
Comparison between Symptomatic and Asymptomatic Mice after Clostridioides difficile Infection Reveals Novel Inflammatory Pathways and Contributing Microbiota

Abstract
:1. Introduction
2. Materials and Methods
2.1. Chemicals and Bacterial Strains
2.2. Mouse Model of C. difficile Infection
2.3. RNA Purification and Transcriptomic Analysis
2.4. Histopathological Analysis of Mice Tissues
2.5. Difference in Microbiome Composition of Infected Symptomatic and Infected Asymptomatic Mice
2.6. Statistical Analysis
3. Results
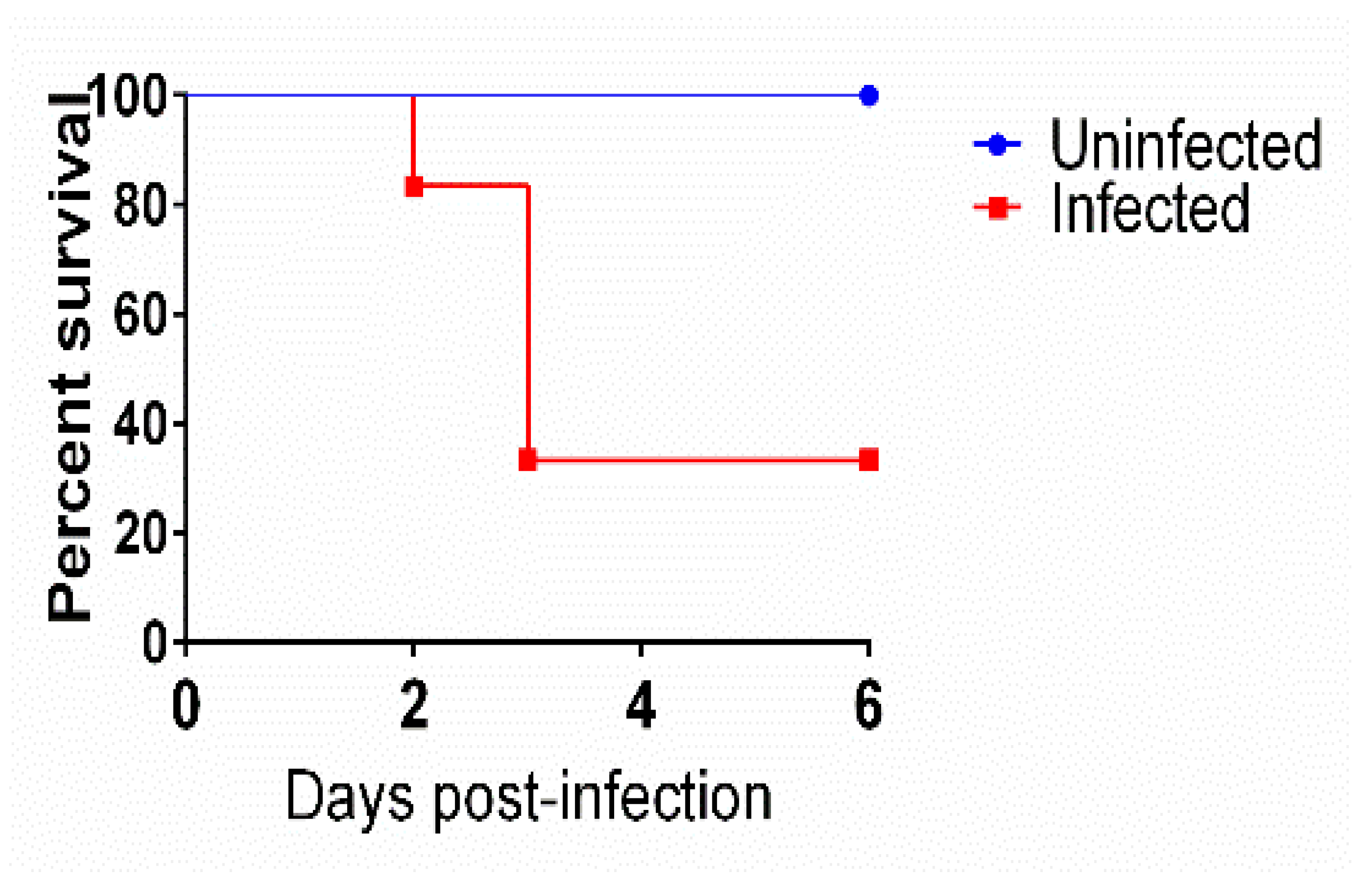
3.1. Mice Survival after Infection with C. difficile
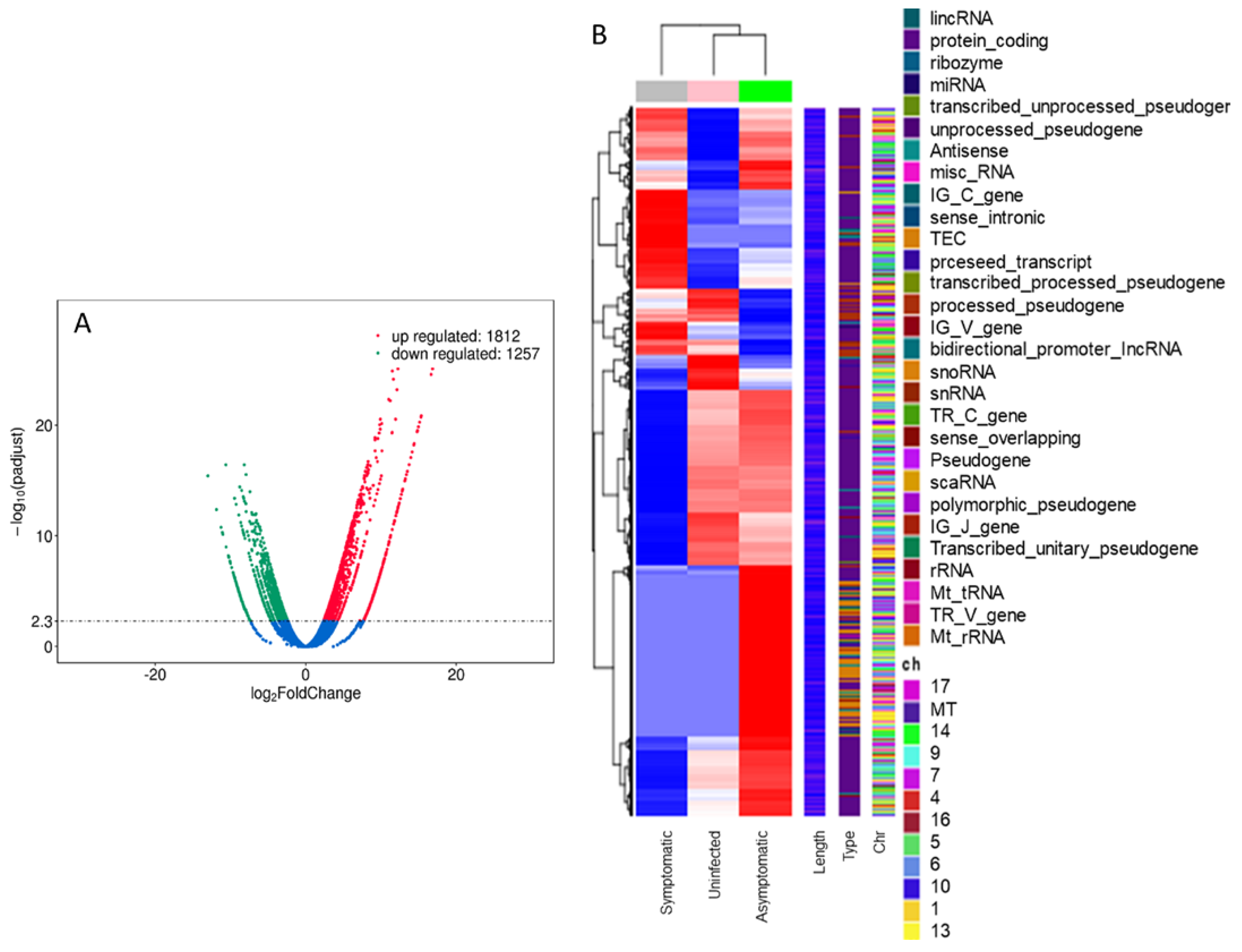
3.2. Transcriptomic Analysis of Symptomatic, Asymptomatic, and Uninfected Mice
3.2.1. RNA-seq Analysis
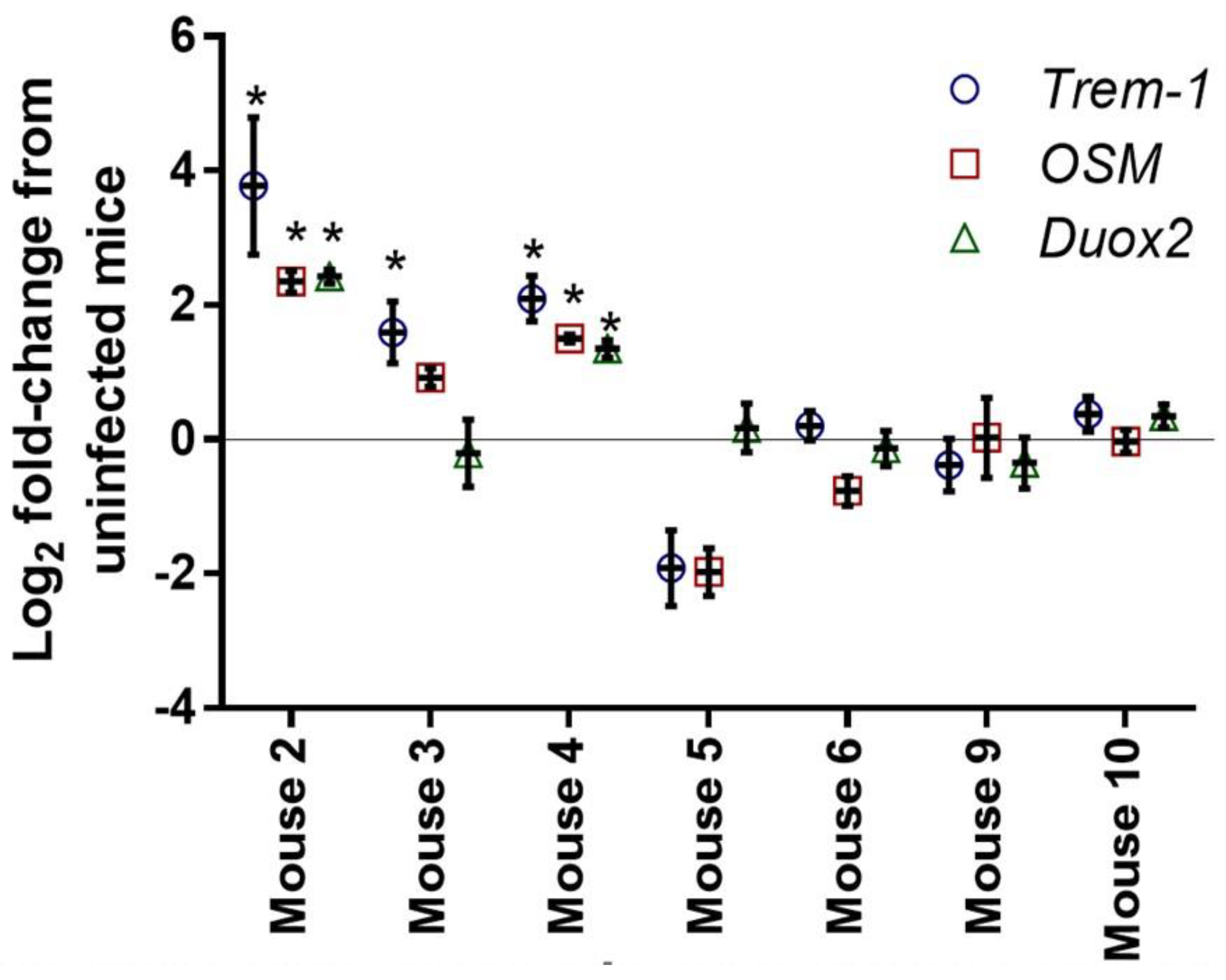
3.2.2. Quantitative PCR
