
Complete Genome Sequence, Molecular Characterization and Phylogenetic Relationships of a Novel Tern Atadenovirus


 , , ,
, , ,  and
and
Abstract
:1. Introduction
2. Materials and Methods
2.1. Sampling
2.2. Sample Preparation and Sequencing
2.3. Assembly and Genome Annotation
2.4. Phylogenetic Analysis
2.5. Comparative Analysis
2.6. Species Delimitation
2.7. Codon-Based Analysis of Positive Selection
2.8. Protein 3D Structure Prediction
3. Results
3.1. Genome of TeAdV-1 and Comparative Analyses
3.2. Evolutionary Relationships of TAdV-1
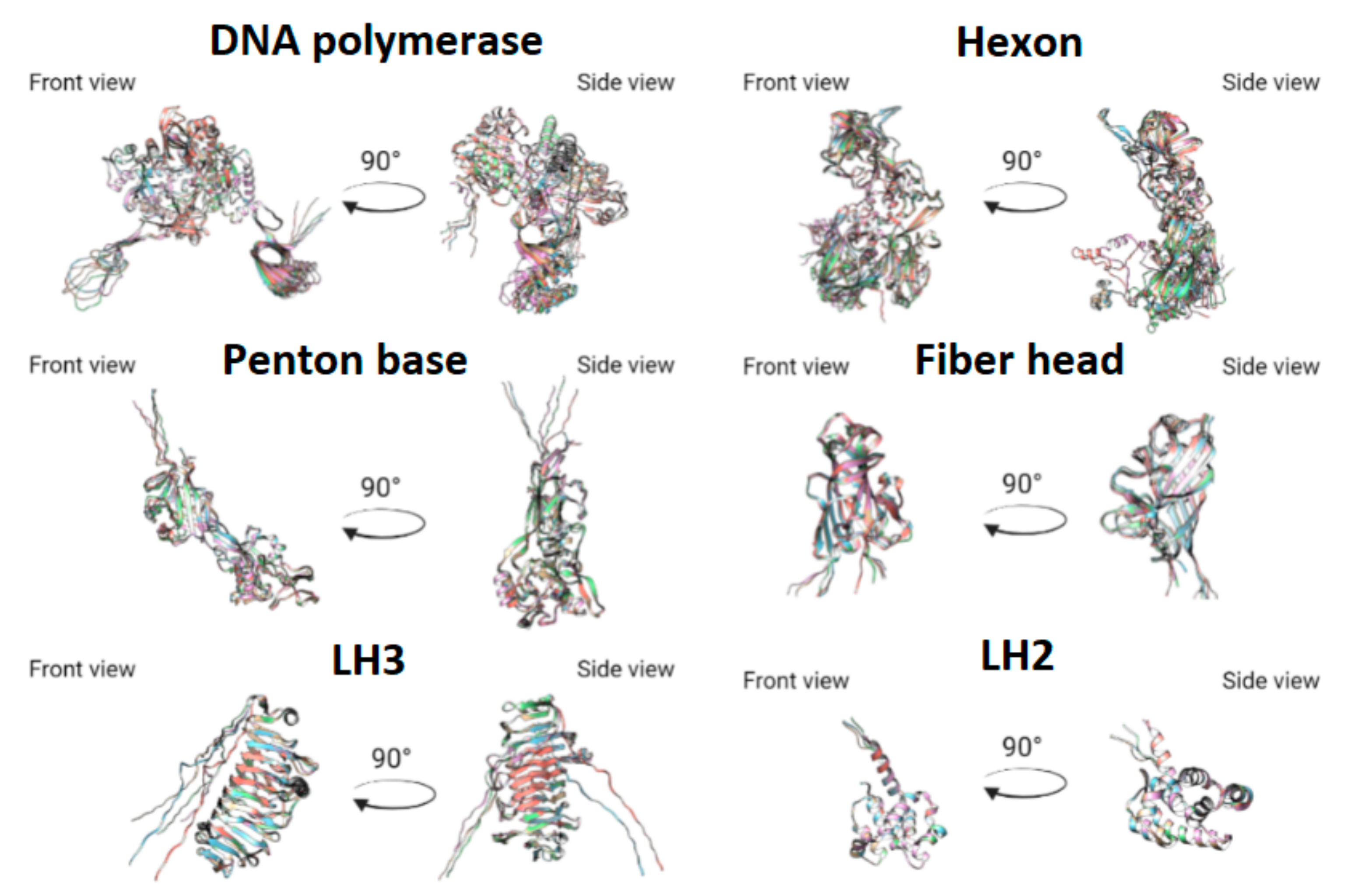
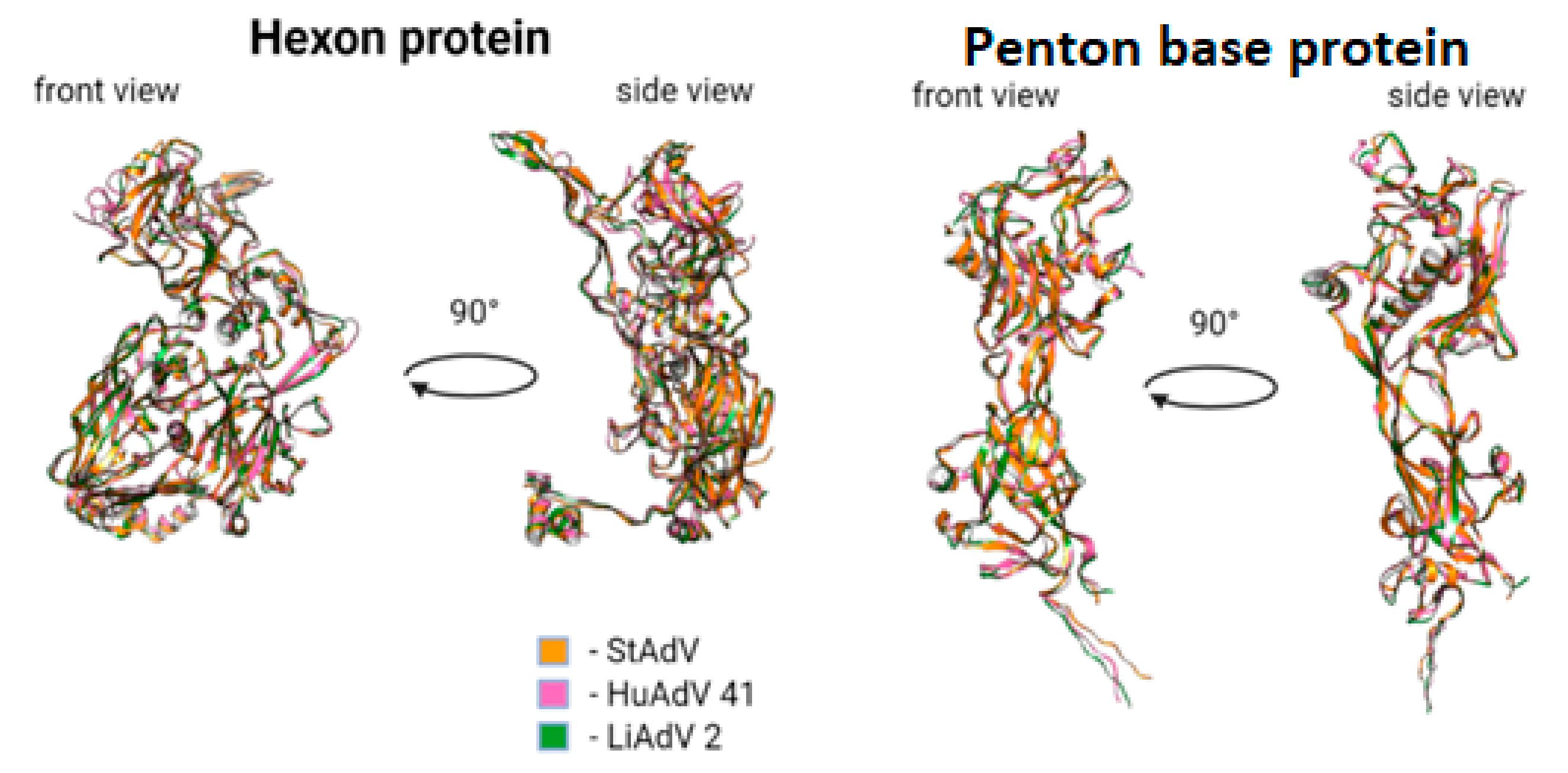
3.3. Protein Prediction
- Absence of alpha-helix, corresponding to Tyr288-Val285 in HAdV-41, lacking in TAdV, lacking in LAdV-2;
- Elongation of alpha-helix Val138-Asn159 (21 aa) in TAdV, which corresponds to Glu173-Ala183 (10 aa) in HAdV-41 and Val138-Gly157 (19 aa) in LAdV-2;
- Presence of beta-sheet-like short structure at Gly208-Asp210 in TAdV, lacking in HAdV-41 and LAdV-2 alike.
- TAdV alpha-helix Thr41-Ser46, presented in HAdV-41 as Asn72-Ala75, lacking in LAdV-2.
- Presence of structure Val233-Leu235 beta-sheet to Tyr236-Ile239 alpha helix, presented in StAdV which is absent in HAdV-41 and LAdV-2.
- Presence of two beta-sheets Glu380-Gly382, Ala400-Ile402, absent in HAdV-41 and LAdV-2;
- Elongation of beta-sheet Gln816-Cys824 (8 aa), corresponding to Val816 –Val823 (7 aa) in LAdV-2 and Ser831-Lys836 (5 aa)
- Presence of beta-sheet Gln229-Leu233, absent in LAdV-2 and HAdV-41.
- Elongation of beta-sheet Ser187-Ile197 (10 aa), corresponding to Arg201-Ile203 (2 aa) in HAdV-41 and absent in LAdV-2
- Presence of alpha-helix Val153-Lys157, absent in HAdV-41 and LAdV-2.
- Elongation of beta-sheet Cys269-Gly273 (4 aa), corresponding to Arg263-Thr265 (2 aa) in LAdV-2 and absent in HAdV-41.
3.4. Detection of Adaptive Evolution Events
3.4.1. Pervasive Positive Selection in the Molecular Evolution of Atadenovirus
3.4.2. Episodic Positive Selection in the Molecular Evolution of TAtV-1
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Harrach, B.; Tarján, Z.L.; Benkő, M. Adenoviruses across the animal kingdom: A walk in the zoo. FEBS Lett. 2019, 593, 3660–3673. [Google Scholar] [CrossRef] [PubMed]
