
First Report and Comparative Genomic Analysis of Mycoplasma capricolum subsp. capricolum HN-B in Hainan Island, China
 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Isolation and Identification of Mcc HN-B
2.2. Mcc HN-B Strain Culture and Genomic DNA Extraction
2.3. ONT Sequencing and Illumina Sequencing
2.4. Genome Assembly and Genome Component Annotation
2.5. Gene Function Analysis
2.6. Collinearity Analysis and SNP/InDel/SV Statistics
2.7. Gene Family Analysis
2.8. Core/Pan-Genome Analysis
2.9. Phylogenetic Tree Analysis and ANI Analysis
2.10. GenBank Accession Number
3. Results and Discussion
3.1. Results of Mcc HN-B Isolation and Identification
3.2. General Characteristics of the Mcc HN-B Genome
3.2.1. GIs
3.2.2. Virulence Factors
3.2.3. Metabolism
3.3. Results of Comparative Genomic Analysis
3.3.1. Collinearity Analysis Results
3.3.2. Statistical Results of SNPs, InDels, and SVs
3.4. Molecular Characterization of the Mm cluster
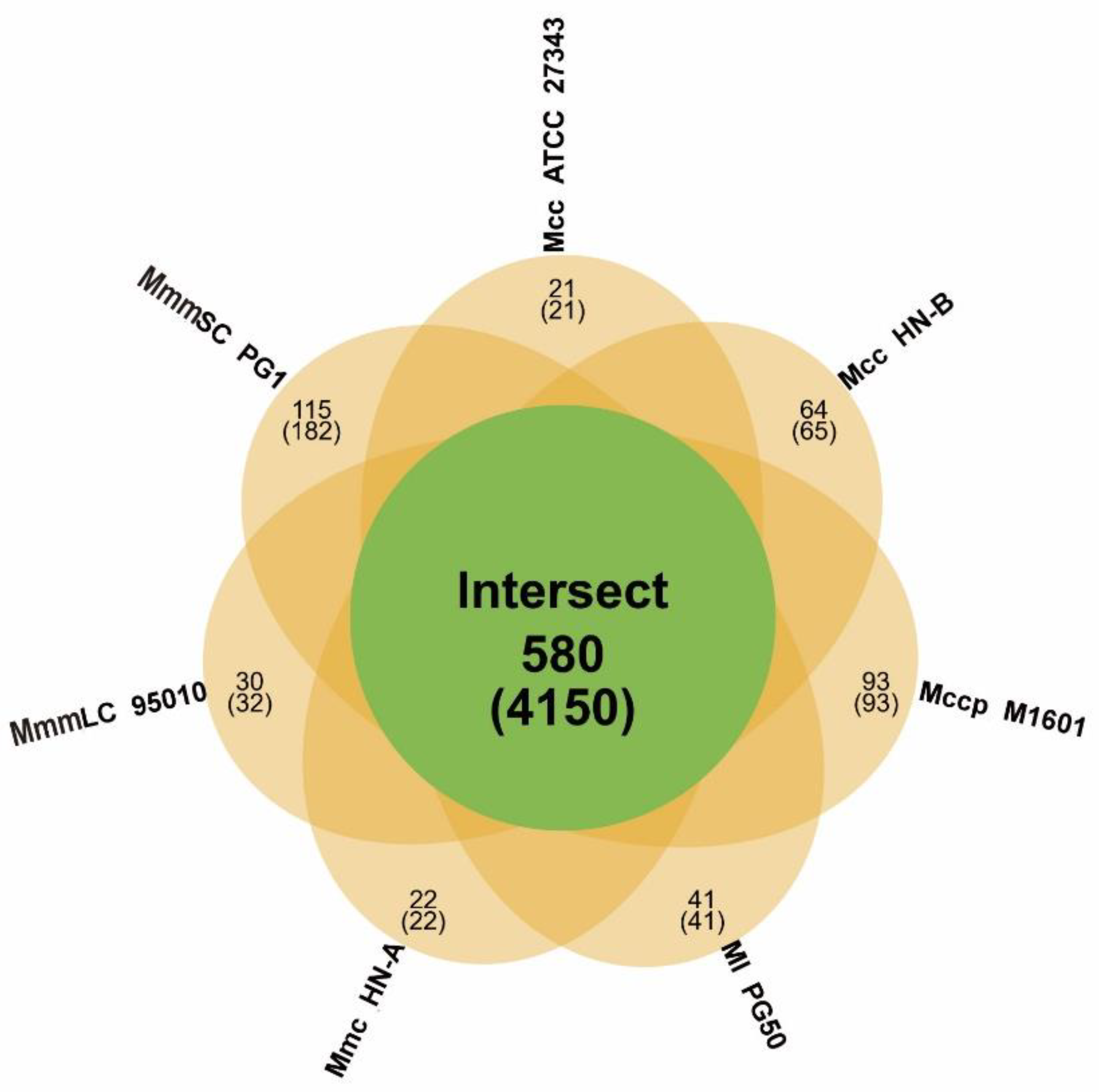
3.4.1. Gene Family Analysis and Core Genome Identification of the Mm Cluster
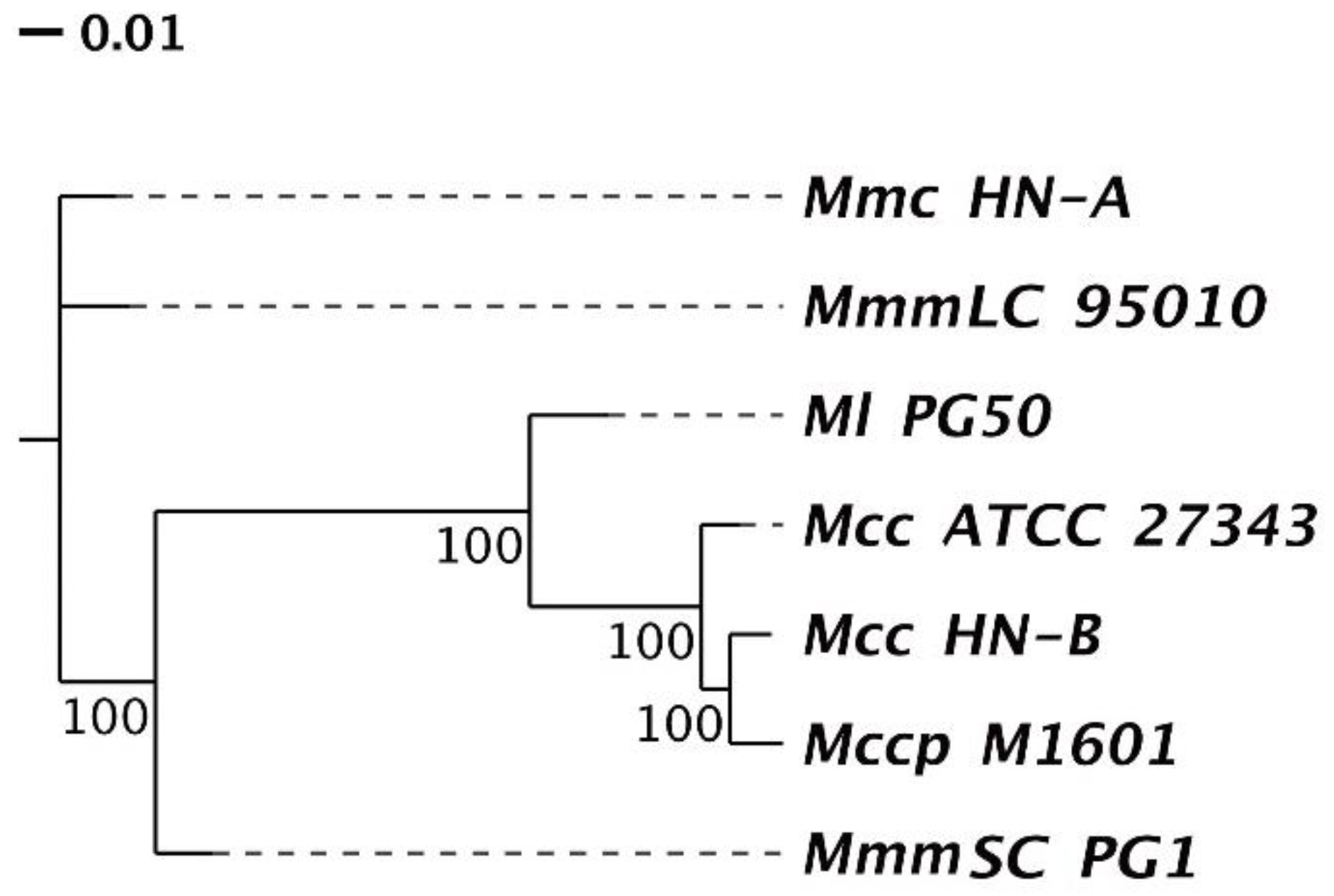
3.4.2. Phylogenetic Tree and ANI Analysis
3.5. Molecular Characterization of the Mycoplasma Capricolum Genome
3.6. Identification of Caprine-Host-Specific Orthologs
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Fischer, A.; Shapiro, B.; Muriuki, C.; Heller, M.; Schnee, C.; Bongcam-Rudloff, E.; Vilei, E.M.; Frey, J.; Jores, J. The origin of the ‘Mycoplasma mycoides cluster’ coincides with domestication of ruminants. PLoS ONE 2012, 7, e36150. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gómez-Martín, A.; Amores, J.; Paterna, A.; De la Fe, C. Contagious agalactia due to Mycoplasma spp. in small dairy ruminants: Epidemiology and prospects for diagnosis and control. Vet. J. 2013, 198, 48–56. [Google Scholar] [CrossRef] [PubMed]
- Cottew, G.S.; Breard, A.; DaMassa, A.J.; Ernø, H.; Leach, R.H.; Lefevre, P.C.; Rodwell, A.W.; Smith, G.R. Taxonomy of the Mycoplasma mycoides cluster. Isr. J. Med. Sci. 1987, 23, 632–635. [Google Scholar] [PubMed]