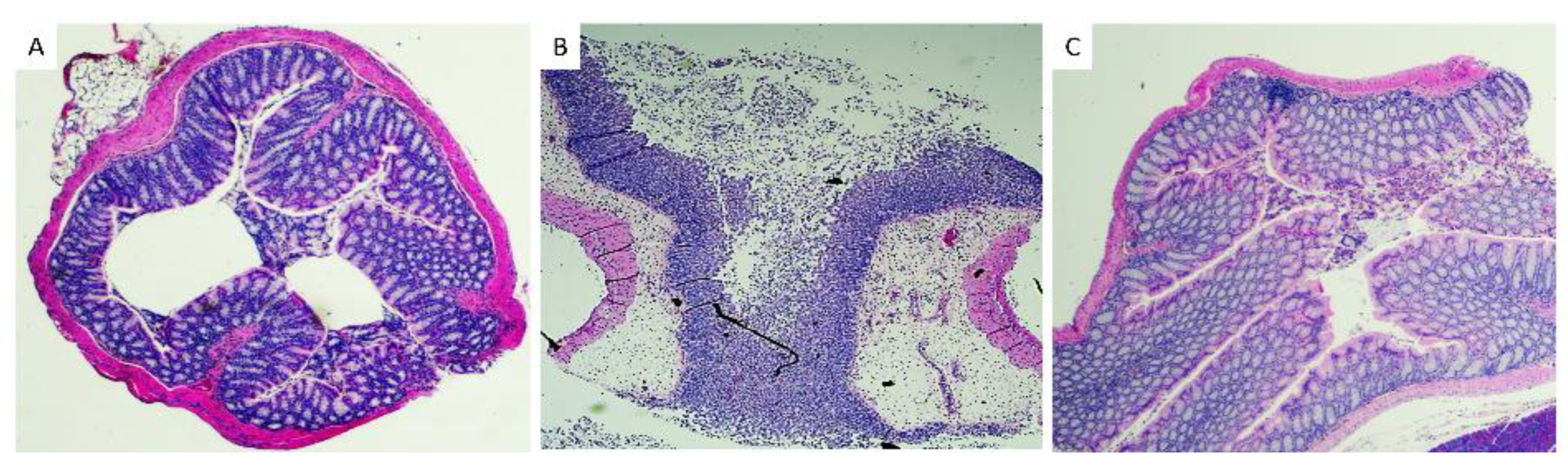
3.3. Histopathological Analysis of Mice Tissues
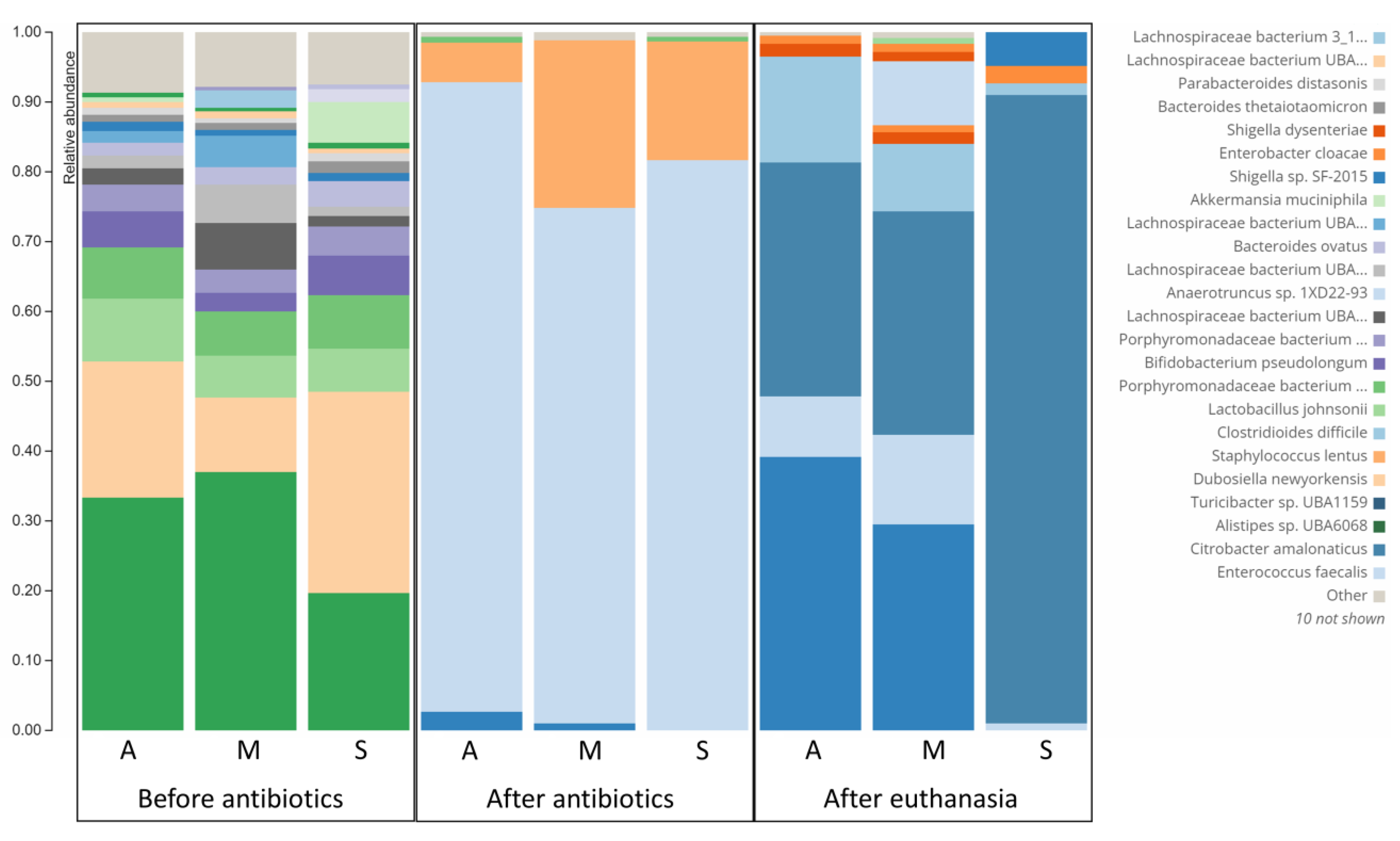
3.4. Difference in Microbiome Composition of Infected Symptomatic and Asymptomatic Mice
4. Discussion
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- McDonald, L.C.; Gerding, D.N.; Johnson, S.; Bakken, J.S.; Carroll, K.C.; Coffin, S.E.; Dubberke, E.R.; Garey, K.W.; Gould, C.V.; Kelly, C.; et al. Clinical Practice Guidelines for Clostridium difficile Infection in Adults and Children: 2017 Update by the Infectious Diseases Society of America (IDSA) and Society for Healthcare Epidemiology of America (SHEA). Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2018, 66, e1–e48. [Google Scholar] [CrossRef] [PubMed]
