Pathogenic Prion Protein Isoforms Are Not Present in Cerebral Organoids Generated from Asymptomatic Donors Carrying the E200K Mutation Associated with Familial Prion Disease

 , ,
, ,
Abstract
:1. Introduction
2. Results
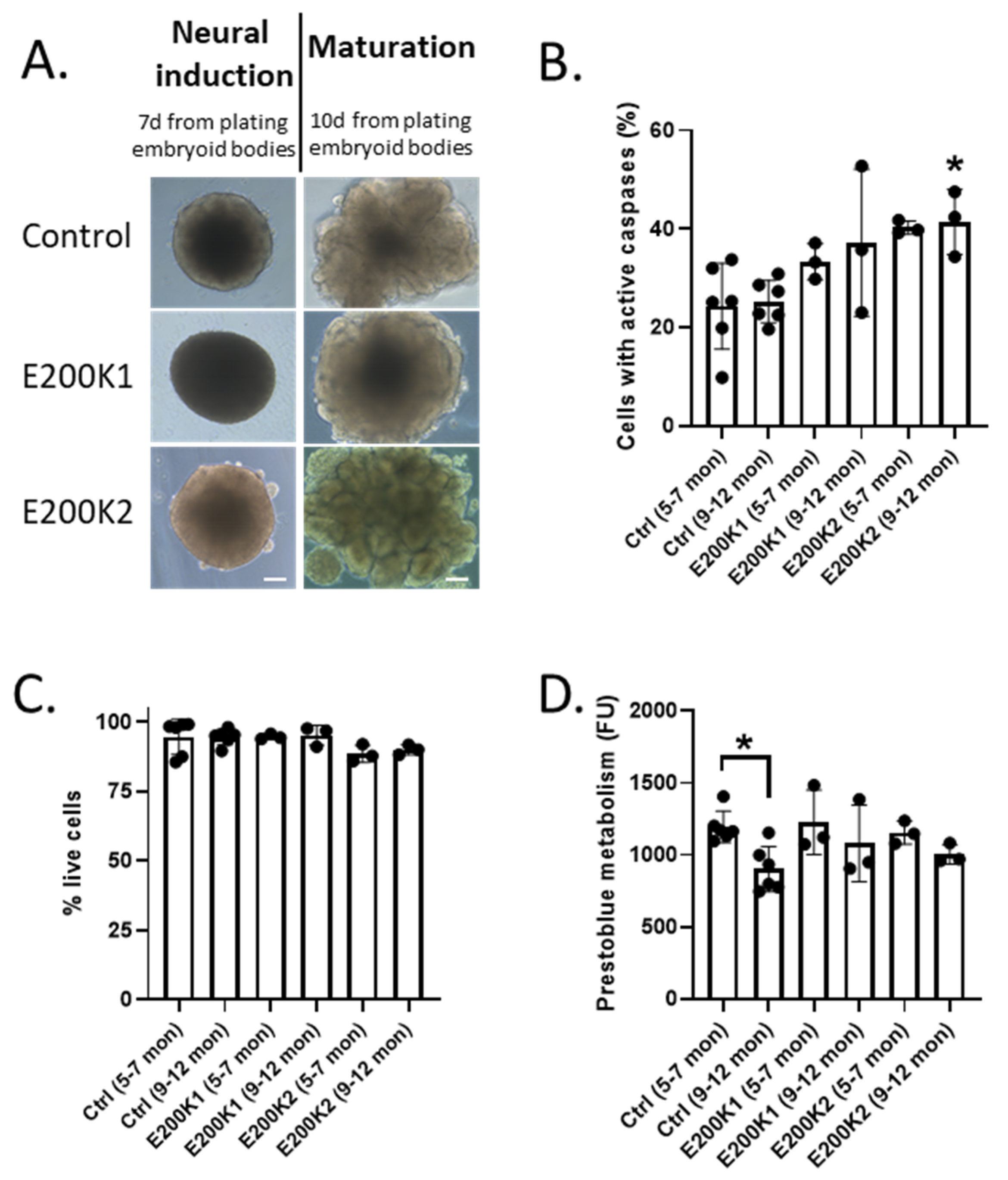
2.1. E200K Organoid Viability
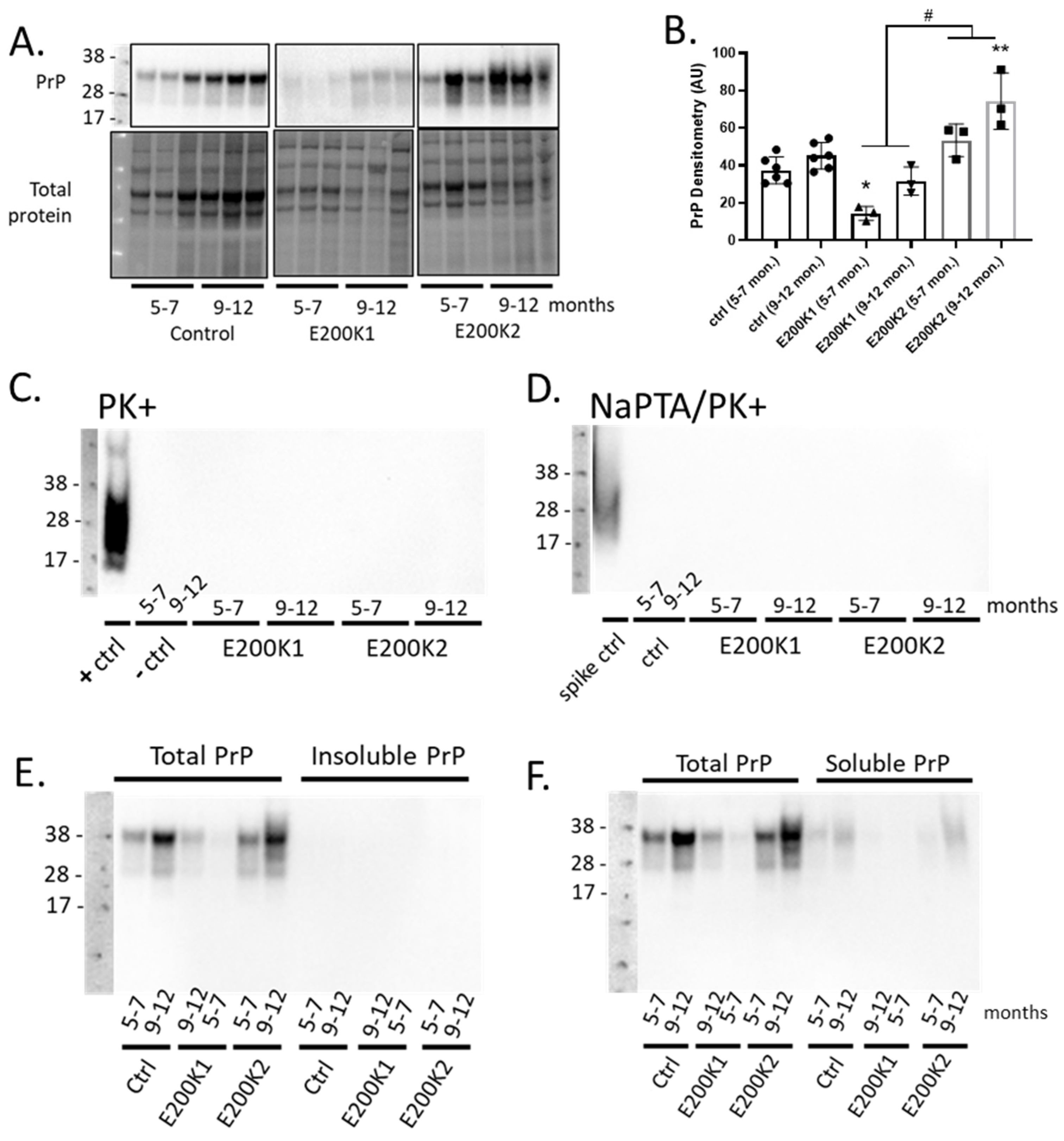
2.2. Expression of PrP Is Different between the Two Lines of E200K Organoids
2.3. E200K COs Do Not Harbor Insoluble and PK-Resistant PrPD
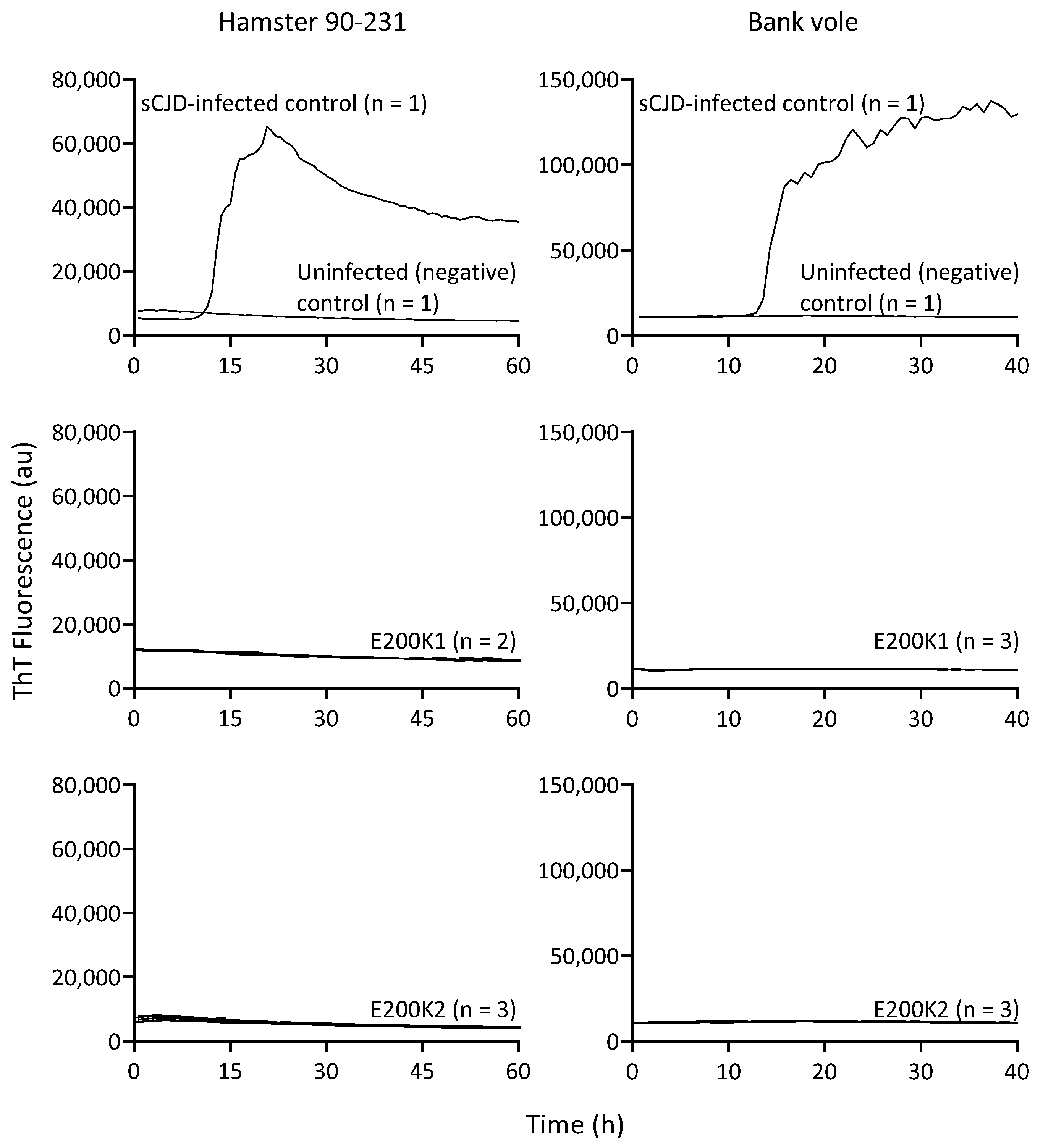
2.4. No PrPD Seeding Activity in E200k Organoid Up to 12 Months Old
3. Discussion
4. Conclusions
5. Materials and Methods
5.1. Human Ethics Statement
5.2. ATCC Human Induced Pluripotent Stem Cells (hiPSCs)
5.3. Generation of E200K(1), E200K(2) and RAH019A hiPSCs
5.4. hiPSC Culture
5.5. Organoid Generation
5.6. Organoid Culture
5.7. Organoid Images
5.8. RT-QuIC
5.9. Sodium Phosphotungstic Acid Precipitation
5.10. Detergent Insolubility Assay
5.11. Active Multicaspase Assay
5.12. Prestoblue Analysis
5.13. PK Digest and Western Blotting
5.14. Data Analysis
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Kim, M.O.; Takada, L.T.; Wong, K.; Forner, S.A.; Geschwind, M.D. Genetic PrP Prion Diseases. Cold Spring Harb. Perspect. Biol. 2018, 10. [Google Scholar] [CrossRef] [PubMed]
- Sandberg, M.K.; Al-Doujaily, H.; Sharps, B.; De Oliveira, M.W.; Schmidt, C.; Richard-Londt, A.; Lyall, S.; Linehan, J.M.; Brandner, S.; Wadsworth, J.D.; et al. Prion neuropathology follows the accumulation of alternate prion protein isoforms after infective titre has peaked. Nat Commun. 2014, 5, 4347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Brazier, M.W.; Lewis, V.; Ciccotosto, G.D.; Klug, G.M.; Lawson, V.A.; Cappai, R.; Ironside, J.W.; Masters, C.L.; Hill, A.F.; White, A.R.; et al. Correlative studies support lipid peroxidation is linked to PrP(res) propagation as an early primary pathogenic event in prion disease. Brain Res. Bull. 2006, 68, 346–354. [Google Scholar] [CrossRef] [PubMed]