- Thiaucourt, F.; Bölske, G. Contagious caprine pleuropneumonia and other pulmonary mycoplasmoses of sheep and goats. Rev. Sci. Tech. 1996, 15, 1397–1414. [Google Scholar] [CrossRef] [Green Version]
- Yatoo, M.L.; Parray, O.R.; Bashir, S.T.; Bhat, R.A.; Gopalakrishnan, A.; Karthik, K.; Dhama, K.; Singh, S.V. Contagious caprine pleuropneumonia—A comprehensive review. Vet. Q. 2019, 39, 1–25. [Google Scholar] [CrossRef]
- Westberg, J.; Persson, A.; Holmberg, A.; Goesmann, A.; Lundeberg, J.; Johansson, K.E.; Pettersson, B.; Uhlén, M. The genome sequence of Mycoplasma mycoides subsp. mycoides SC type strain PG1T, the causative agent of contagious bovine pleuropneumonia (CBPP). Genome Res. 2004, 14, 221–227. [Google Scholar] [CrossRef] [Green Version]
- Bergonier, D.; Berthelot, X.; Poumarat, F. Contagious agalactia of small ruminants: Current knowledge concerning epidemiology, diagnosis and control. Rev. Sci. Tech. 1997, 16, 848–873. [Google Scholar] [CrossRef]
- Rodriguez, J.L.; DaMassa, A.J.; Brooks, D.L. Caprine abortion following exposure to Mycoplasma capricolum subsp. capricolum. J. Vet. Diagn. Investig. 1996, 8, 492–494. [Google Scholar] [CrossRef] [Green Version]
- Nicolas, M.M.; Stalis, I.H.; Clippinger, T.L.; Busch, M.; Nordhausen, R.; Maalouf, G.; Schrenzel, M.D. Systemic disease in Vaal rhebok (Pelea capreolus) caused by mycoplasmas in the mycoides cluster. J. Clin. Microbiol. 2005, 43, 1330–1340. [Google Scholar] [CrossRef] [Green Version]
- De la Fe, C.; Gutiérrez, A.; Poveda, J.B.; Assunção, P.; Ramírez, A.S.; Fabelo, F. First isolation of Mycoplasma capricolum subsp. capricolum, one of the causal agents of caprine contagious agalactia, on the island of Lanzarote (Spain). Vet. J. 2007, 173, 440–442. [Google Scholar] [CrossRef]
