeldorp, J.M.; Martinez, O.M.; Byeon, S.J.; Rha, S.Y.; Kim, S.H.; Kim, Y.S.; Woo, J.H. Epstein-Barr virus BARF1-induced NFκB/miR-146a/SMAD4 alterations in stomach cancer cells. Oncotarget 2016, 7, 82213–82227. [Google Scholar] [CrossRef]
- Danve, C.; Decaussin, G.; Busson, P.; Ooka, T. Growth transformation of primary epithelial cells with a NPC-derived Epstein-Barr virus strain. Virology 2001, 288, 223–235. [Google Scholar] [CrossRef] [Green Version]
- Kikuchi, K.; Taniguchi, A.; Yasumoto, S. Induction of the HPV16 enhancer activity by Jun-B and c-Fos through cooperation of the promoter-proximal AP-1 site and the epithelial cell type—Specific regulatory element in fibroblasts. Virus Genes 1996, 13, 45–52. [Google Scholar] [CrossRef]
- Rahman, R.; Poomsawat, S.; Juengsomjit, R.; Buajeeb, W. Overexpression of Epstein-Barr virus-encoded latent membrane protein-1 (LMP-1) in oral squamous cell carcinoma. BMC Oral Health 2019, 19, 142. [Google Scholar] [CrossRef]
- Dawson, C.W.; Laverick, L.; Morris, M.A.; Tramoutanis, G.; Young, L.S. Epstein-Barr virus-encoded LMP1 regulates epithelial cell motility and invasion via the ERK-MAPK pathway. J. Virol. 2008, 82, 3654–3664. [Google Scholar] [CrossRef] [Green Version]
- Paine, E.; Scheinman, R.I.; Baldwin, A.S.; Raab-Traub, N. Expression of LMP1 in epithelial cells leads to the activation of a select subset of NF-kappa B/Rel family proteins. J. Virol. 1995, 69, 4572–4576. [Google Scholar] [CrossRef] [Green Version]
- Shair, K.H.; Schnegg, C.I.; Raab-Traub, N. Epstein-Barr virus latent membrane protein-1 effects on junctional plakoglobin and induction of a cadherin switch. Cancer Res. 2009, 69, 5734–5742. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fåhraeus, R.; Rymo, L.; Rhim, J.S.; Klein, G. Morphological transformation of human keratinocytes expressing the LMP gene of Epstein-Barr virus. Nature 1990, 345, 447–449. [Google Scholar] [CrossRef] [PubMed]
- Shair, K.H.; Schnegg, C.I.; Raab-Traub, N. EBV latent membrane protein 1 effects on plakoglobin, cell growth, and migration. Cancer Res. 2008, 68, 6997–7005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mainou, B.A.; Everly, D.N.; Raab-Traub, N. Epstein-Barr virus latent membrane protein 1 CTAR1 mediates rodent and human fibroblast transformation through activation of PI3K. Oncogene 2005, 24, 6917–6924. [Google Scholar] [CrossRef] [Green Version]
- Chen, T.; Yu, W.H.; Izard, J.; Baranova, O.V.; Lakshmanan, A.; Dewhirst, F.E. The Human Oral Microbiome Database: A web accessible resource for investigating oral microbe taxonomic and genomic information. Database 2010, 2010, baq013. [Google Scholar] [CrossRef]
- Samaranayake, L.; Matsubara, V.H. Normal Oral Flora and the Oral Ecosystem. Dent. Clin. N. Am. 2017, 61, 199–215. [Google Scholar] [CrossRef]
- Hernández, M.; Dutzan, N.; García-Sesnich, J.; Abusleme, L.; Dezerega, A.; Silva, N.; González, F.E.; Vernal, R.; Sorsa, T.; Gamonal, J. Host-pathogen interactions in progressive chronic periodontitis. J. Dent. Res. 2011, 90, 1164–1170. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Lamont, R.J. Beyond the red complex and into more complexity: The polymicrobial synergy and dysbiosis (PSD) model of periodontal disease etiology. Mol. Oral Microbiol. 2012, 27, 409–419. [Google Scholar] [CrossRef] [Green Version]
- Kawada, M.; Yoshida, A.; Suzuki, N.; Nakano, Y.; Saito, T.; Oho, T.; Koga, T. Prevalence of Porphyromonas gingivalis in relation to periodontal status assessed by real-time PCR. Oral Microbiol. Immunol. 2004, 19, 289–292. [Google Scholar] [CrossRef]