- Lefkowitz, E.J.; Dempsey, D.M.; Hendrickson, R.C.; Orton, R.J.; Siddell, S.G.; Smith, D.B. Virus taxonomy: The database of the International Committee on Taxonomy of Viruses (ICTV). Nucleic Acids Res. 2018, 46, D708–D717. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Benkő, M.; Aoki, K.; Arnberg, N.; Davison, A.J. ICTV Virus Taxonomy Profile: Adenoviridae. J. Gen. Virol. 2021; in press. ISBN 978-0-12-384684-6. [Google Scholar]
- Davison, A.J.; Benko, M.; Harrach, B. Genetic content and evolution of adenoviruses. J. Gen. Virol. 2003, 84, 2895–2908. [Google Scholar] [CrossRef]
- Gorman, J.J.; Wallis, T.P.; Whelan, D.A.; Shaw, J.; Both, G.W. LH3, a “homologue” of the mastadenoviral E1B 55-kDa protein is a structural protein of atadenoviruses. Virology 2005, 342, 159–166. [Google Scholar] [CrossRef] [Green Version]
- Gerlach, H. Viruses. In Avian Medicine: Principles and Application; Ritchie, B.W., Harrison, G.J., Harrison, L.R., Eds.; Wingers Publishing Inc.: Lake Worth, FL, USA, 1994; pp. 862–948. ISBN 978-096369960. [Google Scholar]
- Borkenhagen, L.K.; Fieldhouse, J.K.; Seto, D.; Gray, G.C. Are adenoviruses zoonotic? A systematic review of the evidence. Emerg. Microbes Infect. 2019, 8, 1679–1687. [Google Scholar] [CrossRef] [Green Version]
- Li, K.S.; Guan, Y.; Wang, J.; Smith, G.J.D.; Xu, K.M.; Duan, L.; Rahardjo, A.P.; Puthavathana, P.; Buranathai, C.; Nguyen, T.D.; et al. Genesis of a highly pathogenic and potentially pandemic H5N1 influenza virus in eastern Asia. Nature 2004, 430, 209–213. [Google Scholar] [CrossRef]
- World Health Organization Pandemic H1N1. 2009. Available online: http://apps.who.int/iris/bitstream/handle/10665/78414/9789241503051_eng.pdf?sequence=1 (accessed on 30 November 2021).
- Neumann, G.; Noda, T.; Kawaoka, Y. Emergence and pandemic potential of swine-origin H1N1 influenza virus. Nature 2009, 459, 931–939. [Google Scholar] [CrossRef] [Green Version]
- World Health Organization Statement on the second meeting of the International Health Regulations. 2020. Available online: https://www.who.int/news/item/30-01-2020-statement-on-the-second-meeting-of-the-international-health-regulations-(2005)-emergency-committee-regarding-the-outbreak-of-novel-coronavirus-(2019-ncov) (accessed on 30 November 2021).