- Pinho, L.; Thompson, G.; Machado, M.; Carvalheira, J. Management practices associated with the bulk tank milk prevalence of Mycoplasma spp. in dairy herds in Northwestern Portugal. Prev. Vet. Med. 2013, 108, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, J.L.; Ermel, R.W.; Kenny, T.P.; Brooks, D.L.; DaMassa, A.J. Polymerase chain reaction and restriction endonuclease digestion for selected members of the “Mycoplasma mycoides cluster” and Mycoplasma putrefaciens. J. Vet. Diagn. Investig. 1997, 9, 186–190. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maigre, L.; Citti, C.; Marenda, M.; Poumarat, F.; Tardy, F. Suppression-subtractive hybridization as a strategy to identify taxon-specific sequences within the Mycoplasma mycoides Cluster: Design and validation of an M. capricolum subsp. capricolum-specific PCR assay. J. Clin. Microbiol. 2008, 46, 1307–1316. [Google Scholar] [CrossRef] [Green Version]
- Seersholm, F.V.; Fischer, A.; Heller, M.; Jores, J.; Sachse, K.; Mourier, T.; Hansen, A.J. Draft Genome Sequence of the First Human Isolate of the Ruminant Pathogen Mycoplasma capricolum subsp. capricolum. Genome Announc. 2015, 3, e00583-15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Calcutt, M.J.; Foecking, M.F. Comparative analysis of the Mycoplasma capricolum subsp. capricolum GM508D genome reveals subrogation of phase-vZariable contingency genes and a novel integrated genetic element. Pathog. Dis. 2015, 73, ftv041. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tatay-Dualde, J.; Prats-van der Ham, M.; de la Fe, C.; Paterna, A.; Sánchez, A.; Corrales, J.C.; Contreras, A.; Tola, S.; Gómez-Martin, Á. Antimicrobial susceptibility and multilocus sequence typing of Mycoplasma capricolum subsp. capricolum. PLoS ONE 2017, 12, e0174700. [Google Scholar] [CrossRef] [Green Version]
- Tatay-Dualde, J.; Prats-van der Ham, M.; de la Fe, C.; Paterna, A.; Sánchez, A.; Corrales, J.C.; Contreras, A.; Gómez-Martin, Á. Resistance mechanisms against quinolones in Mycoplasma capricolum subsp. capricolum. Vet. J. 2017, 223, 1–4. [Google Scholar] [CrossRef]
- Eden, P.A.; Schmidt, T.M.; Blakemore, R.P.; Pace, N.R. Phylogenetic analysis of Aquaspirillum magnetotacticum using polymerase chain reaction-amplified 16S rRNA-specific DNA. Int. J. Syst. Bacteriol. 1991, 41, 324–325. [Google Scholar] [CrossRef]