- Alibhai, J.; Blanco, R.A.; Barria, M.A.; Piccardo, P.; Caughey, B.; Perry, V.H.; Freeman, T.C.; Manson, J.C. Distribution of Misfolded Prion Protein Seeding Activity Alone Does Not Predict Regions of Neurodegeneration. PLoS Biol. 2016, 14, e1002579. [Google Scholar] [CrossRef]
- Friedman-Levi, Y.; Meiner, Z.; Canello, T.; Frid, K.; Kovacs, G.G.; Budka, H.; Avrahami, D.; Gabizon, R. Fatal prion disease in a mouse model of genetic E200K Creutzfeldt-Jakob disease. PLoS Pathog. 2011, 7, e1002350. [Google Scholar] [CrossRef] [Green Version]
- Asante, E.A.; Gowland, I.; Grimshaw, A.; Linehan, J.M.; Smidak, M.; Houghton, R.; Osiguwa, O.; Tomlinson, A.; Joiner, S.; Brandner, S.; et al. Absence of spontaneous disease and comparative prion susceptibility of transgenic mice expressing mutant human prion proteins. J. Gen. Virol. 2009, 90, 546–558. [Google Scholar] [CrossRef]
- Groveman, B.R.; Foliaki, S.T.; Orru, C.D.; Zanusso, G.; Carroll, J.A.; Race, B.; Haigh, C.L. Sporadic Creutzfeldt-Jakob disease prion infection of human cerebral organoids. Acta Neuropathol. Commun. 2019, 7, 90. [Google Scholar] [CrossRef] [Green Version]
- Gonzalez, C.; Armijo, E.; Bravo-Alegria, J.; Becerra-Calixto, A.; Mays, C.E.; Soto, C. Modeling amyloid beta and tau pathology in human cerebral organoids. Mol. Psychiatry 2018, 23, 2363–2374. [Google Scholar] [CrossRef] [PubMed]
- Renner, M.; Lancaster, M.A.; Bian, S.; Choi, H.; Ku, T.; Peer, A.; Chung, K.; Knoblich, J.A. Self-organized developmental patterning and differentiation in cerebral organoids. EMBO J. 2017, 36, 1316–1329. [Google Scholar] [CrossRef]
- Carroll, J.A.; Chesebro, B. Neuroinflammation, Microglia, and Cell-Association during Prion Disease. Viruses 2019, 11, 65. [Google Scholar] [CrossRef] [Green Version]
