Pathogenomic Analysis of a Novel Extensively Drug-Resistant Citrobacter freundii Isolate Carrying a blaNDM-1 Carbapenemase in South Africa

 ,
,  ,
,  , ,
, ,  and
and
Abstract
:1. Introduction
2. Results
2.1. Isolation, Phenotypic Confirmation of Carbapenem Resistance, and Antibiotic Susceptibility Testing (AST)
2.2. General Genomic Features of H2730R
2.3. WGS-Based Confirmation and Multilocus Sequence Typing (MLST)
2.4. WGS Analysis of Resistance Genes and Genetic Support
2.5. Pathogenicity, Defence Systems Mechanisms, and Virulome Predictions
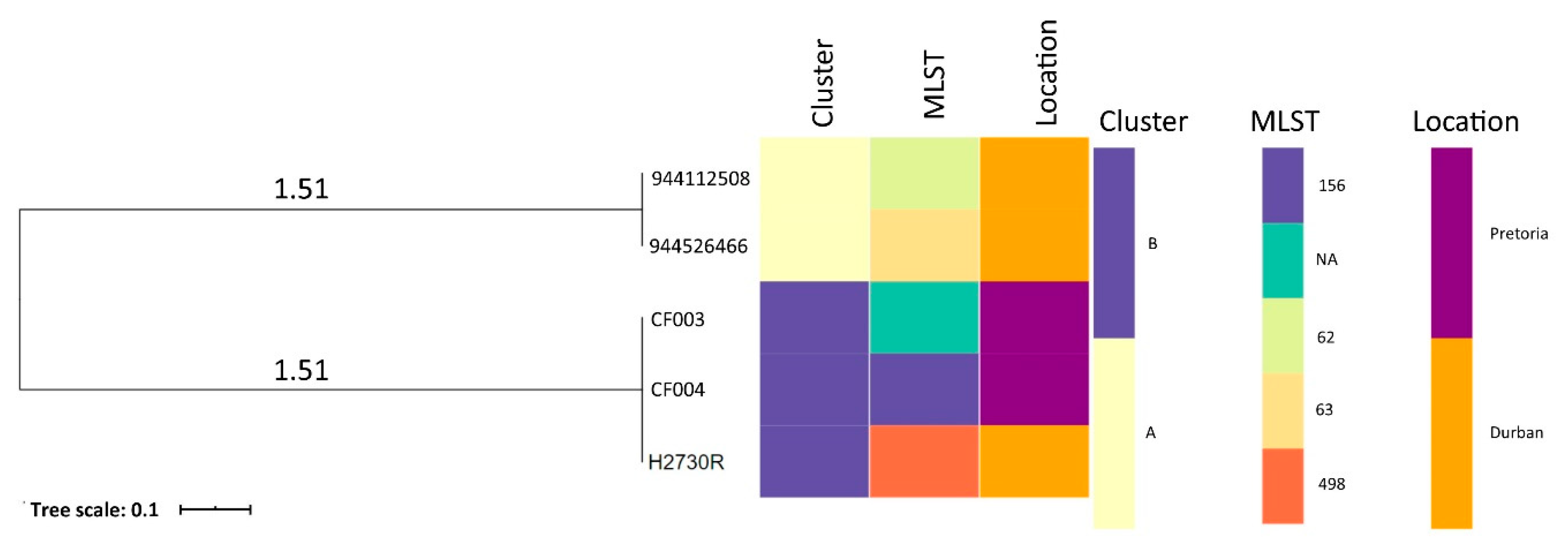
2.6. Phylogenomic Analysis and Metadata Insights of Reported Citrobacter Freundii Isolates from South Africa
3. Discussion
4. Materials and Methods
4.1. Ethical Approval
4.2. Identification of the Isolate
4.3. Antibiotic Susceptibility Testing (AST)
4.4. DNA Purification, Genome Sequencing, and Preprocessing of Sequence Data
4.5. Bioinformatic Analysis
4.5.1. Genome Visualization and Annotation
4.5.2. WGS-Based Confirmation and Molecular Typing
4.5.3. Genomic Identification of the Antibiotic Resistome and Mobile Genetic Elements (MGEs)
4.5.4. Pathogenicity, Defence Systems, and Virulome Predictions
4.6. Phylogenomic Analyses of C. freundii Isolates from South Africa
4.7. Data Availability
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A

Appendix B

{kind=link}
{kind=link}
{kind=link}
| Attribute | Value |
|---|---|
| Sequencing platform | Illumina MiSeq machine |
| Assembler | Skesa (version 2.3) |
| Assembly accession | VWTQ00000000 |
| No. of Contigs | 58 |
| Genome size (bp) | 5,299,408 |
| DNA G + C (%) | 51.8 |
| Genome coverage (X) | 99.0 |
| Number of RNAs genes | 70 |
| Number of tRNAs genes | 12 |
| 23S rRNAs | 7 |
| 5S rRNAs | 5 |
| N50 | 518,368 |
| L50 | 4 |
| Number of subsystems | 396 |
| Number of CDSs | 5135 |
| Genes assigned to COGs | 5006 |
| Pseudo Genes | 129 |
| Isolate | MLST | arcA | aspC | clpX | dnaG | fadD | lysP | mdh |
|---|---|---|---|---|---|---|---|---|
| 944112508 | ST156 | 7 | 15 | 63 | 54 | 13 | 5 | 12 |
| 944526466 | NA | 33 | 49 | 50 | 45 | 57 | 16 | 47 |
| CF003 | ST62 | 32 | 48 | 10 | 6 | 14 | 45 | 13 |
| CF004 | ST63 | 33 | 49 | 50 | 45 | 57 | 16 | 47 |
| H2730R | ST498 | 5 | 16 | 14 | 54 | 103 | 5 | 15 |
References
- Liu, L.; Chen, D.; Liu, L.; Lan, R.; Hao, S.; Jin, W.; Sun, H.; Wang, Y.; Liang, Y.; Xu, J. Genetic Diversity, Multidrug Resistance, and Virulence of Citrobacter freundii From Diarrheal Patients and Healthy Individuals. Front. Microbiol. 2018, 8, 233. [Google Scholar] [CrossRef] [Green Version]
- Heo, G.-J.; Hossain, S.; Wimalasena, S. Virulence Factors and Antimicrobial Resistance Pattern of Citrobacter freundii Isolated from Healthy Pet Turtles and their Environment. Asian J. Anim. Veter- Adv. 2017, 12, 10–16. [Google Scholar] [CrossRef]