- Lin, Y.; Yuan, J.; Kolmogorov, M.; Shen, M.W.; Chaisson, M.; Pevzner, P.A. Assembly of long error-prone reads using de Bruijn graphs. Proc. Natl. Acad. Sci. USA 2016, 113, E8396–E8405. [Google Scholar] [CrossRef] [Green Version]
- Chen, S.F.; Zhou, Y.Q.; Chen, Y.R.; Gu, J. fastp: An ultra-fast all-in-one FASTQ preprocessor. Bioinformatics 2018, 34, 884–890. [Google Scholar] [CrossRef]
- Walker, B.J.; Abeel, T.; Shea, T.; Priest, M.; Abouelliel, A.; Sakthikumar, S.; Cuomo, C.A.; Zeng, Q.D.; Wortman, J.; Young, S.K.; et al. Pilon: An Integrated Tool for Comprehensive Microbial Variant Detection and Genome. PLoS ONE 2014, 9, e112963. [Google Scholar] [CrossRef] [PubMed]
- Tatusova, T.; DiCuccio, M.; Badretdin, A.; Chetvernin, V.; Nawrocki, E.P.; Zaslavsky, L.; Lomsadze, A.; Pruitt, K.D.; Borodovsky, M.; Ostell, J. NCBI prokaryotic genome annotation pipeline. Nucleic Acids Res. 2016, 44, 6614–6624. [Google Scholar] [CrossRef] [PubMed]
- Lagesen, K.; Hallin, P.; Rødland, E.A.; Staerfeldt, H.H.; Rognes, T.; Ussery, D.W. RNAmmer: Consistent and rapid annotation of ribosomal RNA genes. Nucleic Acids Res. 2007, 35, 3100–3108. [Google Scholar] [CrossRef]
- Lowe, T.M.; Chan, P.P. tRNAscan-SE On-line: Integrating search and context for analysis of transfer RNA genes. Nucleic Acids Res. 2016, 44, W54–W57. [Google Scholar] [CrossRef] [PubMed]
- Nawrocki, E.P.; Eddy, S.R. Infernal 1.1: 100-fold faster RNA homology searches. Bioinformatics 2013, 29, 2933–2935. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bertelli, C.; Brinkman, F.S.L. Improved genomic island predictions with IslandPath DIMOB. Bioinformatics 2018, 34, 2161–2167. [Google Scholar] [CrossRef] [PubMed]
- Grissa, I.; Vergnaud, G.; Pourcel, C. CRISPRFinder: A web tool to identify clustered regularly interspaced short palindromic repeats. Nucleic Acids Res. 2007, 35, W52–W57. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Haas, B. TransposonPSI: An Application of PSI-Blast to Mine (Retro-) Transposon ORF Homologies. Available online: http://transposonpsi.sourceforge.net/ (accessed on 1 October 2022 ).