- Trujillo, C.A.; Gao, R.; Negraes, P.D.; Gu, J.; Buchanan, J.; Preissl, S.; Wang, A.; Wu, W.; Haddad, G.G.; Chaim, I.A.; et al. Complex Oscillatory Waves Emerging from Cortical Organoids Model Early Human Brain Network Development. Cell Stem. Cell 2019, 25, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Shalini, S.; Dorstyn, L.; Dawar, S.; Kumar, S. Old, new and emerging functions of caspases. Cell Death Differ. 2015, 22, 526–539. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Foliaki, S.T.; Lewis, V.; Islam, A.M.T.; Ellett, L.J.; Senesi, M.; Finkelstein, D.I.; Roberts, B.; Lawson, V.A.; Adlard, P.A.; Collins, S.J. Early existence and biochemical evolution characterise acutely synaptotoxic PrPSc. PLoS Pathog. 2019, 15, e1007712. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kovacs, G.G.; Seguin, J.; Quadrio, I.; Hoftberger, R.; Kapas, I.; Streichenberger, N.; Biacabe, A.G.; Meyronet, D.; Sciot, R.; Vandenberghe, R.; et al. Genetic Creutzfeldt-Jakob disease associated with the E200K mutation: Characterization of a complex proteinopathy. Acta Neuropathol. 2011, 121, 39–57. [Google Scholar] [CrossRef] [PubMed]
- Bessen, R.A.; Kocisko, D.A.; Raymond, G.J.; Nandan, S.; Lansbury, P.T.; Caughey, B. Non-genetic propagation of strain-specific properties of scrapie prion protein. Nature 1995, 375, 698–700. [Google Scholar] [CrossRef] [PubMed]
- Telling, G.C.; Parchi, P.; DeArmond, S.J.; Cortelli, P.; Montagna, P.; Gabizon, R.; Mastrianni, J.; Lugaresi, E.; Gambetti, P.; Prusiner, S.B. Evidence for the conformation of the pathologic isoform of the prion protein enciphering and propagating prion diversity. Science (New York, N.Y.) 1996, 274, 2079–2082. [Google Scholar] [CrossRef] [Green Version]