- Kageyama, S.; Takeshita, T.; Asakawa, M.; Shibata, Y.; Takeuchi, K.; Yamanaka, W.; Yamashita, Y. Relative abundance of total subgingival plaque-specific bacteria in salivary microbiota reflects the overall periodontal condition in patients with periodontitis. PLoS ONE 2017, 12, e0174782. [Google Scholar] [CrossRef] [Green Version]
- Damgaard, C.; Danielsen, A.K.; Enevold, C.; Massarenti, L.; Nielsen, C.H.; Holmstrup, P.; Belstrøm, D. In saliva associates with chronic and aggressive periodontitis. J. Oral Microbiol. 2019, 11, 1653123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saito, A.; Inagaki, S.; Kimizuka, R.; Okuda, K.; Hosaka, Y.; Nakagawa, T.; Ishihara, K. Fusobacterium nucleatum enhances invasion of human gingival epithelial and aortic endothelial cells by Porphyromonas gingivalis. FEMS Immunol. Med. Microbiol. 2008, 54, 349–355. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Feuille, F.; Ebersole, J.L.; Kesavalu, L.; Stepfen, M.J.; Holt, S.C. Mixed infection with Porphyromonas gingivalis and Fusobacterium nucleatum in a murine lesion model: Potential synergistic effects on virulence. Infect. Immun. 1996, 64, 2094–2100. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Macleod, L.C.; Newsome, E.; Liu, J.; Xu, P. Aggregatibacter actinomycetemcomitans mediates protection of Porphyromonas gingivalis from Streptococcus sanguinis hydrogen peroxide production in multi-species biofilms. Sci. Rep. 2019, 9, 4944. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Isola, G.; Alibrandi, A.; Currò, M.; Matarese, M.; Ricca, S.; Matarese, G.; Ientile, R.; Kocher, T. Evaluation of salivary and serum ADMA levels in patients with periodontal and cardiovascular disease as subclinical marker of cardiovascular risk. J. Periodontol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Isola, G.; Polizzi, A.; Santonocito, S.; Alibrandi, A.; Ferlito, S. Expression of Salivary and Serum Malondialdehyde and Lipid Profile of Patients with Periodontitis and Coronary Heart Disease. Int. J. Mol. Sci. 2019, 20, 6061. [Google Scholar] [CrossRef] [Green Version]
- Isola, G.; Polizzi, A.; Alibrandi, A.; Williams, R.C.; Leonardi, R. Independent impact of periodontitis and cardiovascular disease on elevated soluble urokinase-type plasminogen activator receptor (suPAR) levels. J. Periodontol. 2020. [Google Scholar] [CrossRef]
- Isola, G.; Polizzi, A.; Patini, R.; Ferlito, S.; Alibrandi, A.; Palazzo, G. Association among serum and salivary A. actinomycetemcomitans specific immunoglobulin antibodies and periodontitis. BMC Oral Health 2020, 20, 283. [Google Scholar] [CrossRef]
- Hoare, A.; Soto, C.; Rojas-Celis, V.; Bravo, D. Chronic Inflammation as a Link between Periodontitis and Carcinogenesis. Mediat. Inflamm. 2019, 2019, 1029857. [Google Scholar] [CrossRef] [Green Version]
- Wen, B.W.; Tsai, C.S.; Lin, C.L.; Chang, Y.J.; Lee, C.F.; Hsu, C.H.; Kao, C.H. Cancer risk among gingivitis and periodontitis patients: A nationwide cohort study. QJM 2014, 107, 283–290. [Google Scholar] [CrossRef]
- Michaud, D.S.; Fu, Z.; Shi, J.; Chung, M. Periodontal Disease, Tooth Loss, and Cancer Risk. Epidemiol. Rev. 2017, 39, 49–58. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geng, F.; Liu, J.; Guo, Y.; Li, C.; Wang, H.; Zhao, H.; Pan, Y. Persistent Exposure to Porphyromonas gingivalis promotes proliferative and invasion capabilities, and tumorigenic properties of human immortalized oral epithelial cells. Front. Cell. Infect. Microbiol. 2017, 7, 57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, C.H.; Spooner, R.; DeGuzman, J.; Koutouzis, T.; Ojcius, D.M.; Yilmaz, Ö. Porphyromonas gingivalis-nucleoside-diphosphate-kinase inhibits ATP-induced reactive-oxygen-species via P2X7 receptor/NADPH-oxidase signalling and contributes to persistence. Cell. Microbiol. 2013, 15, 961–976. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 14. [Google Scholar] [CrossRef]