- Andersen, K.G.; Rambaut, A.; Lipkin, W.I.; Holmes, E.C.; Garry, R.F. The proximal origin of SARS-CoV-2. Nat. Med. 2020, 26, 450–452. [Google Scholar] [CrossRef] [Green Version]
- Fritzsche McKay, A.; Hoye, B.J. Are Migratory Animals Superspreaders of Infection?: An Introduction to the Symposium. Integr. Comp. Biol. 2016, 56, 260–267. [Google Scholar] [CrossRef] [Green Version]
- Ayginin, A.A.; Pimkina, E.V.; Matsvay, A.D.; Speranskaya, A.S.; Safonova, M.V.; Blinova, E.A.; Artyushin, I.V.; Dedkov, V.G.; Shipulin, G.A.; Khafizov, K. The Study of Viral RNA Diversity in Bird Samples Using De Novo Designed Multiplex Genus-Specific Primer Panels. Adv. Virol. 2018, 2018, 1–10. [Google Scholar] [CrossRef]
- Bankevich, A.; Nurk, S.; Antipov, D.; Gurevich, A.A.; Dvorkin, M.; Kulikov, A.S.; Lesin, V.M.; Nikolenko, S.I.; Pham, S.; Prjibelski, A.D.; et al. SPAdes: A New Genome Assembly Algorithm and Its Applications to Single-Cell Sequencing. J. Comput. Biol. 2012, 19, 455–477. [Google Scholar] [CrossRef] [Green Version]
- McGinnis, S.; Madden, T.L. BLAST: At the core of a powerful and diverse set of sequence analysis tools. Nucleic Acids Res. 2004, 32, W20–W25. [Google Scholar] [CrossRef]
- Federhen, S. The NCBI Taxonomy database. Nucleic Acids Res. 2012, 40, D136–D143. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinforma. Oxf. Engl. 2009, 25, 1754–1760. [Google Scholar] [CrossRef] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R.; 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinforma. Oxf. Engl. 2009, 25, 2078–2079. [Google Scholar] [CrossRef] [Green Version]
- Milne, I.; Stephen, G.; Bayer, M.; Cock, P.J.A.; Pritchard, L.; Cardle, L.; Shaw, P.D.; Marshall, D. Using Tablet for visual exploration of second-generation sequencing data. Brief. Bioinform. 2013, 14, 193–202. [Google Scholar] [CrossRef]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and open software for comparing large genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [Green Version]
- Zhang, K.-Y.; Gao, Y.-Z.; Du, M.-Z.; Liu, S.; Dong, C.; Guo, F.-B. Vgas: A Viral Genome Annotation System. Front. Microbiol. 2019, 10, 184. [Google Scholar] [CrossRef]
- Emms, D.M.; Kelly, S. OrthoFinder: Phylogenetic orthology inference for comparative genomics. Genome Biol. 2019, 20, 238. [Google Scholar] [CrossRef] [Green Version]
- Reese, M.G.; Eeckman, F.H.; Kulp, D.; Haussler, D. Improved Splice Site Detection in Genie. J. Comput. Biol. 1997, 4, 311–323. [Google Scholar] [CrossRef]
- Cock, P.J.A.; Antao, T.; Chang, J.T.; Chapman, B.A.; Cox, C.J.; Dalke, A.; Friedberg, I.; Hamelryck, T.; Kauff, F.; Wilczynski, B.; et al. Biopython: Freely available Python tools for computational molecular biology and bioinformatics. Bioinforma. Oxf. Engl. 2009, 25, 1422–1423. [Google Scholar] [CrossRef]
- Clark, K.; Karsch-Mizrachi, I.; Lipman, D.J.; Ostell, J.; Sayers, E.W. GenBank. Nucleic Acids Res. 2016, 44, D67–D72. [Google Scholar] [CrossRef] [Green Version]
- Larkin, M.A.; Blackshields, G.; Brown, N.P.; Chenna, R.; McGettigan, P.A.; McWilliam, H.; Valentin, F.; Wallace, I.M.; Wilm, A.; Lopez, R.; et al. Clustal W and Clustal X version 2.0. Bioinforma. Oxf. Engl. 2007, 23, 2947–2948. [Google Scholar] [CrossRef] [Green Version]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Lanfear, R.; Frandsen, P.B.; Wright, A.M.; Senfeld, T.; Calcott, B. PartitionFinder 2: New Methods for Selecting Partitioned Models of Evolution for Molecular and Morphological Phylogenetic Analyses. Mol. Biol. Evol. 2017, 34, 772–773. [Google Scholar] [CrossRef] [Green Version]
- Kozlov, A.M.; Darriba, D.; Flouri, T.; Morel, B.; Stamatakis, A. RAxML-NG: A fast, scalable and user-friendly tool for maximum likelihood phylogenetic inference. Bioinformatics 2019, 35, 4453–4455. [Google Scholar] [CrossRef] [Green Version]
- Rambaut, A. FigTree v. 1.4.3. 2018. Available online: http://tree.bio.ed.ac.uk/software/figtree/ (accessed on 30 November 2021).