- Mohanty, S.; Singhal, R.; Sood, S.; Dhawan, B.; Kapil, A.; Das, B.K. Citrobacter infections in a tertiary care hospital in Northern India. J. Infect. 2007, 54, 58–64. [Google Scholar] [CrossRef]
- Pepperell, C.; Kus, J.V.; Gardam, M.A.; Humar, A.; Burrows, L.L. Low-Virulence Citrobacter Species Encode Resistance to Multiple Antimicrobials. Antimicrob. Agents Chemother. 2002, 46, 3555–3560. [Google Scholar] [CrossRef] [Green Version]
- Gupta, N.; Yadav, A.; Choudhary, U.; Aropa, D.R. Citrobacter Bacteremia in a Tertiary Care Hospital. Scand. J. Infect. Dis. 2003, 35, 765–768. [Google Scholar] [CrossRef]
- Divyashree, M.; Kumar, D.V.; Ballamoole, K.K.; Shetty, V.; Chakraborty, A.; Karunasagar, I. Occurrence of antibiotic resistance among gram-negative bacteria isolated from effluents of fish processing plants in and around Mangalore. Int. J. Environ. Heal. Res. 2019, 1–8. [Google Scholar] [CrossRef]
- Park, Y.-J.; Park, S.Y.; Oh, E.-J.; Park, J.-J.; Lee, K.-Y.; Woo, G.-J.; Lee, K. Occurrence of extended-spectrum β-lactamases among chromosomal AmpC-producing Enterobacter cloacae, Citrobacter freundii, and Serratia marcescens in Korea and investigation of screening criteria. Diagn. Microbiol. Infect. Dis. 2005, 51, 265–269. [Google Scholar] [CrossRef]
- Majewski, P.; Wieczorek, P.; Łapuć, I.; Ojdana, D.; Sieńko, A.; Sacha, P.; Kłoczko, J.; Tryniszewska, E. Emergence of a multidrug-resistant Citrobacter freundii ST8 harboring an unusual VIM-4 gene cassette in Poland. Int. J. Infect. Dis. 2017, 61, 70–73. [Google Scholar] [CrossRef]
- Yang, L.; Li, P.; Liang, B.; Hu, X.; Li, J.; Xie, J.; Yang, C.; Hao, R.; Wang, L.; Jia, L.; et al. Multidrug-resistant Citrobacter freundii ST139 co-producing NDM-1 and CMY-152 from China. Sci. Rep. 2018, 8, 10653. [Google Scholar] [CrossRef]
- Aminov, R.I. Horizontal Gene Exchange in Environmental Microbiota. Front. Microbiol. 2011, 2, 1–19. [Google Scholar] [CrossRef] [Green Version]
- Stokes, H.W.; Gillings, M.R. Gene flow, mobile genetic elements and the recruitment of antibiotic resistance genes into Gram-negative pathogens. FEMS Microbiol. Rev. 2011, 35, 790–819. [Google Scholar] [CrossRef] [PubMed]
- Partridge, S.R.; Kwong, S.M.; Firth, N.; Jensen, S.O. Mobile Genetic Elements Associated with Antimicrobial Resistance. Clin. Microbiol. Rev. 2018, 31, 1–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lerminiaux, N.A.; Cameron, A.D.S. Horizontal transfer of antibiotic resistance genes in clinical environments. Can. J. Microbiol. 2019, 65, 34–44. [Google Scholar] [CrossRef]
- Magiorakos, A.-P.; Srinivasan, A.; Carey, R.; Carmeli, Y.; Falagas, M.E.; Giske, C.; Harbarth, S.; Hindler, J.; Kahlmeter, G.; Olsson-Liljequist, B.; et al. Multidrug-resistant, extensively drug-resistant and pandrug-resistant bacteria: an international expert proposal for interim standard definitions for acquired resistance. Clin. Microbiol. Infect. 2012, 18, 268–281. [Google Scholar] [CrossRef] [Green Version]