- Poetsch, A.R. The genomics of oxidative DNA damage, repair, and resulting mutagenesis. Comput. Struct. Biotechnol. J. 2020, 18, 207–219. [Google Scholar] [CrossRef]
- Liu, J.; Tang, X.; Li, C.; Pan, C.; Li, Q.; Geng, F.; Pan, Y. Porphyromonas gingivalis promotes the cell cycle and inflammatory cytokine production in periodontal ligament fibroblasts. Arch. Oral Biol. 2015, 60, 1153–1161. [Google Scholar] [CrossRef]
- Sztukowska, M.N.; Ojo, A.; Ahmed, S.; Carenbauer, A.L.; Wang, Q.; Shumway, B.; Jenkinson, H.F.; Wang, H.; Darling, D.S.; Lamont, R.J. Porphyromonas gingivalis initiates a mesenchymal-like transition through ZEB1 in gingival epithelial cells. Cell. Microbiol. 2016, 18, 844–858. [Google Scholar] [CrossRef] [Green Version]
- Maekawa, T.; Krauss, J.L.; Abe, T.; Jotwani, R.; Triantafilou, M.; Triantafilou, K.; Hashim, A.; Hoch, S.; Curtis, M.A.; Nussbaum, G.; et al. Porphyromonas gingivalis manipulates complement and TLR signaling to uncouple bacterial clearance from inflammation and promote dysbiosis. Cell Host Microbe 2014, 15, 768–778. [Google Scholar] [CrossRef] [Green Version]
- Groeger, S.; Jarzina, F.; Domann, E.; Meyle, J. Porphyromonas gingivalis activates NFκB and MAPK pathways in human oral epithelial cells. BMC Immunol. 2017, 18, 1. [Google Scholar] [CrossRef] [Green Version]
- Yilmaz, O.; Sater, A.A.; Yao, L.; Koutouzis, T.; Pettengill, M.; Ojcius, D.M. ATP-dependent activation of an inflammasome in primary gingival epithelial cells infected by Porphyromonas gingivalis. Cell. Microbiol. 2010, 12, 188–198. [Google Scholar] [CrossRef] [Green Version]
- Utispan, K.; Pugdee, K.; Koontongkaew, S. Porphyromonas gingivalis lipopolysaccharide-induced macrophages modulate proliferation and invasion of head and neck cancer cell lines. Biomed. Pharm. 2018, 101, 988–995. [Google Scholar] [CrossRef] [PubMed]
- Singh, A.; Wyant, T.; Anaya-Bergman, C.; Aduse-Opoku, J.; Brunner, J.; Laine, M.L.; Curtis, M.A.; Lewis, J.P. The capsule of Porphyromonas gingivalis leads to a reduction in the host inflammatory response, evasion of phagocytosis, and increase in virulence. Infect. Immun. 2011, 79, 4533–4542. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brunner, J.; Scheres, N.; El Idrissi, N.B.; Deng, D.M.; Laine, M.L.; van Winkelhoff, A.J.; Crielaard, W. The capsule of Porphyromonas gingivalis reduces the immune response of human gingival fibroblasts. BMC Microbiol. 2010, 10, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aoki, Y.; Tabeta, K.; Murakami, Y.; Yoshimura, F.; Yamazaki, K. Analysis of immunostimulatory activity of Porphyromonas gingivalis fimbriae conferred by Toll-like receptor 2. Biochem. Biophys. Res. Commun. 2010, 398, 86–91. [Google Scholar] [CrossRef] [PubMed]
- Hajishengallis, G.; Wang, M.; Liang, S. Induction of distinct TLR2-mediated proinflammatory and proadhesive signaling pathways in response to Porphyromonas gingivalis fimbriae. J. Immunol. 2009, 182, 6690–6696. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Baba, A.; Abe, N.; Kadowaki, T.; Nakanishi, H.; Ohishi, M.; Asao, T.; Yamamoto, K. Arg-gingipain is responsible for the degradation of cell adhesion molecules of human gingival fibroblasts and their death induced by Porphyromonas gingivalis. Biol. Chem. 2001, 382, 817–824. [Google Scholar] [CrossRef] [PubMed]