- Vindigni, S.M.; Surawicz, C.M. C. difficile Infection: Changing Epidemiology and Management Paradigms. Clin. Transl. Gastroenterol. 2015, 6, e99. [Google Scholar] [CrossRef] [PubMed]
- Noren, T. Clostridium difficile and the disease it causes. Methods Mol. Biol. 2010, 646, 9–35. [Google Scholar] [CrossRef] [PubMed]
- Edwards, A.N.; Karim, S.T.; Pascual, R.A.; Jowhar, L.M.; Anderson, S.E.; McBride, S.M. Chemical and Stress Resistances of Clostridium difficile Spores and Vegetative Cells. Front. Microbiol. 2016, 7, 1698. [Google Scholar] [CrossRef] [Green Version]
- Heinlen, L.; Ballard, J.D. Clostridium difficile infection. Am. J. Med. Sci. 2010, 340, 247–252. [Google Scholar] [CrossRef]
- Paredes-Sabja, D.; Shen, A.; Sorg, J.A. Clostridium difficile spore biology: Sporulation, germination, and spore structural proteins. Trends Microbiol. 2014, 22, 406–416. [Google Scholar] [CrossRef] [Green Version]
- Isidro, J.; Mendes, A.L.; Serrano, M.; Henriques, A.O.; Oleastro, M. Henriques and Mónica Oleastro. Overview of Clostridium difficile Infection: Life Cycle, Epidemiology, Antimicrobial Resistance and Treatment. In Clostridium Difficile-A Comprehensive Overview; Enany, S., Ed.; IntechOpen: London, UK, 2017. [Google Scholar] [CrossRef] [Green Version]
- Smits, W.K.; Lyras, D.; Lacy, D.B.; Wilcox, M.H.; Kuijper, E.J. Clostridium difficile infection. Nat. Rev. Dis. Prim. 2016, 2, 16020. [Google Scholar] [CrossRef] [Green Version]
- Spigaglia, P. Clostridioides difficile infection (CDI) during the COVID-19 pandemic. Anaerobe 2022, 74, 102518. [Google Scholar] [CrossRef]