- Sekyere, J.O.; Amoako, D.G. Genomic and phenotypic characterisation of fluoroquinolone resistance mechanisms in Enterobacteriaceae in Durban, South Africa. PLOS ONE 2017, 12, e0178888. [Google Scholar] [CrossRef] [Green Version]
- Mbelle, N.M.; Maningi, N.E.; Tshisevhe, V.; Modipane, L.; Amoako, D.G.; Sekyere, J.O. Draft Genome Sequence of a Clinical Enterococcus faecium Sequence Type 18 Strain from South Africa. Genome Announc. 2017, 5, e01381-17. [Google Scholar] [CrossRef] [Green Version]
- Agyepong, N.; Govinden, U.; Owusu-Ofori, A.; Amoako, D.G.; Allam, M.; Janice, J.; Pedersen, T.; Sundsfjord, A.; Essack, S. Genomic characterization of multidrug-resistant ESBL-producing Klebsiella pneumoniae isolated from a Ghanaian teaching hospital. Int. J. Infect. Dis. 2019, 85, 117–123. [Google Scholar] [CrossRef] [Green Version]
- Sekyere, J.O.; Amoako, D.G. Carbonyl Cyanide m-Chlorophenylhydrazine (CCCP) Reverses Resistance to Colistin, but Not to Carbapenems and Tigecycline in Multidrug-Resistant Enterobacteriaceae. Front. Microbiol. 2017, 8, 228. [Google Scholar] [CrossRef] [Green Version]
- Mbelle, N.M.; Sekyere, J.O.; Amoako, D.G.; Maningi, N.E.; Modipane, L.; Essack, S.Y.; Feldman, C. Genomic analysis of a multidrug-resistant clinical Providencia rettgeri (PR002) strain with the novel integron ln1483 and an A/C plasmid replicon. Ann. New York Acad. Sci. 2019. [Google Scholar] [CrossRef]
- Pedersen, T.; Sekyere, J.O.; Govinden, U.; Moodley, K.; Sivertsen, A.; Samuelsen, Ø.; Essack, S.Y.; Sundsfjord, A. Spread of Plasmid-Encoded NDM-1 and GES-5 Carbapenemases among Extensively Drug-Resistant and Pandrug-Resistant Clinical Enterobacteriaceae in Durban, South Africa. Antimicrob. Agents Chemother. 2018, 62, e02178-17. [Google Scholar] [CrossRef] [Green Version]
- Ramsamy, Y.; Mlisana, K.P.; Allam, M.; Amoako, D.G.; Abia, A.L.K.; Ismail, A.; Singh, R.; Kisten, T.; Essack, S.Y. Genomic Analysis of Carbapenemase-Producing Extensively Drug-Resistant Klebsiella pneumoniae Isolates Reveals the Horizontal Spread of p18–43 _ 01 Plasmid Encoding bla NDM-1 in South Africa. Microorganisms 2020, 8, 137. [Google Scholar] [CrossRef] [Green Version]
- Hartl, R.; Kerschner, H.; Gattringer, R.; Lepuschitz, S.; Allerberger, F.; Sorschag, S.; Ruppitsch, W.; Apfalter, P. Whole-Genome Analysis of a Human Enterobacter mori Isolate Carrying a blaIMI-2 Carbapenemase in Austria. Microb. Drug Resist. 2019, 25, 94–96. [Google Scholar] [CrossRef]