- O’Brien-Simpson, N.M.; Paolini, R.A.; Hoffmann, B.; Slakeski, N.; Dashper, S.G.; Reynolds, E.C. Role of RgpA, RgpB, and Kgp proteinases in virulence of Porphyromonas gingivalis W50 in a murine lesion model. Infect. Immun. 2001, 69, 7527–7534. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tanaka, T.; Tanaka, M. Oral carcinogenesis and oral cancer chemoprevention: A review. Pathol. Res. Int. 2011, 2011, 431246. [Google Scholar] [CrossRef]
- Lin, T.; Zhang, B.; He, H. Identification of genes correlated with oral squamous cell carcinoma. J. Cancer Res. Ther. 2018, 14, S675–S679. [Google Scholar] [CrossRef]
- Nakagaki, T.; Tamura, M.; Kobashi, K.; Koyama, R.; Fukushima, H.; Ohashi, T.; Idogawa, M.; Ogi, K.; Hiratsuka, H.; Tokino, T.; et al. Profiling cancer-related gene mutations in oral squamous cell carcinoma from Japanese patients by targeted amplicon sequencing. Oncotarget 2017, 8, 59113–59122. [Google Scholar] [CrossRef] [Green Version]
- Nakagaki, T.; Tamura, M.; Kobashi, K.; Omori, A.; Koyama, R.; Idogawa, M.; Ogi, K.; Hiratsuka, H.; Tokino, T.; Sasaki, Y. Targeted next-generation sequencing of 50 cancer-related genes in Japanese patients with oral squamous cell carcinoma. Tumor Biol. 2018, 40. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, T.; Kon, N.; Jiang, L.; Tan, M.; Ludwig, T.; Zhao, Y.; Baer, R.; Gu, W. Tumor suppression in the absence of p53-mediated cell-cycle arrest, apoptosis, and senescence. Cell 2012, 149, 1269–1283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Senturk, E.; Manfredi, J.J. p53 and cell cycle effects after DNA damage. Methods Mol. Biol. 2013, 962, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hsieh, L.L.; Wang, P.F.; Chen, I.H.; Liao, C.T.; Wang, H.M.; Chen, M.C.; Chang, J.T.; Cheng, A.J. Characteristics of mutations in the p53 gene in oral squamous cell carcinoma associated with betel quid chewing and cigarette smoking in Taiwanese. Carcinogenesis 2001, 22, 1497–1503. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lapke, N.; Lu, Y.J.; Liao, C.T.; Lee, L.Y.; Lin, C.Y.; Wang, H.M.; Ng, S.H.; Chen, S.J.; Yen, T.C. Missense mutations in the TP53 DNA-binding domain predict outcomes in patients with advanced oral cavity squamous cell carcinoma. Oncotarget 2016, 7, 44194–44210. [Google Scholar] [CrossRef] [Green Version]
- Zhou, G.; Liu, Z.; Myers, J.N. TP53 Mutations in Head and Neck Squamous Cell Carcinoma and Their Impact on Disease Progression and Treatment Response. J. Cell. Biochem. 2016, 117, 2682–2692. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Acin, S.; Li, Z.; Mejia, O.; Roop, D.R.; El-Naggar, A.K.; Caulin, C. Gain-of-function mutant p53 but not p53 deletion promotes head and neck cancer progression in response to oncogenic K-ras. J. Pathol. 2011, 225, 479–489. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Göhler, T.; Jäger, S.; Warnecke, G.; Yasuda, H.; Kim, E.; Deppert, W. Mutant p53 proteins bind DNA in a DNA structure-selective mode. Nucleic Acids Res. 2005, 33, 1087–1100. [Google Scholar] [CrossRef] [Green Version]
- Costa, V.; Kowalski, L.P.; Coutinho-Camillo, C.M.; Begnami, M.D.; Calsavara, V.F.; Neves, J.I.; Kaminagakura, E. EGFR amplification and expression in oral squamous cell carcinoma in young adults. Int. J. Oral Maxillofac. Surg. 2018, 47, 817–823. [Google Scholar] [CrossRef] [Green Version]
- Huang, S.F.; Cheng, S.D.; Chien, H.T.; Liao, C.T.; Chen, I.H.; Wang, H.M.; Chuang, W.Y.; Wang, C.Y.; Hsieh, L.L. Relationship between epidermal growth factor receptor gene copy number and protein expression in oral cavity squamous cell carcinoma. Oral Oncol. 2012, 48, 67–72. [Google Scholar] [CrossRef]