- Abadi, S.; Azouri, D.; Pupko, T.; Mayrose, I. Model selection may not be a mandatory step for phylogeny reconstruction. Nat. Commun. 2019, 10, 934. [Google Scholar] [CrossRef] [Green Version]
- Spielman, S.J. Relative Model Fit Does Not Predict Topological Accuracy in Single-Gene Protein Phylogenetics. Mol. Biol. Evol. 2020, 37, 2110–2123. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive Tree Of Life (iTOL) v5: An online tool for phylogenetic tree display and annotation. Nucleic Acids Res. 2021, 49, W293–W296. [Google Scholar] [CrossRef] [PubMed]
- Katoh, K. MAFFT: A novel method for rapid multiple sequence alignment based on fast Fourier transform. Nucleic Acids Res. 2002, 30, 3059–3066. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhang, J.; Kapli, P.; Pavlidis, P.; Stamatakis, A. A general species delimitation method with applications to phylogenetic placements. Bioinformatics 2013, 29, 2869–2876. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fujisawa, T.; Barraclough, T.G. Delimiting Species Using Single-Locus Data and the Generalized Mixed Yule Coalescent Approach: A Revised Method and Evaluation on Simulated Data Sets. Syst. Biol. 2013, 62, 707–724. [Google Scholar] [CrossRef] [Green Version]
- Pons, J.; Barraclough, T.G.; Gomez-Zurita, J.; Cardoso, A.; Duran, D.P.; Hazell, S.; Kamoun, S.; Sumlin, W.D.; Vogler, A.P. Sequence-Based Species Delimitation for the DNA Taxonomy of Undescribed Insects. Syst. Biol. 2006, 55, 595–609. [Google Scholar] [CrossRef] [Green Version]
- Fontaneto, D.; Herniou, E.A.; Boschetti, C.; Caprioli, M.; Melone, G.; Ricci, C.; Barraclough, T.G. Independently Evolving Species in Asexual Bdelloid Rotifers. PLoS Biol. 2007, 5, e87. [Google Scholar] [CrossRef]
- Birky, C.W., Jr.; Ricci, C.; Melone, G.; Fontaneto, D. Integrating DNA and morphological taxonomy to describe diversity in poorly studied microscopic animals: New species of the genus Abrochtha Bryce, 1910 (Rotifera: Bdelloidea: Philodinavidae): NEW CRYPTIC ROTIFER SPECIES. Zool. J. Linn. Soc. 2011, 161, 723–734. [Google Scholar] [CrossRef] [Green Version]
- Yule, G.U. II.—A mathematical theory of evolution, based on the conclusions of Dr. J. C. Willis, F.R.S. Philos. Trans. R. Soc. Lond. Ser. B Contain. Pap. Biol. Character 1925, 213, 21–87. [Google Scholar] [CrossRef]
- Hudson, R. Gene genealogies and the coalescent process. Oxf. Surv. Evol. Biol. 1990, 7, 1–44. [Google Scholar]
- Tamura, K.; Battistuzzi, F.U.; Billing-Ross, P.; Murillo, O.; Filipski, A.; Kumar, S. Estimating divergence times in large molecular phylogenies. Proc. Natl. Acad. Sci. USA 2012, 109, 19333–19338. [Google Scholar] [CrossRef] [Green Version]
- Tamura, K.; Tao, Q.; Kumar, S. Theoretical Foundation of the RelTime Method for Estimating Divergence Times from Variable Evolutionary Rates. Mol. Biol. Evol. 2018, 35, 1770–1782. [Google Scholar] [CrossRef]
- Thomas, R.H. Molecular Evolution and Phylogenetics. Heredity 2001, 86, 385. [Google Scholar] [CrossRef]
- Puillandre, N.; Brouillet, S.; Achaz, G. ASAP: Assemble species by automatic partitioning. Mol. Ecol. Resour. 2021, 21, 609–620. [Google Scholar] [CrossRef]
- Boc, A.; Diallo, A.B.; Makarenkov, V. T-REX: A web server for inferring, validating and visualizing phylogenetic trees and networks. Nucleic Acids Res. 2012, 40, W573–W579. [Google Scholar] [CrossRef] [Green Version]
- Birky, C.W.; Adams, J.; Gemmel, M.; Perry, J. Using Population Genetic Theory and DNA Sequences for Species Detection and Identification in Asexual Organisms. PLoS ONE 2010, 5, e10609. [Google Scholar] [CrossRef] [Green Version]
- Kosakovsky Pond, S.L.; Posada, D.; Gravenor, M.B.; Woelk, C.H.; Frost, S.D.W. GARD: A genetic algorithm for recombination detection. Bioinformatics 2006, 22, 3096–3098. [Google Scholar] [CrossRef] [Green Version]
- Pond, S.L.K.; Muse, S.V. HyPhy: Hypothesis Testing Using Phylogenies. In Statistical Methods in Molecular Evolution; Springer: New York, NY, USA, 2005; pp. 125–181. ISBN 978-0-387-22333-9. [Google Scholar]
- Yang, Z. PAML 4: Phylogenetic Analysis by Maximum Likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [Green Version]
- Kosakovsky Pond, S.L.; Frost, S.D.W. Not So Different After All: A Comparison of Methods for Detecting Amino Acid Sites Under Selection. Mol. Biol. Evol. 2005, 22, 1208–1222. [Google Scholar] [CrossRef] [Green Version]
- Zhang, J. Evaluation of an Improved Branch-Site Likelihood Method for Detecting Positive Selection at the Molecular Level. Mol. Biol. Evol. 2005, 22, 2472–2479. [Google Scholar] [CrossRef] [Green Version]
- Jumper, J.; Evans, R.; Pritzel, A.; Green, T.; Figurnov, M.; Ronneberger, O.; Tunyasuvunakool, K.; Bates, R.; Žídek, A.; Potapenko, A.; et al. Highly accurate protein structure prediction with AlphaFold. Nature 2021, 596, 583–589. [Google Scholar] [CrossRef]
- Pettersen, E.F.; Goddard, T.D.; Huang, C.C.; Couch, G.S.; Greenblatt, D.M.; Meng, E.C.; Ferrin, T.E. UCSF Chimera?A visualization system for exploratory research and analysis. J. Comput. Chem. 2004, 25, 1605–1612. [Google Scholar] [CrossRef] [Green Version]