- Chen, N. Using RepeatMasker to identify repetitive elements in genomic sequences. Curr. Protoc. Bioinform. 2004, 4, 4.10.1–4.10.14. [Google Scholar]
- Benson, G. Tandem repeats finder: A program to analyze DNA sequences. Nucleic Acids Res. 1999, 27, 573–580. [Google Scholar] [CrossRef] [Green Version]
- Fouts, D.E. Phage_Finder: Automated identification and classification of prophage regions in complete bacterial genome sequences. Nucleic Acids Res. 2006, 34, 5839–5851. [Google Scholar] [CrossRef]
- Petersen, T.N.; Brunak, S.; Von Heijne, G.; Nielsen, H. SignalP 4.0: Discriminating Signal Peptides from Transmembrane Regions. Nat. Methods 2011, 8, 785–786. [Google Scholar] [CrossRef] [PubMed]
- Krogh, A.; Larsson, B.; von Heijne, G.; Sonnhammer, E.L. Predicting Transmembrane Protein Topology with a Hidden Markov Model: Application to Complete Genomes. J. Mol. Biol. 2001, 305, 567–580. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Arnold, R.; Brandmaier, S.; Kleine, F.; Tischler, P.; Heinz, E.; Behrens, S.; Niinikoski, A.; Mewes, H.W.; Horn, M.; Rattei, T. Sequence-Based Prediction of Type III Secreted Proteins. PLoS Pathog. 2009, 5, e1000376. [Google Scholar] [CrossRef]
- Blin, K.; Wolf, T.; Chevrette, M.G.; Lu, X.; Schwalen, C.J.; Kautsar, S.A.; Suarez Duran, H.G.; de Los Santos, E.L.C.; Kim, H.U.; Nave, M.; et al. AntiSMASH 4.0-Improvements in Chemistry Rrediction and Gene Cluster Boundary Identification. Nucleic Acids Res. 2017, 45, W36–W41. [Google Scholar] [CrossRef] [PubMed]
- Kurtz, S.; Phillippy, A.; Delcher, A.L.; Smoot, M.; Shumway, M.; Antonescu, C.; Salzberg, S.L. Versatile and Open Software for Comparing Large Genomes. Genome Biol. 2004, 5, R12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Goel, M.; Sun, H.; Jiao, W.B.; Schneeberger, K. SyRI: Finding Genomic Rearrangements and Local Sequence Differences from Whole-Genome Assemblies. Genome Biol. 2019, 20, 277. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Buchfink, B.; Reuter, K.; Drost, H.G. Sensitive Protein Alignments at Tree-of-Life Scale using DIAMOND. Nat. Methods 2021, 18, 366–368. [Google Scholar] [CrossRef]
- Li, L.; Stoeckert, C.J., Jr.; Roos, D.S. OrthoMCL: Identification of Ortholog Groups for Eukaryotic Genomes. Genome Res. 2003, 13, 2178–2189. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. MUSCLE: A multiple sequence alignment method with reduced time and space complexity. BMC Bioinform. 2004, 5, 113. [Google Scholar] [CrossRef] [Green Version]
- Castresana, J. Selection of Conserved Blocks from Multiple Alignments for Their Use in Phylogenetic Analysis. Mol. Biol. Evol. 2000, 17, 540–552. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, L.T.; Schmidt, H.A.; von Haeseler, A.; Minh, B.Q. IQ-TREE: A fast and effective stochastic algorithm for estimating maximum-likelihood phylogenies. Mol. Biol. Evol. 2015, 32, 268–274. [Google Scholar] [CrossRef] [PubMed]
- Pritchard, L.; Glover, R.H.; Humphris, S.; Elphinstone, J.G.; Toth, I.K. Genomics and taxonomy in diagnostics for food security: Soft-rotting enterobacterial plant pathogens. Anal. Methods 2016, 8, 12–24. [Google Scholar] [CrossRef]