- Chiang, W.F.; Liu, S.Y.; Yen, C.Y.; Lin, C.N.; Chen, Y.C.; Lin, S.C.; Chang, K.W. Association of epidermal growth factor receptor (EGFR) gene copy number amplification with neck lymph node metastasis in areca-associated oral carcinomas. Oral Oncol. 2008, 44, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Xia, W.; Lau, Y.K.; Zhang, H.Z.; Xiao, F.Y.; Johnston, D.A.; Liu, A.R.; Li, L.; Katz, R.L.; Hung, M.C. Combination of EGFR, HER-2/neu, and HER-3 is a stronger predictor for the outcome of oral squamous cell carcinoma than any individual family members. Clin. Cancer Res. 1999, 5, 4164–4174. [Google Scholar] [PubMed]
- Szabó, B.; Nelhubel, G.A.; Kárpáti, A.; Kenessey, I.; Jóri, B.; Székely, C.; Peták, I.; Lotz, G.; Hegedus, Z.; Hegedus, B.; et al. Clinical significance of genetic alterations and expression of epidermal growth factor receptor (EGFR) in head and neck squamous cell carcinomas. Oral Oncol. 2011, 47, 487–496. [Google Scholar] [CrossRef] [PubMed]
- Chung, C.H.; Ely, K.; McGavran, L.; Varella-Garcia, M.; Parker, J.; Parker, N.; Jarrett, C.; Carter, J.; Murphy, B.A.; Netterville, J.; et al. Increased epidermal growth factor receptor gene copy number is associated with poor prognosis in head and neck squamous cell carcinomas. J. Clin. Oncol. 2006, 24, 4170–4176. [Google Scholar] [CrossRef]
- Temam, S.; Kawaguchi, H.; El-Naggar, A.K.; Jelinek, J.; Tang, H.; Liu, D.D.; Lang, W.; Issa, J.P.; Lee, J.J.; Mao, L. Epidermal growth factor receptor copy number alterations correlate with poor clinical outcome in patients with head and neck squamous cancer. J. Clin. Oncol. 2007, 25, 2164–2170. [Google Scholar] [CrossRef]
- Rajalingam, K.; Schreck, R.; Rapp, U.R.; Albert, S. Ras oncogenes and their downstream targets. Biochim. Biophys. Acta 2007, 1773, 1177–1195. [Google Scholar] [CrossRef] [Green Version]
- Lee, H.W.; Kyung, T.; Yoo, J.; Kim, T.; Chung, C.; Ryu, J.Y.; Lee, H.; Park, K.; Lee, S.; Jones, W.D.; et al. Real-time single-molecule co-immunoprecipitation analyses reveal cancer-specific Ras signalling dynamics. Nat. Commun. 2013, 4, 1505. [Google Scholar] [CrossRef] [Green Version]
- Krishna, A.; Singh, S.; Singh, V.; Kumar, V.; Singh, U.S.; Sankhwar, S.N. Does Harvey-Ras gene expression lead to oral squamous cell carcinoma? A clinicopathological aspect. J. Oral Maxillofac. Pathol. 2018, 22, 65–72. [Google Scholar] [CrossRef]
- Das, N.; Majumder, J.; DasGupta, U.B. ras gene mutations in oral cancer in eastern India. Oral Oncol. 2000, 36, 76–80. [Google Scholar] [CrossRef]
- Saranath, D.; Chang, S.E.; Bhoite, L.T.; Panchal, R.G.; Kerr, I.B.; Mehta, A.R.; Johnson, N.W.; Deo, M.G. High frequency mutation in codons 12 and 61 of H-ras oncogene in chewing tobacco-related human oral carcinoma in India. Br. J. Cancer 1991, 63, 573–578. [Google Scholar] [CrossRef] [Green Version]
- Wang, J.; Wang, W.; Li, J.; Wu, L.; Song, M.; Meng, Q. miR182 activates the Ras-MEK-ERK pathway in human oral cavity squamous cell carcinoma by suppressing RASA1 and SPRED1. Onco Targets Ther. 2017, 10, 667–679. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Aggarwal, S.; John, S.; Sapra, L.; Sharma, S.C.; Das, S.N. Targeted disruption of PI3K/Akt/mTOR signaling pathway, via PI3K inhibitors, promotes growth inhibitory effects in oral cancer cells. Cancer Chemother. Pharmacol. 2019, 83, 451–461. [Google Scholar] [CrossRef] [PubMed]