- Wilham, J.M.; Orru, C.D.; Bessen, R.A.; Atarashi, R.; Sano, K.; Race, B.; Meade-White, K.D.; Taubner, L.M.; Timmes, A.; Caughey, B. Rapid end-point quantitation of prion seeding activity with sensitivity comparable to bioassays. PLoS Pathog. 2010, 6, e1001217. [Google Scholar] [CrossRef] [Green Version]
- Atarashi, R.; Sano, K.; Satoh, K.; Nishida, N. Real-time quaking-induced conversion: A highly sensitive assay for prion detection. Prion 2011, 5, 150–153. [Google Scholar] [CrossRef] [Green Version]
- Peden, A.H.; McGuire, L.I.; Appleford, N.E.J.; Mallinson, G.; Wilham, J.M.; Orrú, C.D.; Caughey, B.; Ironside, J.W.; Knight, R.S.; Will, R.G.; et al. Sensitive and specific detection of sporadic Creutzfeldt-Jakob disease brain prion protein using real-time quaking-induced conversion. J. Gen. Virol. 2012, 93, 438–449. [Google Scholar] [CrossRef]
- Orru, C.D.; Bongianni, M.; Tonoli, G.; Ferrari, S.; Hughson, A.G.; Groveman, B.R.; Fiorini, M.; Pocchiari, M.; Monaco, S.; Caughey, B.; et al. A test for Creutzfeldt-Jakob disease using nasal brushings. N. Engl. J. Med. 2014, 371, 519–529. [Google Scholar] [CrossRef] [Green Version]
- Orrù, C.D.; Wilham, J.M.; Vascellari, S.; Hughson, A.G.; Caughey, B. New generation QuIC assays for prion seeding activity. Prion 2012, 6, 147–152. [Google Scholar] [CrossRef] [Green Version]
- Orru, C.D.; Groveman, B.R.; Raymond, L.D.; Hughson, A.G.; Nonno, R.; Zou, W.; Ghetti, B.; Gambetti, P.; Caughey, B. Bank Vole Prion Protein As an Apparently Universal Substrate for RT-QuIC-Based Detection and Discrimination of Prion Strains. PLoS Pathog. 2015, 11, e1004983. [Google Scholar] [CrossRef]
- Whitehouse, I.J.; Miners, J.S.; Glennon, E.B.; Kehoe, P.G.; Love, S.; Kellett, K.A.; Hooper, N.M. Prion protein is decreased in Alzheimer’s brain and inversely correlates with BACE1 activity, amyloid-beta levels and Braak stage. PLoS ONE 2013, 8, e59554. [Google Scholar] [CrossRef] [PubMed]