- Marabini, R.; Condezo, G.N.; Krupovic, M.; Menéndez-Conejero, R.; Gómez-Blanco, J.; San Martín, C. Near-atomic structure of an atadenovirus reveals a conserved capsid-binding motif and intergenera variations in cementing proteins. Sci. Adv. 2021, 7, eabe6008. [Google Scholar] [CrossRef]
- Kundhavai Natchiar, S.; Venkataraman, S.; Mullen, T.-M.; Nemerow, G.R.; Reddy, V.S. Revised Crystal Structure of Human Adenovirus Reveals the Limits on Protein IX Quasi-Equivalence and on Analyzing Large Macromolecular Complexes. J. Mol. Biol. 2018, 430, 4132–4141. [Google Scholar] [CrossRef]
- To, K.K.W.; Tse, H.; Chan, W.-M.; Choi, G.K.Y.; Zhang, A.J.X.; Sridhar, S.; Wong, S.C.Y.; Chan, J.F.W.; Chan, A.S.F.; Woo, P.C.Y.; et al. A Novel Psittacine Adenovirus Identified During an Outbreak of Avian Chlamydiosis and Human Psittacosis: Zoonosis Associated with Virus-Bacterium Coinfection in Birds. PLoS Negl. Trop. Dis. 2014, 8, e3318. [Google Scholar] [CrossRef]
- Harrach, B. Adenoviruses: General Features. In Reference Module in Biomedical Sciences; Elsevier: Amsterdam, The Netherlands, 2014; p. B978012801238302523X. ISBN 978-0-12-801238-3. [Google Scholar]
- Cepko, C.L.; Sharp, P.A. Analysis of Ad5 Hexon and 100K is mutants using conformation-specific monoclonal antibodies. Virology 1983, 129, 137–154. [Google Scholar] [CrossRef]
- Russell, W.C. Adenoviruses: Update on structure and function. J. Gen. Virol. 2009, 90, 1–20. [Google Scholar] [CrossRef]
- Kulanayake, S.; Tikoo, S.K. Adenovirus Core Proteins: Structure and Function. Viruses 2021, 13, 388. [Google Scholar] [CrossRef]
- Parker, E. Adenovirus DNA polymerase: Domain organisation and interaction with preterminal protein. Nucleic Acids Res. 1998, 26, 1240–1247. [Google Scholar] [CrossRef] [Green Version]
- Vellinga, J.; Van der Heijdt, S.; Hoeben, R.C. The adenovirus capsid: Major progress in minor proteins. J. Gen. Virol. 2005, 86, 1581–1588. [Google Scholar] [CrossRef]
- Duarte, M.A.; Silva, J.M.F.; Brito, C.R.; Teixeira, D.S.; Melo, F.L.; Ribeiro, B.M.; Nagata, T.; Campos, F.S. Faecal Virome Analysis of Wild Animals from Brazil. Viruses 2019, 11, 803. [Google Scholar] [CrossRef] [Green Version]
- Shriner, D.; Nickle, D.C.; Jensen, M.A.; Mullins, J.I. Potential impact of recombination on sitewise approaches for detecting positive natural selection. Genet. Res. 2003, 81, 115–121. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vrati, S.; Brookes, D.E.; Strike, P.; Khatri, A.; Boyle, D.B.; Both, G.W. Unique Genome Arrangement of an Ovine Adenovirus: Identification of New Proteins and Proteinase Cleavage Sites. Virology 1996, 220, 186–199. [Google Scholar] [CrossRef] [PubMed]
- Pénzes, J.J.; Szirovicza, L.; Harrach, B. The complete genome sequence of bearded dragon adenovirus 1 harbors three genes encoding proteins of the C-type lectin-like domain superfamily. Infect. Genet. Evol. 2020, 83, 104321. [Google Scholar] [CrossRef] [PubMed]
- Athukorala, A.; Forwood, J.K.; Phalen, D.N.; Sarker, S. Molecular Characterisation of a Novel and Highly Divergent Passerine Adenovirus 1. Viruses 2020, 12, 1036. [Google Scholar] [CrossRef]
- Benkö, M.; Harrach, B. A proposal for a new (third) genus within the family Adenoviridae. Arch. Virol. 1998, 143, 829–837. [Google Scholar] [CrossRef]
- Simón, D.; Cristina, J.; Musto, H. Nucleotide Composition and Codon Usage Across Viruses and Their Respective Hosts. Front. Microbiol. 2021, 12, 646300. [Google Scholar] [CrossRef]
- Maeda, K.; Hondo, E.; Terakawa, J.; Kiso, Y.; Nakaichi, N.; Endoh, D.; Sakai, K.; Morikawa, S.; Mizutani, T. Isolation of Novel Adenovirus from Fruit Bat (Pteropus dasymallus yayeyamae). Emerg. Infect. Dis. 2008, 14, 347–349. [Google Scholar] [CrossRef]
- Prado-Irwin, S.R.; van de Schoot, M.; Geneva, A.J. Detection and phylogenetic analysis of adenoviruses occurring in a single anole species. PeerJ 2018, 6, e5521. [Google Scholar] [CrossRef]
- Wellehan, J.F.X.; Greenacre, C.B.; Fleming, G.J.; Stetter, M.D.; Childress, A.L.; Terrell, S.P. Siadenovirus infection in two psittacine bird species. Avian Pathol. 2009, 38, 413–417. [Google Scholar] [CrossRef]
- Conrardy, C.; Tao, Y.; Kuzmin, I.V.; Niezgoda, M.; Agwanda, B.; Breiman, R.F.; Anderson, L.J.; Rupprecht, C.E.; Tong, S. Molecular Detection of Adenoviruses, Rhabdoviruses, and Paramyxoviruses in Bats from Kenya. Am. J. Trop. Med. Hyg. 2014, 91, 258–266. [Google Scholar] [CrossRef] [Green Version]
- Geisbert, T.W.; Bailey, M.; Hensley, L.; Asiedu, C.; Geisbert, J.; Stanley, D.; Honko, A.; Johnson, J.; Mulangu, S.; Pau, M.G.; et al. Recombinant Adenovirus Serotype 26 (Ad26) and Ad35 Vaccine Vectors Bypass Immunity to Ad5 and Protect Nonhuman Primates against Ebolavirus Challenge. J. Virol. 2011, 85, 4222–4233. [Google Scholar] [CrossRef] [Green Version]
- Adenoviridae. In Virus Taxonomy; Elsevier: Amsterdam, The Netherlands, 2012; pp. 125–141. ISBN 978-0-12-384684-6.