- Lui, V.W.; Hedberg, M.L.; Li, H.; Vangara, B.S.; Pendleton, K.; Zeng, Y.; Lu, Y.; Zhang, Q.; Du, Y.; Gilbert, B.R.; et al. Frequent mutation of the PI3K pathway in head and neck cancer defines predictive biomarkers. Cancer Discov. 2013, 3, 761–769. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cai, Y.; Dodhia, S.; Su, G.H. Dysregulations in the PI3K pathway and targeted therapies for head and neck squamous cell carcinoma. Oncotarget 2017, 8, 22203–22217. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ozes, O.N.; Mayo, L.D.; Gustin, J.A.; Pfeffer, S.R.; Pfeffer, L.M.; Donner, D.B. NF-kappaB activation by tumour necrosis factor requires the Akt serine-threonine kinase. Nature 1999, 401, 82–85. [Google Scholar] [CrossRef] [PubMed]
- Freudlsperger, C.; Burnett, J.R.; Friedman, J.A.; Kannabiran, V.R.; Chen, Z.; Van Waes, C. EGFR-PI3K-AKT-mTOR signaling in head and neck squamous cell carcinomas: Attractive targets for molecular-oriented therapy. Expert Opin. Ther. Targets 2011, 15, 63–74. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Qiu, W.; Schönleben, F.; Li, X.; Ho, D.J.; Close, L.G.; Manolidis, S.; Bennett, B.P.; Su, G.H. PIK3CA mutations in head and neck squamous cell carcinoma. Clin. Cancer Res. 2006, 12, 1441–1446. [Google Scholar] [CrossRef] [Green Version]
- Kozaki, K.; Imoto, I.; Pimkhaokham, A.; Hasegawa, S.; Tsuda, H.; Omura, K.; Inazawa, J. PIK3CA mutation is an oncogenic aberration at advanced stages of oral squamous cell carcinoma. Cancer Sci. 2006, 97, 1351–1358. [Google Scholar] [CrossRef]
- Kurasawa, Y.; Shiiba, M.; Nakamura, M.; Fushimi, K.; Ishigami, T.; Bukawa, H.; Yokoe, H.; Uzawa, K.; Tanzawa, H. PTEN expression and methylation status in oral squamous cell carcinoma. Oncol. Rep. 2008, 19, 1429–1434. [Google Scholar]
- Gasparotto, D.; Vukosavljevic, T.; Piccinin, S.; Barzan, L.; Sulfaro, S.; Armellin, M.; Boiocchi, M.; Maestro, R. Loss of heterozygosity at 10q in tumors of the upper respiratory tract is associated with poor prognosis. Int. J. Cancer 1999, 84, 432–436. [Google Scholar] [CrossRef]
- Yoshida, R.; Nagata, M.; Nakayama, H.; Niimori-Kita, K.; Hassan, W.; Tanaka, T.; Shinohara, M.; Ito, T. The pathological significance of Notch1 in oral squamous cell carcinoma. Lab. Investig. 2013, 93, 1068–1081. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Osathanon, T.; Nowwarote, N.; Pavasant, P. Expression and influence of Notch signaling in oral squamous cell carcinoma. J. Oral Sci. 2016, 58, 283–294. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Zheng, G.; Zhou, L.; Li, P.; Yun, M.; Shi, Q.; Wang, T.; Wu, X. Notch signalling induces epithelial-mesenchymal transition to promote metastasis in oral squamous cell carcinoma. Int. J. Mol. Med. 2018, 42, 2276–2284. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Roshan, V.G.D.; Sinto, M.S.; Thomas, S.; Kannan, S. Cyclin D1 overexpression associated with activation of STAT3 in oral carcinoma patients from South India. J. Cancer Res. Ther. 2018, 14, 403–408. [Google Scholar] [CrossRef]
- Ni, Y.; Zhang, X.; Wan, Y.; Dun Tang, K.; Xiao, Y.; Jing, Y.; Song, Y.; Huang, X.; Punyadeera, C.; Hu, Q. Relationship between p16 expression and prognosis in different anatomic subsites of OSCC. Cancer Biomark. 2019, 26, 375–383. [Google Scholar] [CrossRef]
- Padhi, S.S.; Roy, S.; Kar, M.; Saha, A.; Adhya, A.; Baisakh, M.; Banerjee, B. Role of CDKN2A/p16 expression in the prognostication of oral squamous cell carcinoma. Oral Oncol. 2017, 73, 27–35. [Google Scholar] [CrossRef]
- Juneja, S.; Chaitanya, N.B.; Agarwal, M. Immunohistochemical expression of Bcl-2 in oral epithelial dysplasia and oral squamous cell carcinoma. Indian J. Cancer 2015, 52, 505–510. [Google Scholar] [CrossRef]