- Hammerum, A.M.; Hansen, F.; Olesen, B.; Struve, C.; Holzknecht, B.J.; Andersen, P.S.; Thye, A.-M.; Jakobsen, L.; Røder, B.L.; Stegger, M.; et al. Investigation of a possible outbreak of NDM-5-producing ST16 Klebsiella pneumoniae among patients in Denmark with no history of recent travel using whole-genome sequencing. J. Glob. Antimicrob. Resist. 2015, 3, 219–221. [Google Scholar] [CrossRef]
- Nordmann, P.; Naas, T.; Poirel, L. Global Spread of Carbapenemase-producing Enterobacteriaceae. Emerg. Infect. Dis. 2011, 17, 1791–1798. [Google Scholar] [CrossRef]
- Vandecraen, J.; Chandler, M.; Aertsen, A.; Van Houdt, R. The impact of insertion sequences on bacterial genome plasticity and adaptability. Crit. Rev. Microbiol. 2017, 43, 709–730. [Google Scholar] [CrossRef]
- Chen, Y.-G.; Zhang, Y.; Yu, Y.-S.; Qu, T.-T.; Wei, Z.-Q.; Shen, P.; Li, L.-J. In vivo development of carbapenem resistance in clinical isolates of Enterobacter aerogenes producing multiple β-lactamases. Int. J. Antimicrob. Agents 2008, 32, 302–307. [Google Scholar] [CrossRef]
- Fortier, L.-C.; Sekulovic, O. Importance of prophages to evolution and virulence of bacterial pathogens. Virulence 2013, 4, 354–365. [Google Scholar] [CrossRef]
- Mottawea, W.; Duceppe, M.-O.; Dupras, A.A.; Usongo, V.; Jeukens, J.; Freschi, L.; Emond-Rheault, J.-G.; Hamel, J.; Kukavica-Ibrulj, I.; Boyle, B.; et al. Salmonella enterica Prophage Sequence Profiles Reflect Genome Diversity and Can Be Used for High Discrimination Subtyping. Front. Microbiol. 2018, 9, 836. [Google Scholar] [CrossRef] [Green Version]
- Sultan, I.; Rahman, S.; Jan, A.T.; Siddiqui, M.T.; Mondal, A.H.; Haq, Q.M.R. Antibiotics, Resistome and Resistance Mechanisms: A Bacterial Perspective. Front. Microbiol. 2018, 9, 2066. [Google Scholar] [CrossRef] [Green Version]
- Shabbir, M.A.B.; Hao, H.; Shabbir, M.Z.; Hussain, H.I.; Iqbal, Z.; Ahmed, S.; Sattar, A.; Iqbal, M.; Li, J.; Yuan, Z. Survival and Evolution of CRISPR–Cas System in Prokaryotes and Its Applications. Front. Immunol. 2016, 7, 511. [Google Scholar] [CrossRef] [Green Version]
- Pleška, M.; Qian, L.; Okura, R.; Bergmiller, T.; Wakamoto, Y.; Kussell, E.; Guet, C.C. Bacterial Autoimmunity Due to a Restriction-Modification System. Curr. Boil. 2016, 26, 404–409. [Google Scholar] [CrossRef] [Green Version]
- Clements, A.; Young, J.C.; Constantinou, N.; Frankel, G. Infection strategies of enteric pathogenic Escherichia coli. Gut Microbes 2012, 3, 71–87. [Google Scholar] [CrossRef] [Green Version]
- Amoako, D.G.; Somboro, A.M.; Abia, A.L.K.; Allam, M.; Ismail, A.; Bester, L.A.; Essack, S.Y. Genome Mining and Comparative Pathogenomic Analysis of An Endemic Methicillin-Resistant Staphylococcus Aureus (MRSA) Clone, ST612-CC8-t1257-SCCmec_IVd(2B), Isolated in South Africa. Pathogens 2019, 8, 166. [Google Scholar] [CrossRef] [Green Version]