- Capesius, I.; Bopp, M. New classification of liverworts based on molecular and morphological data. Plant Syst. Evol. 1997, 207, 87–97. [Google Scholar] [CrossRef]
- Nei, M.; Kumar, S.; Takahashi, K. The optimization principle in phylogenetic analysis tends to give incorrect topologies when the number of nucleotides or amino acids used is small. Proc. Natl. Acad. Sci. USA 1998, 95, 12390–12397. [Google Scholar] [CrossRef] [Green Version]
- Poe, S.; Swofford, D.L. Taxon sampling revisited. Nature 1999, 398, 299–300. [Google Scholar] [CrossRef]
- Nickrent, D.L.; Parkinson, C.L.; Palmer, J.D.; Duff, R.J. Multigene Phylogeny of Land Plants with Special Reference to Bryophytes and the Earliest Land Plants. Mol. Biol. Evol. 2000, 17, 1885–1895. [Google Scholar] [CrossRef]
- Hervé, P. Opinion: Long Branch Attraction and Protist Phylogeny. Protist 2000, 151, 307–316. [Google Scholar] [CrossRef]
- Hoef-Emden, K.; Marin, B.; Melkonian, M. Nuclear and Nucleomorph SSU rDNA Phylogeny in the Cryptophyta and the Evolution of Cryptophyte Diversity. J. Mol. Evol. 2002, 55, 161–179. [Google Scholar] [CrossRef]
- Singh, G.; Robinson, C.M.; Dehghan, S.; Schmidt, T.; Seto, D.; Jones, M.S.; Dyer, D.W.; Chodosh, J. Overreliance on the Hexon Gene, Leading to Misclassification of Human Adenoviruses: Fig 1. J. Virol. 2012, 86, 4693–4695. [Google Scholar] [CrossRef] [Green Version]
- Hillis, D.M. Inferring complex phytogenies. Nature 1996, 383, 130–131. [Google Scholar] [CrossRef]
- Gontcharov, A.A. Are Combined Analyses Better Than Single Gene Phylogenies? A Case Study Using SSU rDNA and rbcL Sequence Comparisons in the Zygnematophyceae (Streptophyta). Mol. Biol. Evol. 2003, 21, 612–624. [Google Scholar] [CrossRef]
- Young, A.D.; Gillung, J.P. Phylogenomics—Principles, opportunities and pitfalls of big-data phylogenetics. Syst. Entomol. 2020, 45, 225–247. [Google Scholar] [CrossRef] [Green Version]
- Teng, J.L.L.; Tang, Y.; Huang, Y.; Guo, F.-B.; Wei, W.; Chen, J.H.K.; Wong, S.S.Y.; Lau, S.K.P.; Woo, P.C.Y. Phylogenomic Analyses and Reclassification of Species within the Genus Tsukamurella: Insights to Species Definition in the Post-genomic Era. Front. Microbiol. 2016, 7, 1137. [Google Scholar] [CrossRef] [Green Version]
- Mahadevan, P. An Analysis of Adenovirus Genomes Using Whole Genome Software Tools. Bioinformation 2016, 12, 301–310. [Google Scholar] [CrossRef] [Green Version]
- Konstantinidis, K.T.; Tiedje, J.M. Genomic insights that advance the species definition for prokaryotes. Proc. Natl. Acad. Sci. USA 2005, 102, 2567–2572. [Google Scholar] [CrossRef] [Green Version]
- Arrigoni, R.; Berumen, M.L.; Chen, C.A.; Terraneo, T.I.; Baird, A.H.; Payri, C.; Benzoni, F. Species delimitation in the reef coral genera Echinophyllia and Oxypora (Scleractinia, Lobophylliidae) with a description of two new species. Mol. Phylogenet. Evol. 2016, 105, 146–159. [Google Scholar] [CrossRef]
- Renner, M.A.M.; Heslewood, M.M.; Patzak, S.D.F.; Schäfer-Verwimp, A.; Heinrichs, J. By how much do we underestimate species diversity of liverworts using morphological evidence? An example from Australasian Plagiochila (Plagiochilaceae: Jungermanniopsida). Mol. Phylogenet. Evol. 2017, 107, 576–593. [Google Scholar] [CrossRef] [PubMed]
- Nieto-Montes de Oca, A.; Barley, A.J.; Meza-Lázaro, R.N.; García-Vázquez, U.O.; Zamora-Abrego, J.G.; Thomson, R.C.; Leaché, A.D. Phylogenomics and species delimitation in the knob-scaled lizards of the genus Xenosaurus (Squamata: Xenosauridae) using ddRADseq data reveal a substantial underestimation of diversity. Mol. Phylogenet. Evol. 2017, 106, 241–253. [Google Scholar] [CrossRef] [PubMed]
- Tang, C.Q.; Humphreys, A.M.; Fontaneto, D.; Barraclough, T.G. Effects of phylogenetic reconstruction method on the robustness of species delimitation using single-locus data. Methods Ecol. Evol. 2014, 5, 1086–1094. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ducasse, J.; Ung, V.; Lecointre, G.; Miralles, A. LIMES: A tool for comparing species partition. Bioinformatics 2020, 36, 2282–2283. [Google Scholar] [CrossRef] [PubMed]
- International Committee on Taxonomy of Viruses Virus Taxonomy: The ICTV Report on Virus Classification and Taxon Nomenclature. Available online: https://talk.ictvonline.org/ictv-reports/ictv_online_report/ (accessed on 30 November 2021).
- Spielman, S.J.; Weaver, S.; Shank, S.D.; Magalis, B.R.; Li, M.; Kosakovsky Pond, S.L. Evolution of Viral Genomes: Interplay Between Selection, Recombination, and Other Forces. In Evolutionary Genomics; Anisimova, M., Ed.; Springer: New York, NY, USA, 2019; Volume 1910, pp. 427–468. ISBN 978-1-4939-9073-3. [Google Scholar]
- Van Valen, L. Molecular evolution as predicted by natural selection. J. Mol. Evol. 1974, 3, 89–101. [Google Scholar] [CrossRef]
- Stenseth, N.; Smith, J.M. Coevolution in Ecosystems: Red Queen Evolution or Stasis? Evolution 1984, 38, 870. [Google Scholar] [CrossRef] [Green Version]
- Hedrick, P.W. Evolutionary Genetics of the Major Histocompatibility Complex. Am. Nat. 1994, 143, 945–964. [Google Scholar] [CrossRef]
- Obbard, D.J.; Jiggins, F.M.; Halligan, D.L.; Little, T.J. Natural Selection Drives Extremely Rapid Evolution in Antiviral RNAi Genes. Curr. Biol. 2006, 16, 580–585. [Google Scholar] [CrossRef] [Green Version]