- Garewal, J.; Garewal, R.; Sircar, K. Expression of Bcl-2 and MIB-1 Markers in Oral Squamous Cell Carcinoma (OSCC)- A Comparative Study. J. Clin. Diagn Res. 2014, 8, QC01. [Google Scholar] [CrossRef]
- Jordan, R.C.; Catzavelos, G.C.; Barrett, A.W.; Speight, P.M. Differential expression of bcl-2 and bax in squamous cell carcinomas of the oral cavity. Eur. J. Cancer B Oral Oncol. 1996, 32B, 394–400. [Google Scholar] [CrossRef]
- Chen, K.; Hu, Z.; Wang, L.E.; Sturgis, E.M.; El-Naggar, A.K.; Zhang, W.; Wei, Q. Single-nucleotide polymorphisms at the TP53-binding or responsive promoter regions of BAX and BCL2 genes and risk of squamous cell carcinoma of the head and neck. Carcinogenesis 2007, 28, 2008–2012. [Google Scholar] [CrossRef]
- Alam, M.; Kashyap, T.; Mishra, P.; Panda, A.K.; Nagini, S.; Mishra, R. Role and regulation of proapoptotic Bax in oral squamous cell carcinoma and drug resistance. Head Neck 2019, 41, 185–197. [Google Scholar] [CrossRef] [Green Version]
- Singh, R.; Lillard, J.W.; Singh, S. Chemokines: Key players in cancer progression and metastasis. Front. Biosci. 2011, 3, 1569–1582. [Google Scholar] [CrossRef] [Green Version]
- Yanjia, H.; Xinchun, J. The role of epithelial-mesenchymal transition in oral squamous cell carcinoma and oral submucous fibrosis. Clin. Chim. Acta 2007, 383, 51–56. [Google Scholar] [CrossRef] [PubMed]
- Kalluri, R.; Weinberg, R.A. The basics of epithelial-mesenchymal transition. J. Clin. Investig. 2009, 119, 1420–1428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Impola, U.; Uitto, V.J.; Hietanen, J.; Hakkinen, L.; Zhang, L.; Larjava, H.; Isaka, K.; Saarialho-Kere, U. Differential expression of matrilysin-1 (MMP-7), 92 kD gelatinase (MMP-9), and metalloelastase (MMP-12) in oral verrucous and squamous cell cancer. J. Pathol. 2004, 202, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Saygun, I.; Kubar, A.; Ozdemir, A.; Yapar, M.; Slots, J. Herpesviral-bacterial interrelationships in aggressive periodontitis. J. Periodontal. Res. 2004, 39, 207–212. [Google Scholar] [CrossRef] [PubMed]
- Kato, A.; Imai, K.; Sato, H.; Ogata, Y. Prevalence of Epstein-Barr virus DNA and Porphyromonas gingivalis in Japanese peri-implantitis patients. BMC Oral Health 2017, 17, 148. [Google Scholar] [CrossRef]
- Jakovljevic, A.; Andric, M.; Knezevic, A.; Milicic, B.; Beljic-Ivanovic, K.; Perunovic, N.; Nikolic, N.; Milasin, J. Herpesviral-bacterial co-infection in mandibular third molar pericoronitis. Clin. Oral Investig. 2017, 21, 1639–1646. [Google Scholar] [CrossRef]
- Kato, A.; Imai, K.; Ochiai, K.; Ogata, Y. Prevalence and quantitative analysis of Epstein-Barr virus DNA and Porphyromonas gingivalis associated with Japanese chronic periodontitis patients. Clin. Oral Investig. 2015, 19, 1605–1610. [Google Scholar] [CrossRef] [Green Version]
- Sugano, N.; Ikeda, K.; Oshikawa, M.; Idesawa, M.; Tanaka, H.; Sato, S.; Ito, K. Relationship between Porphyromonas gingivalis, Epstein-Barr virus infection and reactivation in periodontitis. J. Oral Sci. 2004, 46, 203–206. [Google Scholar] [CrossRef] [Green Version]