- Johnson, L.; Horsman, S.R.; Charron-Mazenod, L.; Turnbull, A.L.; Mulcahy, H.; Surette, M.G.; Lewenza, S. Extracellular DNA-induced antimicrobial peptide resistance in Salmonella enterica serovar Typhimurium. BMC Microbiol. 2013, 13, 115. [Google Scholar] [CrossRef] [Green Version]
- Amoako, D.G.; Somboro, A.M.; Abia, A.L.K.; Allam, M.; Ismail, A.; Bester, L.; Essack, S.Y. Genomic analysis of methicillin-resistant Staphylococcus aureus isolated from poultry and occupational farm workers in Umgungundlovu District, South Africa. Sci. Total Environ. 2019, 670, 704–716. [Google Scholar] [CrossRef]
- Clinical and Laboratory Standards Institute. Performance Standards for Antimicrobial Susceptibility Testing: 27th Edition Informational Supplement M100-S27; CLSI: Wayne, PA, USA, 2017. [Google Scholar]
- Parks, D.H.; Imelfort, M.; Skennerton, C.T.; Hugenholtz, P.; Tyson, G.W. CheckM: assessing the quality of microbial genomes recovered from isolates, single cells, and metagenomes. Genome Res. 2015, 25, 1043–1055. [Google Scholar] [CrossRef] [Green Version]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Boil. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef]
- Aziz, R.K.; Bartels, D.; A Best, A.; DeJongh, M.; Disz, T.; A Edwards, R.; Formsma, K.; Gerdes, S.; Glass, E.M.; Kubal, M.; et al. The RAST Server: Rapid Annotations using Subsystems Technology. BMC Genom. 2008, 9, 75. [Google Scholar] [CrossRef] [Green Version]
- Neher, R.A.; Bedford, T. Real-Time Analysis and Visualization of Pathogen Sequence Data. J. Clin. Microbiol. 2018, 56, 11. [Google Scholar] [CrossRef] [Green Version]
- Larsen, M.V.; Cosentino, S.; Rasmussen, S.; Friis, C.; Hasman, H.; Marvig, R.L.; Jelsbak, L.; Sicheritz-Pontén, T.; Ussery, D.W.; Aarestrup, F.M.; et al. Multilocus Sequence Typing of Total-Genome-Sequenced Bacteria. J. Clin. Microbiol. 2012, 50, 1355–1361. [Google Scholar] [CrossRef] [Green Version]
- Feil, E.J.; Li, B.C.; Aanensen, D.M.; Hanage, W.P.; Spratt, B.G. eBURST: Inferring Patterns of Evolutionary Descent among Clusters of Related Bacterial Genotypes from Multilocus Sequence Typing Data. J. Bacteriol. 2004, 186, 1518–1530. [Google Scholar] [CrossRef] [Green Version]
- Zankari, E.; Hasman, H.; Cosentino, S.; Vestergaard, M.; Rasmussen, S.; Lund, O.; Aarestrup, F.M.; Larsen, M.V. Identification of acquired antimicrobial resistance genes. J. Antimicrob. Chemother. 2012, 67, 2640–2644. [Google Scholar] [CrossRef]
- Gupta, S.K.; Padmanabhan, B.R.; Diene, S.M.; Lopez-Rojas, R.; Kempf, M.; Landraud, L.; Rolain, J.M. ARG-annot, a new bioinformatic tool to discover antibiotic resistance genes in bacterial genomes. Antimicrob. Agents Chemother. 2014, 58, 212–220. [Google Scholar] [CrossRef] [Green Version]
- Jia, B.; Raphenya, A.R.; Alcock, B.; Waglechner, N.; Guo, P.; Tsang, K.K.; Lago, B.A.; Dave, B.M.; Pereira, S.; Sharma, A.N.; et al. CARD 2017: expansion and model-centric curation of the comprehensive antibiotic resistance database. Nucleic Acids Res. 2017, 45, D566–D573. [Google Scholar] [CrossRef]