- Lapucci, C.; Romano, N.; Boffa, G.; Saitta, L.; Nobili, F.; Mancardi, G.L.; Mandich, P.; Grandis, M. E200k Familial Creutzfeldt-Jakob Disease Presenting with Subacute Multiple Cranial Neuropathy. Open Neurol. J. 2019, 13, 72–75. [Google Scholar] [CrossRef] [Green Version]
- Poleggi, A.; van der Lee, S.; Capellari, S.; Puopolo, M.; Ladogana, A.; De Pascali, E.; Lia, D.; Formato, A.; Bartoletti-Stella, A.; Parchi, P.; et al. Age at onset of genetic (E200K) and sporadic Creutzfeldt-Jakob diseases is modulated by the CYP4X1 gene. J. Neurol. Neurosurg. Psychiatry 2018, 89, 1243–1249. [Google Scholar] [CrossRef] [PubMed]
- Frid, K.; Binyamin, O.; Fainstein, N.; Keller, G.; Ben-Hur, T.; Gabizon, R. Autologous neural progenitor cell transplantation into newborn mice modeling for E200K genetic prion disease delays disease progression. Neurobiol. Aging 2018, 65, 192–200. [Google Scholar] [CrossRef]
- Capellari, S.; Parchi, P.; Russo, C.M.; Sanford, J.; Sy, M.-S.; Gambetti, P.; Petersen, R.B. Effect of the E200K Mutation on Prion Protein Metabolism. Am. J. Pathol. 2000, 157, 613–622. [Google Scholar] [CrossRef]
- Rosenmann, H.; Talmor, G.; Halimi, M.; Yanai, A.; Gabizon, R.; Meiner, Z. Prion protein with an E200K mutation displays properties similar to those of the cellular isoform PrP(C). J. Neurochem. 2001, 76, 1654–1662. [Google Scholar] [CrossRef] [PubMed]
- Matamoros-Angles, A.; Gayosso, L.M.; Richaud-Patin, Y.; di Domenico, A.; Vergara, C.; Hervera, A.; Sousa, A.; Fernandez-Borges, N.; Consiglio, A.; Gavin, R.; et al. iPS Cell Cultures from a Gerstmann-Straussler-Scheinker Patient with the Y218N PRNP Mutation Recapitulate tau Pathology. Mol. Neurobiol. 2018, 55, 3033–3048. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mammana, A.; Baiardi, S.; Rossi, M.; Franceschini, A.; Donadio, V.; Capellari, S.; Caughey, B.; Parchi, P. Detection of prions in skin punch biopsies of Creutzfeldt-Jakob disease patients. Ann. Clin. Transl. Neurol. 2020, 7, 559–564. [Google Scholar] [CrossRef] [PubMed]