- Simpson, D.R.; Mell, L.K.; Cohen, E.E. Targeting the PI3K/AKT/mTOR pathway in squamous cell carcinoma of the head and neck. Oral Oncol. 2015, 51, 291–298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kennedy, S.G.; Kandel, E.S.; Cross, T.K.; Hay, N. Akt/Protein kinase B inhibits cell death by preventing the release of cytochrome c from mitochondria. Mol. Cell. Biol. 1999, 19, 5800–5810. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, Y.; Liu, X.; Wang, H.; Liu, S.; Hu, N.; Li, X. Akt Regulated Phosphorylation of GSK-3β/Cyclin D1, p21 and p27 Contributes to Cell Proliferation Through Cell Cycle Progression From G1 to S/G2M Phase in Low-Dose Arsenite Exposed HaCat Cells. Front. Pharmacol. 2019, 10, 1176. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hay, N.; Sonenberg, N. Upstream and downstream of mTOR. Genes Dev. 2004, 18, 1926–1945. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Imai, K.; Inoue, H.; Tamura, M.; Cueno, M.E.; Takeichi, O.; Kusama, K.; Saito, I.; Ochiai, K. The periodontal pathogen Porphyromonas gingivalis induces the Epstein-Barr virus lytic switch transactivator ZEBRA by histone modification. Biochimie 2012, 94, 839–846. [Google Scholar] [CrossRef] [PubMed]
- Murata, T.; Kondo, Y.; Sugimoto, A.; Kawashima, D.; Saito, S.; Isomura, H.; Kanda, T.; Tsurumi, T. Epigenetic histone modification of Epstein-Barr virus BZLF1 promoter during latency and reactivation in Raji cells. J. Virol. 2012, 86, 4752–4761. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Riggs, M.G.; Whittaker, R.G.; Neumann, J.R.; Ingram, V.M. n-Butyrate causes histone modification in HeLa and Friend erythroleukaemia cells. Nature 1977, 268, 462–464. [Google Scholar] [CrossRef] [PubMed]
- Countryman, J.K.; Gradoville, L.; Miller, G. Histone hyperacetylation occurs on promoters of lytic cycle regulatory genes in Epstein-Barr virus-infected cell lines which are refractory to disruption of latency by histone deacetylase inhibitors. J. Virol. 2008, 82, 4706–4719. [Google Scholar] [CrossRef] [Green Version]
- Shirasugi, M.; Nakagawa, M.; Nishioka, K.; Yamamoto, T.; Nakaya, T.; Kanamura, N. Relationship between periodontal disease and butyric acid produced by periodontopathic bacteria. Inflamm. Regen. 2018, 38, 23. [Google Scholar] [CrossRef]
- Martel-Renoir, D.; Grunewald, V.; Touitou, R.; Schwaab, G.; Joab, I. Qualitative analysis of the expression of Epstein-Barr virus lytic genes in nasopharyngeal carcinoma biopsies. J Gen. Virol. 1995, 76, 1401–1408. [Google Scholar] [CrossRef]
- Al Tabaa, Y.; Tuaillon, E.; Jeziorski, E.; Ouedraogo, D.E.; Bolloré, K.; Rubbo, P.A.; Foulongne, V.; Rodière, M.; Vendrell, J.P. B-cell polyclonal activation and Epstein-Barr viral abortive lytic cycle are two key features in acute infectious mononucleosis. J. Clin. Virol. 2011, 52, 33–37. [Google Scholar] [CrossRef] [PubMed]
- Gölz, L.; Memmert, S.; Rath-Deschner, B.; Jäger, A.; Appel, T.; Baumgarten, G.; Götz, W.; Frede, S. LPS from P. gingivalis and hypoxia increases oxidative stress in periodontal ligament fibroblasts and contributes to periodontitis. Mediat. Inflamm. 2014, 2014, 986264. [Google Scholar] [CrossRef] [PubMed] [Green Version]


Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Núñez-Acurio, D.; Bravo, D.; Aguayo, F. Epstein–Barr Virus—Oral Bacterial Link in the Development of Oral Squamous Cell Carcinoma. Pathogens 2020, 9, 1059. https://doi.org/10.3390/pathogens9121059
Núñez-Acurio D, Bravo D, Aguayo F. Epstein–Barr Virus—Oral Bacterial Link in the Development of Oral Squamous Cell Carcinoma. Pathogens. 2020; 9(12):1059. https://doi.org/10.3390/pathogens9121059
Chicago/Turabian StyleNúñez-Acurio, Daniela, Denisse Bravo, and Francisco Aguayo. 2020. "Epstein–Barr Virus—Oral Bacterial Link in the Development of Oral Squamous Cell Carcinoma" Pathogens 9, no. 12: 1059. https://doi.org/10.3390/pathogens9121059
APA StyleNúñez-Acurio, D., Bravo, D., & Aguayo, F. (2020). Epstein–Barr Virus—Oral Bacterial Link in the Development of Oral Squamous Cell Carcinoma. Pathogens, 9(12), 1059. https://doi.org/10.3390/pathogens9121059