- Drosophila 12 Genomes Consortium Evolution of genes and genomes on the Drosophila phylogeny. Nature 2007, 450, 203–218. [CrossRef]
- Blanc, G.; Ngwamidiba, M.; Ogata, H.; Fournier, P.-E.; Claverie, J.-M.; Raoult, D. Molecular Evolution of Rickettsia Surface Antigens: Evidence of Positive Selection. Mol. Biol. Evol. 2005, 22, 2073–2083. [Google Scholar] [CrossRef] [Green Version]
- Mu, J.; Awadalla, P.; Duan, J.; McGee, K.M.; Keebler, J.; Seydel, K.; McVean, G.A.T.; Su, X. Genome-wide variation and identification of vaccine targets in the Plasmodium falciparum genome. Nat. Genet. 2007, 39, 126–130. [Google Scholar] [CrossRef]
- Barrett, L.G.; Thrall, P.H.; Dodds, P.N.; van der Merwe, M.; Linde, C.C.; Lawrence, G.J.; Burdon, J.J. Diversity and Evolution of Effector Loci in Natural Populations of the Plant Pathogen Melampsora lini. Mol. Biol. Evol. 2009, 26, 2499–2513. [Google Scholar] [CrossRef] [Green Version]
- Streicker, D.G.; Altizer, S.M.; Velasco-Villa, A.; Rupprecht, C.E. Variable evolutionary routes to host establishment across repeated rabies virus host shifts among bats. Proc. Natl. Acad. Sci. USA 2012, 109, 19715–19720. [Google Scholar] [CrossRef] [Green Version]
- Brockhurst, M.A.; Chapman, T.; King, K.C.; Mank, J.E.; Paterson, S.; Hurst, G.D.D. Running with the Red Queen: The role of biotic conflicts in evolution. Proc. R. Soc. B Biol. Sci. 2014, 281, 20141382. [Google Scholar] [CrossRef] [Green Version]
- Frank, H.K.; Enard, D.; Boyd, S.D. Exceptional diversity and selection pressure on SARS-CoV and SARS-CoV-2 host receptor in bats compared to other mammals. bioRxiv 2020. [Google Scholar] [CrossRef]
- Shih, A.C.-C.; Hsiao, T.-C.; Ho, M.-S.; Li, W.-H. Simultaneous amino acid substitutions at antigenic sites drive influenza A hemagglutinin evolution. Proc. Natl. Acad. Sci. USA 2007, 104, 6283–6288. [Google Scholar] [CrossRef] [Green Version]
- Bhatt, S.; Holmes, E.C.; Pybus, O.G. The Genomic Rate of Molecular Adaptation of the Human Influenza A Virus. Mol. Biol. Evol. 2011, 28, 2443–2451. [Google Scholar] [CrossRef] [Green Version]
- Strelkowa, N.; Lässig, M. Clonal Interference in the Evolution of Influenza. Genetics 2012, 192, 671–682. [Google Scholar] [CrossRef]
- Illingworth, C.J.R.; Mustonen, V. Components of Selection in the Evolution of the Influenza Virus: Linkage Effects Beat Inherent Selection. PLoS Pathog. 2012, 8, e1003091. [Google Scholar] [CrossRef] [Green Version]
- Meyer, A.G.; Dawson, E.T.; Wilke, C.O. Cross-species comparison of site-specific evolutionary-rate variation in influenza haemagglutinin. Philos. Trans. R. Soc. B Biol. Sci. 2013, 368, 20120334. [Google Scholar] [CrossRef] [Green Version]
- Nozawa, M.; Suzuki, Y.; Nei, M. Reliabilities of identifying positive selection by the branch-site and the site-prediction methods. Proc. Natl. Acad. Sci. USA 2009, 106, 6700–6705. [Google Scholar] [CrossRef] [Green Version]
- Longdon, B.; Day, J.P.; Alves, J.M.; Smith, S.C.L.; Houslay, T.M.; McGonigle, J.E.; Tagliaferri, L.; Jiggins, F.M. Host shifts result in parallel genetic changes when viruses evolve in closely related species. PLoS Pathog. 2018, 14, e1006951. [Google Scholar] [CrossRef] [Green Version]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| TeAdV-1 | Gene | Strand | Size (aa) | DAdV-1 | PsAdV-3 | |
|---|---|---|---|---|---|---|
| p32 K | 236 | 1174 | − | 312 | p32 K | p32 K |
| LH2 | 1209 | 1625 | + | 138 | LH2 | E1B protein, small T-antigen |
| LH1 | 1656 | 2804 | + | 382 | E1B 55 K | |
| IVa2 protein | 2875 | 3696 | − | 296 | IVa2 protein | IVa2 protein |
| 4614 | 4682 | |||||
| DNA polymerase | 3945 | 7181 | − | 1078 | DNA polymerase | DNA polymerase |
| pTP | 7157 | 8950 | − | 602 | pTP | pTP |
| 11680 | 11694 | |||||
| 52 K protein | 8985 | 9971 | + | 328 | 52 K protein | 52 K protein |
| pIIIa protein | 9955 | 11664 | + | 569 | pIIIa protein | pIIIa protein |
| penton base protein | 11704 | 13062 | + | 452 | penton base protein | penton base protein |
| pVII protein | 13104 | 13559 | + | 151 | pVII | pVII |
| pX protein | 13568 | 13765 | + | 65 | pX | pX |
| pVI protein | 13800 | 14435 | + | 211 | pVI | pVI |
| hexon protein | 14456 | 17188 | + | 910 | hexon protein | hexon protein |
| protease | 17185 | 17790 | + | 201 | protease | protease |
| DNA-binding protein | 17809 | 18951 | − | 380 | DNA-binding protein | DNA-binding protein |
| 100 K protein | 19005 | 20924 | + | 639 | 100 K protein | 100 K protein |
| 22 K protein | 20758 | 20982 | + | 75 | ||
| 33 K protein | 20758 | 20973 | + | 150 | 33 K protein | 33 K protein |
| 21059 | 21292 | |||||
| pVIII protein | 21323 | 22129 | + | 268 | pVIII protein | pVIII protein |
| U-exon | 22142 | 22306 | − | 54 | U-exon | U-exon |
| fiber protein | 22324 | 24369 | + | 681 | fiber protein | fiber 2 protein |
| E4.3 protein | 24383 | 25273 | − | 296 | 34 K-2 | E4.3 protein |
| E4.2 protein | 25221 | 26027 | − | 268 | 34 K-1 | E4.2 protein |
| E4.1 protein | 25948 | 26385 | − | 145 | E4.1 protein | |
| ORF8 | 26628 | 26870 | − | 80 | ||
| ORF7 | 26888 | 27451 | − | 187 | ||
| ORF1 | 27508 | 27966 | + | 152 | ||
| ORF2 | 28072 | 28359 | + | 95 | ||
| ORF3 | 28366 | 28698 | + | 110 | ||
| ORF6 | 28872 | 29558 | − | 228 | ||
| ORF5 | 29658 | 30296 | − | 212 | ||
| ORF4 | 30424 | 31146 | + | 240 | ||