- Orrú, C.D.; Yuan, J.; Appleby, B.S.; Li, B.; Li, Y.; Winner, D.; Wang, Z.; Zhan, Y.A.; Rodgers, M.; Rarick, J.; et al. Prion seeding activity and infectivity in skin samples from patients with sporadic Creutzfeldt-Jakob disease. Sci. Transl. Med. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, Z.; Manca, M.; Foutz, A.; Camacho, M.V.; Raymond, G.J.; Race, B.; Orru, C.D.; Yuan, J.; Shen, P.; Li, B.; et al. Early preclinical detection of prions in the skin of prion-infected animals. Nat. Commun. 2019, 10, 247. [Google Scholar] [CrossRef] [PubMed]
- Lancaster, M.A.; Knoblich, J.A. Generation of cerebral organoids from human pluripotent stem cells. Nat. Protoc. 2014, 9, 2329–2340. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Orru, C.D.; Hughson, A.G.; Groveman, B.R.; Campbell, K.J.; Anson, K.J.; Manca, M.; Kraus, A.; Caughey, B. Factors That Improve RT-QuIC Detection of Prion Seeding Activity. Viruses 2016, 8, 140. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wiegmans, A.P.; Saunus, J.M.; Ham, S.; Lobb, R.; Kutasovic, J.R.; Dalley, A.J.; Miranda, M.; Atkinson, C.; Foliaki, S.T.; Ferguson, K.; et al. Secreted cellular prion protein binds doxorubicin and correlates with anthracycline resistance in breast cancer. JCI Insight 2019, 5. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saijo, E.; Ghetti, B.; Zanusso, G.; Oblak, A.; Furman, J.L.; Diamond, M.I.; Kraus, A.; Caughey, B. Ultrasensitive and selective detection of 3-repeat tau seeding activity in Pick disease brain and cerebrospinal fluid. Acta Neuropathol. 2017, 133, 751–765. [Google Scholar] [CrossRef]
- Foliaki, S.T.; Lewis, V.; Finkelstein, D.I.; Lawson, V.; Coleman, H.A.; Senesi, M.; Islam, A.M.T.; Chen, F.; Sarros, S.; Roberts, B.; et al. Prion acute synaptotoxicity is largely driven by protease-resistant PrPSc species. PLoS Pathog. 2018, 14, e1007214. [Google Scholar] [CrossRef] [Green Version]