- Qiu, X.B.; Shao, Y.M.; Miao, S.; Wang, L. The diversity of the DnaJ/Hsp40 family, the crucial partners for Hsp70 chaperones. Cell Mol. Life Sci. 2006, 63, 2560–2570. [Google Scholar] [CrossRef] [PubMed]
- Xie, Z.; Tang, H. ISEScan: Automated identification of insertion sequence elements in prokaryotic genomes. Bioinformatics 2017, 33, 3340–3347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sirover, M.A. New insights into an old protein: The functional diversity of mammalian glyceraldehyde-3-phosphate dehydrogenase. Biochim. Biophys Acta 1999, 1432, 159–184. [Google Scholar] [CrossRef]
- Hoelzle, L.E.; Hoelzle, K.; Helbling, M.; Aupperle, H.; Schoon, H.A.; Ritzmann, M.; Heinritzi, K.; Felder, K.M.; Wittenbrink, M.M. MSG1, a surface-localised protein of Mycoplasma suis is involved in the adhesion to erythrocytes. Microbes Infect. 2007, 9, 466–474. [Google Scholar] [CrossRef] [PubMed]
- Goebel, W.; Chakraborty, T.; Kreft, J. Bacterial hemolysins as virulence factors. Antonie Van Leeuwenhoek 1988, 54, 453–463. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mengaud, J.; Vicente, M.F.; Chenevert, J.; Pereira, J.M.; Geoffroy, C.; Gicquel-Sanzey, B.; Baquero, F.; Perez-Diaz, J.C.; Cossart, P. Expression in Escherichia coli and sequence analysis of the listeriolysin O determinant of Listeria monocytogenes. Infect Immun. 1988, 56, 766–772. [Google Scholar] [CrossRef] [Green Version]
- Alm, R.A.; Stroeher, U.H.; Manning, P.A. Extracellular proteins of Vibrio cholerae: Nucleotide sequence of the structural gene (hlyA) for the haemolysin of the haemolytic El Tor strain 017 and characterization of the hlyA mutation in the non-haemolytic classical strain 569B. Mol. Microbiol. 1988, 2, 481–488. [Google Scholar]
- Li, Y.; Wang, R.; Sun, W.; Song, Z.; Bai, F.; Zheng, H.; Xin, J. Comparative genomics analysis of Mycoplasma capricolum subsp. capripneumoniae 87001. Genomics 2020, 112, 615–620. [Google Scholar] [CrossRef]
- Balasubramanian, S.; Kannan, T.R.; Baseman, J.B. The surface-exposed carboxyl region of Mycoplasma pneumoniae elongation factor Tu interacts with fibronectin. Infect. Immun. 2008, 76, 3116–3123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pinto, P.M.; Chemale, G.; de Castro, L.A.; Costa, A.P.; Kich, J.D.; Vainstein, M.H.; Zaha, A.; Ferreira, H.B. Proteomic survey of the pathogenic Mycoplasma hyopneumoniae strain 7448 and identification of novel post-translationally modified and antigenic proteins. Vet. Microbiol. 2007, 121, 83–93. [Google Scholar] [CrossRef] [PubMed]
- Dallo, S.F.; Kannan, T.R.; Blaylock, M.W.; Baseman, J.B. Elongation factor Tu and E1 beta subunit of pyruvate dehydrogenase complex act as fibronectin binding proteins in Mycoplasma pneumoniae. Mol. Microbiol. 2002, 46, 1041–1051. [Google Scholar] [CrossRef] [PubMed]
- Thomas, C.; Jacobs, E.; Dumke, R. Characterization of pyruvate dehydrogenase subunit B and enolase as plasminogen-binding proteins in Mycoplasma pneumoniae. Microbiology 2013, 159, 352–365. [Google Scholar] [CrossRef]
- Hoelzle, L.E. Haemotrophic mycoplasmas: Recent advances in Mycoplasma suis. Vet. Microbiol. 2008, 130, 215–226. [Google Scholar] [CrossRef]