| Gene | Annotation |
|---|---|
| 100 K protein | participation in the transport of hexon monomers to the nucleus and trimerization [61] |
| 23 K protein (endopeptidase, protease) | participation in the cleavage of some AdV precursor proteins [62,63] |
| 52 K protein | participation in the packaging of the viral DNA into the capsid [62,63] |
| DBP (DNA-binding protein) | participation in the elongation phase of AdV DNA replication by unwinding the template [64] |
| hexon | major capsid protein [62,63] |
| III (penton base) | major capsid protein [62,63] |
| pIIIa | minor capsid protein [62,63,65] |
| IVa2 | participation in the packaging of the viral DNA into the capsid [62,63] |
| Pol (DNA polymerase) | participation in the elongation phase of AdV DNA replication [64] |
| pTP (preterminal protein) | the protein primer for AdV DNA replication [64] |
| pVI | minor capsid protein [62,63,65] |
| pVIII | minor capsid protein [62,63,65] |
| ASAP | PTP | GMYC (Single-Threshold) | GMYC (Multiple-Threshold) |
|---|---|---|---|
| TAdV-1 | TAdV-1 | TAdV-1 | TAdV-1 |
| LC606503.1 BoAdV-F LC597488.1 BoAdV-F MN901942.2 BoAdV-F | LC606503.1 BoAdV-F LC597488.1 BoAdV-F MN901942.2 BoAdV-F | LC606503.1 BoAdV-F LC597488.1 BoAdV-F MN901942.2 BoAdV-F | LC606503.1 BoAdV-F LC597488.1 BoAdV-F MN901942.2 BoAdV-F |
| U40839.3 OvAdV-D | U40839.3 OvAdV-D | U40839.3 OvAdV-D | U40839.3 OvAdV-D |
| MK537328.1 OdAdV-A KY748210.1 OdAdV-A KY468403.1 OdAdV-A KY468402.1 OdAdV-A MK343439.1 OdAdV-A KY468406.1 OdAdV-A KY468407.1 OdAdV-A KY468404.1 OdAdV-A KY468405.1 OdAdV-A | MK537328.1 OdAdV-A KY748210.1 OdAdV-A KY468403.1 OdAdV-A KY468402.1 OdAdV-A MK343439.1 OdAdV-A KY468406.1 OdAdV-A KY468407.1 OdAdV-A KY468404.1 OdAdV-A KY468405.1 OdAdV-A | MK537328.1 OdAdV-A KY748210.1 OdAdV-A KY468403.1 OdAdV-A KY468402.1 OdAdV-A MK343439.1 OdAdV-A KY468406.1 OdAdV-A KY468407.1 OdAdV-A KY468404.1 OdAdV-A KY468405.1 OdAdV-A | MK537328.1 OdAdV-A KY748210.1 OdAdV-A KY468403.1 OdAdV-A KY468402.1 OdAdV-A MK343439.1 OdAdV-A KY468406.1 OdAdV-A KY468407.1 OdAdV-A KY468404.1 OdAdV-A KY468405.1 OdAdV-A |
| AF036092.3 BoAdV-D JQ345700.1 BoAdV-E | AF036092.3 BoAdV-D | AF036092.3 BoAdV-D JQ345700.1 BoAdV-E | AF036092.3 BoAdV-D |
| JQ345700.1 BoAdV-E | JQ345700.1 BoAdV-E | ||
| MT050041.1 LiAdV-B | MT050041.1 LiAdV-B | MT050041.1 LiAdV-B | MT050041.1 LiAdV-B |
| KJ156523.1 LiAdV-A | KJ156523.1 LiAdV-A | KJ156523.1 LiAdV-A | KJ156523.1 LiAdV-A |
| KJ675568.1 PsAdV-A MN025529.1 PsAdV-A | KJ675568.1 PsAdV-A | KJ675568.1 PsAdV-A MN025529.1 PsAdV-A | KJ675568.1 PsAdV-A |
| MN025529.1 PsAdV-A | MN025529.1 PsAdV-A | ||
| KJ452170.1 DAdV-A KJ452171.1 DAdV-A | KJ452170.1 DAdV-A KJ452171.1 DAdV-A | KJ452170.1 DAdV-A KJ452171.1 DAdV-A | KJ452170.1 DAdV-A KJ452171.1 DAdV-A |
| KF286430.1 DAdV-A KJ452172.1 DAdV-A MT646045.1 DAdV-A MN310513.1 DAdV-A | KF286430.1 DAdV-A KJ452172.1 DAdV-A MT646045.1 DAdV-A MN310513.1 DAdV-A | KF286430.1 DAdV-A KJ452172.1 DAdV-A MT646045.1 DAdV-A MN310513.1 DAdV-A | KF286430.1 DAdV-A KJ452172.1 DAdV-A |
| MT646045.1 DAdV-A | |||
| MN310513.1 DAdV-A | |||
| 9 | 11 | 8 | 14 |
| Protein | Amino Acid Coordinate | PP (CODEML) | PP (HyPhy) |
|---|---|---|---|
| 100 K protein | 118 | 0.886 | 0.962 |
| 230 | 0.807 | 0.951 | |
| 450 | 0.905 | 0.972 | |
| 96 | 0.87 | 0.972 | |
| 13 | 0.903 | 0.955 | |
| 162 | 0.904 | 0.950 | |
| 180 | 0.936 | 0.980 | |
| 192 | 0.974 | 0.990 | |
| 35 | 0.953 | 0.946 | |
| pIVa2 | 123 | 0.937 | 0.983 |
| 137 | 0.894 | 0.982 | |
| 152 | 0.946 | 0.971 | |
| 94 | 0.935 | 0.964 | |
| DNA polymerase | 1044 | 0.862 | 0.951 |
| 366 | 0.934 | 0.973 | |
| 367 | 0.927 | 0.962 | |
| 773 | 0.941 | 0.961 | |
| pTP | 145 | 0.85 | 0.961 |
| 187 | 0.946 | 0.966 | |
| 297 | 0.859 | 0.975 | |
| 405 | 0.926 | 0.952 | |
| 44 | 0.946 | 0.972 | |
| 445 | 0.939 | 0.966 | |
| 565 | 0.795 | 0.950 | |
| 89 | 0.93 | 0.966 | |
| 96 | 0.939 | 0.988 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2021 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Matsvay, A.; Dyachkova, M.; Mikhaylov, I.; Kiselev, D.; Say, A.; Burskaia, V.; Artyushin, I.; Khafizov, K.; Shipulin, G. Complete Genome Sequence, Molecular Characterization and Phylogenetic Relationships of a Novel Tern Atadenovirus. Microorganisms 2022, 10, 31. https://doi.org/10.3390/microorganisms10010031
Matsvay A, Dyachkova M, Mikhaylov I, Kiselev D, Say A, Burskaia V, Artyushin I, Khafizov K, Shipulin G. Complete Genome Sequence, Molecular Characterization and Phylogenetic Relationships of a Novel Tern Atadenovirus. Microorganisms. 2022; 10(1):31. https://doi.org/10.3390/microorganisms10010031
Chicago/Turabian StyleMatsvay, Alina, Marina Dyachkova, Ivan Mikhaylov, Daniil Kiselev, Anna Say, Valentina Burskaia, Ilya Artyushin, Kamil Khafizov, and German Shipulin. 2022. "Complete Genome Sequence, Molecular Characterization and Phylogenetic Relationships of a Novel Tern Atadenovirus" Microorganisms 10, no. 1: 31. https://doi.org/10.3390/microorganisms10010031
APA StyleMatsvay, A., Dyachkova, M., Mikhaylov, I., Kiselev, D., Say, A., Burskaia, V., Artyushin, I., Khafizov, K., & Shipulin, G. (2022). Complete Genome Sequence, Molecular Characterization and Phylogenetic Relationships of a Novel Tern Atadenovirus. Microorganisms, 10(1), 31. https://doi.org/10.3390/microorganisms10010031