{kind=link}
{kind=link}
{kind=link}
| Days Old | Substrate | RT-QuIC Positive | |
|---|---|---|---|
| E200K1 | E200K2 | ||
| 35 | bank vole | 0/3 | ND |
| 84 | hamster 90–231 | 0/3 | ND |
| 112 | hamster 90–231 | 0/3 | ND |
| 140 | bank vole | 0/3 | ND |
| 180 | hamster 90–231 | ND | 0/3 |
| >365 | hamster 90–231 | 0/2 | 0/3 |
| >365 | bank vole | 0/3 | 0/3 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Foliaki, S.T.; Groveman, B.R.; Yuan, J.; Walters, R.; Zhang, S.; Tesar, P.; Zou, W.; Haigh, C.L. Pathogenic Prion Protein Isoforms Are Not Present in Cerebral Organoids Generated from Asymptomatic Donors Carrying the E200K Mutation Associated with Familial Prion Disease. Pathogens 2020, 9, 482. https://doi.org/10.3390/pathogens9060482
Foliaki ST, Groveman BR, Yuan J, Walters R, Zhang S, Tesar P, Zou W, Haigh CL. Pathogenic Prion Protein Isoforms Are Not Present in Cerebral Organoids Generated from Asymptomatic Donors Carrying the E200K Mutation Associated with Familial Prion Disease. Pathogens. 2020; 9(6):482. https://doi.org/10.3390/pathogens9060482
Chicago/Turabian StyleFoliaki, Simote T., Bradley R. Groveman, Jue Yuan, Ryan Walters, Shulin Zhang, Paul Tesar, Wenquan Zou, and Cathryn L. Haigh. 2020. "Pathogenic Prion Protein Isoforms Are Not Present in Cerebral Organoids Generated from Asymptomatic Donors Carrying the E200K Mutation Associated with Familial Prion Disease" Pathogens 9, no. 6: 482. https://doi.org/10.3390/pathogens9060482
APA StyleFoliaki, S. T., Groveman, B. R., Yuan, J., Walters, R., Zhang, S., Tesar, P., Zou, W., & Haigh, C. L. (2020). Pathogenic Prion Protein Isoforms Are Not Present in Cerebral Organoids Generated from Asymptomatic Donors Carrying the E200K Mutation Associated with Familial Prion Disease. Pathogens, 9(6), 482. https://doi.org/10.3390/pathogens9060482