- Esgleas, M.; Li, Y.; Hancock, M.A.; Harel, J.; Dubreuil, J.D.; Gottschalk, M. Isolation and characterization of alpha-enolase, a novel fibronectin-binding protein from Streptococcus suis. Microbiology 2008, 154, 2668–2679. [Google Scholar] [CrossRef] [Green Version]
- Bergmann, S.; Rohde, M.; Chhatwal, G.S.; Hammerschmidt, S. α-Enolase of Streptococcus pneumoniae is a plasmin(ogen)-binding protein displayed on the bacterial cell surface. Mol. Microbiol. 2001, 40, 1273–1287. [Google Scholar] [CrossRef]
- Allam, A.B.; Brown, M.B.; Reyes, L. Disruption of the S41 peptidase gene in Mycoplasma mycoides capri impacts proteome profile, H(2)O(2) production, and sensitivity to heat shock. PLoS ONE 2012, 7, e51345. [Google Scholar] [CrossRef] [Green Version]
- De Ste Croix, M.; Vacca, I.; Kwun, M.J.; Ralph, J.D.; Bentley, S.D.; Haigh, R.; Croucher, N.J.; Oggioni, M.R. Phase-variable methylation and epigenetic regulation by type I restriction-modification systems. FEMS Microbiol. Rev. 2017, 41, S3–S15. [Google Scholar] [CrossRef]
- Doberenz, S.; Eckweiler, D.; Reichert, O.; Jensen, V.; Bunk, B.; Spröer, C.; Kordes, A.; Frangipani, E.; Luong, K.; Korlach, J.; et al. Identification of a Pseudomonas aeruginosa PAO1 DNA methyltransferase, its targets, and physiological roles. mBio 2017, 8, e02312-16. [Google Scholar] [CrossRef]
- Nye, T.M.; Jacob, K.M.; Holley, E.K.; Nevarez, J.M.; Dawid, S.; Simmons, L.A.; Watson, M.E. DNA methylation from a Type I restriction modification system influences gene expression and virulence in Streptococcus pyogenes. PLoS Pathog. 2019, 15, e1007841. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kundig, W.; Ghosh, S.; Roseman, S. Phosphate Bound to Histidine in a Protein as an Intermediate in a Novel Phospho-Transferase System. Proc. Natl. Acad. Sci. USA 1964, 52, 1067–1074. [Google Scholar] [CrossRef]
- Deutscher, J.; Francke, C.; Postma, P.W. How phosphotransferase system-related protein phosphorylation regulates carbohydrate metabolism in bacteria. Microbiol. Mol. Biol. Rev. 2006, 70, 939–1031. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Crigler, J.; Bannerman-Akwei, L.; Cole, A.E.; Eiteman, M.A.; Altman, E. Glucose can be transported and utilized in Escherichia coli by an altered or overproduced N-acetylglucosamine phosphotransferase system (PTS). Microbiology 2018, 164, 163–172. [Google Scholar] [CrossRef] [PubMed]
- Voigt, C.; Bahl, H.; Fischer, R.J. Identification of PTS(Fru) as the major fructose uptake system of Clostridium acetobutylicum. Appl. Microbiol. Biotechnol. 2014, 98, 7161–7172. [Google Scholar] [CrossRef]










{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
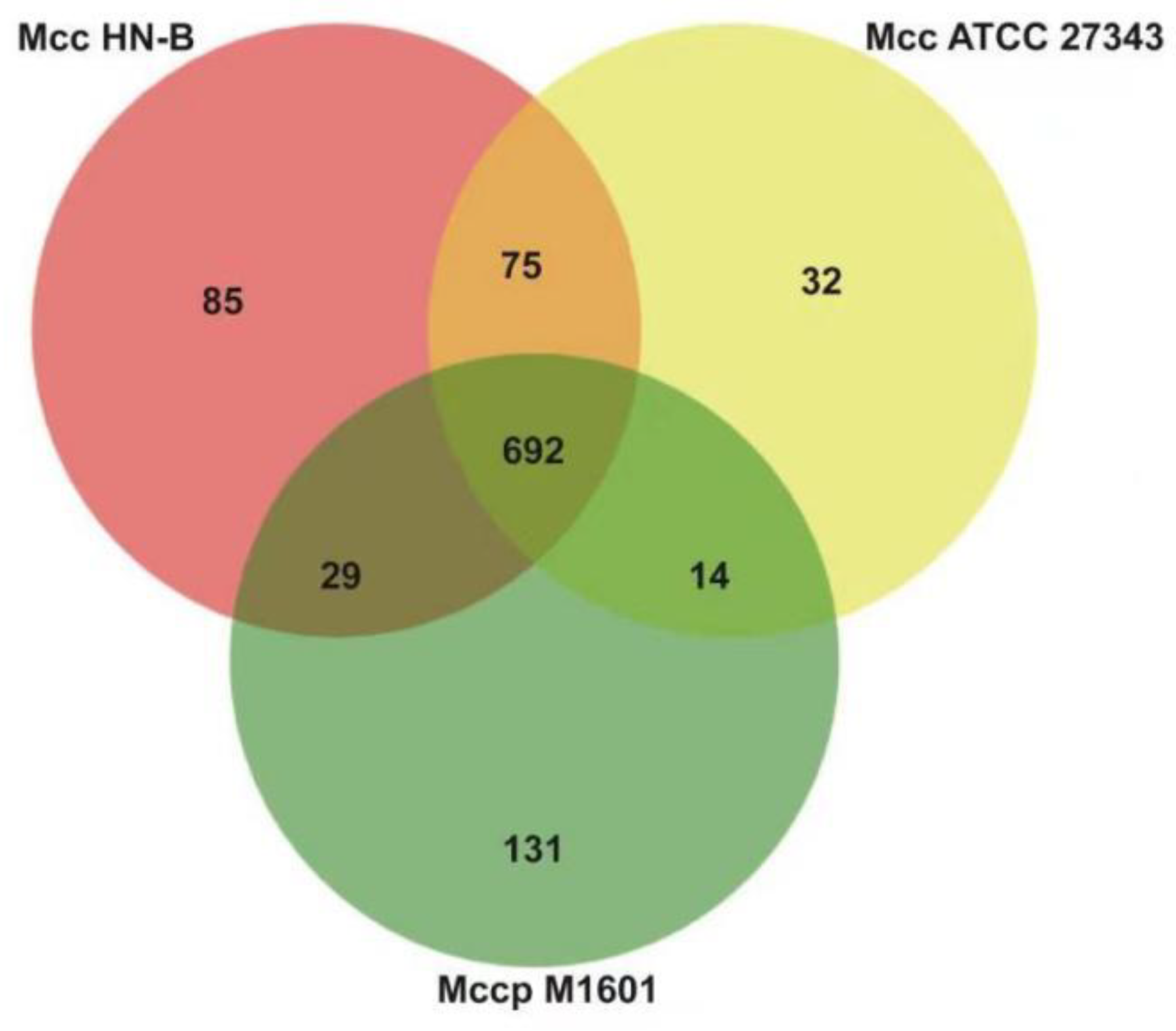{kind=link}
{kind=link}
| Item | Number | Item | Number |
|---|---|---|---|
| Genome size (bp) | 1,117,925 | Number of rRNA genes | 6 |
| Genome GC content (%) | 23.79 | Number of tRNA genes | 31 |
| Number of genes | 912 | Number of sRNA genes | 1 |
| Length of gene (bp) | 45–5,895 | Number of interspersed repeats | 6 |
| Total length of gene (bp) | 983,931 | Number of short interspersed elements | 4 |
| Average length of gene (bp) | 1078.87 | Number of long interspersed repeated sequences | 1 |
| GC content of gene region (%) | 23.95 | Number of DNA elements | 1 |
| Total length of gene/genome (%) | 88.01 | Number of tandem repeats | 175 |