- Carattoli, A.; Zankari, E.; García-Fernández, A.; Larsen, M.V.; Lund, O.; Villa, L.; Aarestrup, F.M.; Hasman, H. In Silico Detection and Typing of Plasmids using PlasmidFinder and Plasmid Multilocus Sequence Typing. Antimicrob. Agents Chemother. 2014, 58, 3895–3903. [Google Scholar] [CrossRef] [Green Version]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: a fast phage search tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef]
- Zhao, X.; Yu, Z.; Xu, Z. Study the Features of 57 Confirmed CRISPR Loci in 38 Strains of Staphylococcus aureus. Front. Microbiol. 2018, 9, 1–14. [Google Scholar] [CrossRef] [Green Version]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol Biol Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef] [Green Version]
- Cosentino, S.; Larsen, M.V.; Aarestrup, F.M.; Lund, O. PathogenFinder - Distinguishing Friend from Foe Using Bacterial Whole Genome Sequence Data. PLOS ONE 2013, 8, e77302. [Google Scholar] [CrossRef]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: a web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef] [Green Version]
- Camacho, C.; Coulouris, G.; Avagyan, V.; Ma, N.; Papadopoulos, J.; Bealer, K.; Madden, T.L. BLAST+: Architecture and applications. BMC Bioinformat. 2009, 10, 421. [Google Scholar] [CrossRef] [Green Version]
- Liu, B.; Zheng, D.; Jin, Q.; Chen, L.; Yang, J. VFDB 2019: a comparative pathogenomic platform with an interactive web interface. Nucleic Acids Res. 2019, 47, D687–D692. [Google Scholar] [CrossRef]
- Ahrenfeldt, J.; Skaarup, C.; Hasman, H.; Pedersen, A.G.; Aarestrup, F.M.; Lund, O. Bacterial whole genome-based phylogeny: construction of a new benchmarking dataset and assessment of some existing methods. BMC Genom. 2017, 18, 19. [Google Scholar] [CrossRef] [Green Version]
- Hadfield, J.; Croucher, N.J.; Goater, R.J.; AbuDahab, K.; Aanensen, D.M.; Harris, S.R. Phandango: An interactive viewer for bacterial population genomics. Bioinformatics 2017, 34, 292–293. [Google Scholar] [CrossRef]


| Antibiotic | AST Profile | Minimum Inhibitory Concentrations (mg/L) |
|---|---|---|
| Amoxicillin-clavulanic acid | R | (32/16) |
| Piperacillin-tazobactam | R | (128/4) |
| Cefuroxime | R | (32) |
| Cefotaxime | R | (4) |
| Ceftriaxone | R | (4) |
| Ceftazidime | R | (16) |
| Cefepime | R | (16) |
| Cefoxitin | R | (32) |
| Imipenem | R | (4) |
| Meropenem | R | (4) |
| Ciprofloxacin | R | (1) |
| Gentamicin | R | (16) |
| Amikacin | R | (64) |
| Nitrofurantoin | R | (128) |
| Trimethoprim-sulfamethoxazole | R | (4/76) |
| Tigecycline | S | (2*) |
| Antibiotic class | Genes | Genomic location |
|---|---|---|
| β-lactams | blaNDM-1, blaCMY-48, blaCTX-M-15, blaOXA-10, OXA-1, blaTEM-1B | Plasmid |