| Total length of intergenic region/genome (%) | 11.99 | Total bases in tandem repeats | 20,145 |
| GIs number | 3 | Number of prophages | 0 |
| Total GI length (bp) | 69,172 | Average length (bp) | 23,057.33 |
| Mcc HN-B | Mcc ATCC 27343 | Mccp M1601 | Mmc HN-A | MmmLC 95010 | MmmSC PG1 | Ml PG50 | |
|---|---|---|---|---|---|---|---|
| Accession number | CP093215 | CP000123.1 | CP017125.1 | CP093215 | FQ377874.1 | BX293980.2 | CP002108.1 |
| Isolation place | China | USA | China | China | France | Sweden | USA |
| Host | goat | goat | goat | goat | goat | cattle | cattle |
| Collection date | 2022 | 2005 | 2016 | 2021 | 1995 | 2003 | 2010 |
| Size (bp) | 1,117,925 | 1,010,023 | 1,016,707 | 1,084,691 | 1,153,998 | 1,211,703 | 1,008,951 |
| G + C (%) | 23.79 | 23.8 | 23.67 | 23.76 | 23.81 | 24.0 | 23.8 |
| SNPs | Insertions | Deletions | SVs | |
|---|---|---|---|---|
| Mcc ATCC 27343 | 17,885 | 500 | 497 | 98 |
| Mccp M1601 | 19,302 | 634 | 652 | 104 |
| Mmc HN-A | 32,999 | 806 | 841 | 123 |
| MmmLC 95010 | 33,126 | 849 | 912 | 122 |
| MmmSC PG1 | 25,573 | 613 | 683 | 141 |
| Ml PG50 | 33,196 | 703 | 792 | 97 |
| Species | Total Genes | Gene in Families | Unclustered Genes | Families | Unique Families |
|---|---|---|---|---|---|
| Mcc HN-B | 912 | 849 | 63 | 818 | 64 |
| Mcc ATCC 27343 | 825 | 804 | 21 | 792 | 21 |
| Mccp M1601 | 889 | 796 | 93 | 773 | 93 |
| Mmc HN-A | 848 | 826 | 22 | 802 | 22 |
| MmmLC 95010 | 927 | 899 | 28 | 837 | 30 |
| MmmSC PG1 | 1,017 | 919 | 98 | 777 | 115 |
| Ml PG50 | 850 | 809 | 41 | 789 | 41 |
| Species | Total Genes | Shared Gene Number | Specific Gene Number |
|---|---|---|---|
| Mcc HN-B | 912 | 847 | 65 |
| Mcc ATCC 27343 | 825 | 804 | 21 |
| Mccp M1601 | 889 | 796 | 93 |
| Mmc HN-A | 848 | 826 | 22 |
| MmmLC 95010 | 927 | 895 | 32 |
| MmmSC PG1 | 1,017 | 835 | 182 |
| Ml PG50 | 850 | 809 | 41 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhang, Z.; He, M.; Jiang, J.; Li, X.; Li, H.; Zhang, W.; Chen, S.; Du, L.; Man, C.; Chen, Q.; et al. First Report and Comparative Genomic Analysis of Mycoplasma capricolum subsp. capricolum HN-B in Hainan Island, China. Microorganisms 2022, 10, 2298. https://doi.org/10.3390/microorganisms10112298
Zhang Z, He M, Jiang J, Li X, Li H, Zhang W, Chen S, Du L, Man C, Chen Q, et al. First Report and Comparative Genomic Analysis of Mycoplasma capricolum subsp. capricolum HN-B in Hainan Island, China. Microorganisms. 2022; 10(11):2298. https://doi.org/10.3390/microorganisms10112298
Chicago/Turabian StyleZhang, Zhenxing, Meirong He, Junming Jiang, Xubo Li, Haoyang Li, Wencan Zhang, Si Chen, Li Du, Churiga Man, Qiaoling Chen, and et al. 2022. "First Report and Comparative Genomic Analysis of Mycoplasma capricolum subsp. capricolum HN-B in Hainan Island, China" Microorganisms 10, no. 11: 2298. https://doi.org/10.3390/microorganisms10112298
APA StyleZhang, Z., He, M., Jiang, J., Li, X., Li, H., Zhang, W., Chen, S., Du, L., Man, C., Chen, Q., Gao, H., & Wang, F. (2022). First Report and Comparative Genomic Analysis of Mycoplasma capricolum subsp. capricolum HN-B in Hainan Island, China. Microorganisms, 10(11), 2298. https://doi.org/10.3390/microorganisms10112298