| Aminoglycosides | aph(3')-Ia, aac(6')-Ib-cr, aac(3)-Iia, aph(6)-Id, aadA1, aac(3)-Iid, rmtC, aph(3'')-Ib | Plasmid |
| Fluroquinolones | aac(6')-Ib-cr, qnrB1, GyrA(S83I) | Plasmid and Chromosome (GyrA) |
| Fosfomycin | fosA | Chromosome |
| Trimethoprim | dfrA23, dfrA7, dfrA14 | Plasmid |
| Rifampicin | ARR-2 | Plasmid |
| Phenicols | catB3, cmlA1 | Plasmid |
| Sulphonamides | sul2, sul1 | Plasmid |
| Tetracycline | tet(A) | Plasmid |
| Efflux pump systems | ||
| ATP-binding cassette (ABC) | msbA | Chromosome |
| Major facilitator superfamily (MFS) | mdfA, mdtG | Chromosome |
| Resistance– nodulation–division (RND) | marA, H-NS, mdtC, baeR, acrA, acrB, CRP | Chromosome |
| No. | Putative Virulence Factors | Genes | Organisms [Highest Homology] |
|---|---|---|---|
| 1 | Fimbrial adherence determinants | csgA, csgB, csgC, csgD, csgE, csgF, csgG fimA, fimC, fimD, fimF, fimH, fimI, fimW, fimZ, lpfC, pegB, staB, staC, stcA, stcC, stgA, stgB, stkA, stkB, stkC, stkD, stkE and StkF | Salmonella enterica |
| 2 | Nonfimbrial adherence determinants | misL, ratB, shdA and sinH | Salmonella enterica |
| 3 | Regulation | phoP and phoQ | Salmonella typhimurium |
| 4 | Toxin | usp | Escherichia coli |
| 5 | Motility | flaA | Bordetella bronchiseptica |
| 6 | Antiphagocytosis | uge | Klebsiella pneumoniae |
| 7 | Invasion | ibeB | Escherichia coli |
| 8 | Biofilm formation | pgaC | Acinetobacter baumannii |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ramsamy, Y.; Mlisana, K.P.; Amoako, D.G.; Allam, M.; Ismail, A.; Singh, R.; Abia, A.L.K.; Essack, S.Y. Pathogenomic Analysis of a Novel Extensively Drug-Resistant Citrobacter freundii Isolate Carrying a blaNDM-1 Carbapenemase in South Africa. Pathogens 2020, 9, 89. https://doi.org/10.3390/pathogens9020089
Ramsamy Y, Mlisana KP, Amoako DG, Allam M, Ismail A, Singh R, Abia ALK, Essack SY. Pathogenomic Analysis of a Novel Extensively Drug-Resistant Citrobacter freundii Isolate Carrying a blaNDM-1 Carbapenemase in South Africa. Pathogens. 2020; 9(2):89. https://doi.org/10.3390/pathogens9020089
Chicago/Turabian StyleRamsamy, Yogandree, Koleka P. Mlisana, Daniel G. Amoako, Mushal Allam, Arshad Ismail, Ravesh Singh, Akebe Luther King Abia, and Sabiha Y. Essack. 2020. "Pathogenomic Analysis of a Novel Extensively Drug-Resistant Citrobacter freundii Isolate Carrying a blaNDM-1 Carbapenemase in South Africa" Pathogens 9, no. 2: 89. https://doi.org/10.3390/pathogens9020089
APA StyleRamsamy, Y., Mlisana, K. P., Amoako, D. G., Allam, M., Ismail, A., Singh, R., Abia, A. L. K., & Essack, S. Y. (2020). Pathogenomic Analysis of a Novel Extensively Drug-Resistant Citrobacter freundii Isolate Carrying a blaNDM-1 Carbapenemase in South Africa. Pathogens, 9(2), 89. https://doi.org/10.3390/pathogens9020089