- Sandhu, A.; Tillotson, G.; Polistico, J.; Salimnia, H.; Cranis, M.; Moshos, J.; Cullen, L.; Jabbo, L.; Diebel, L.; Chopra, T. Clostridioides difficile in COVID-19 Patients, Detroit, Michigan, USA, March-April 2020. Emerg. Infect. Dis. 2020, 26, 2272–2274. [Google Scholar] [CrossRef]
- Paramo-Zunzunegui, J.; Ortega-Fernandez, I.; Calvo-Espino, P.; Diego-Hernandez, C.; Ariza-Ibarra, I.; Otazu-Canals, L.; Danes-Grases, J.E.; Menchero-Sanchez, A. Severe Clostridium difficile colitis as potential late complication associated with COVID-19. Ann. R. Coll. Surg. Engl. 2020, 102, e176–e179. [Google Scholar] [CrossRef]
- Chandrasekaran, R.; Lacy, D.B. The role of toxins in Clostridium difficile infection. FEMS Microbiol. Rev. 2017, 41, 723–750. [Google Scholar] [CrossRef] [Green Version]
- Di Bella, S.; Ascenzi, P.; Siarakas, S.; Petrosillo, N.; di Masi, A. Clostridium difficile Toxins A and B: Insights into Pathogenic Properties and Extraintestinal Effects. Toxins 2016, 8, 134. [Google Scholar] [CrossRef] [Green Version]
- McVey, D.C.; Vigna, S.R. The role of leukotriene B4 in Clostridium difficile toxin A-induced ileitis in rats. Gastroenterology 2005, 128, 1306–1316. [Google Scholar] [CrossRef]
- Madan, R.; Petri, W.A., Jr. Immune responses to Clostridium difficile infection. Trends Mol. Med. 2012, 18, 658–666. [Google Scholar] [CrossRef] [Green Version]
- Solomon, K. The host immune response to Clostridium difficile infection. Ther. Adv. Infect. Dis. 2013, 1, 19–35. [Google Scholar] [CrossRef] [Green Version]
- Fletcher, J.R.; Pike, C.M.; Parsons, R.J.; Rivera, A.J.; Foley, M.H.; McLaren, M.R.; Montgomery, S.A.; Theriot, C.M. Clostridioides difficile exploits toxin-mediated inflammation to alter the host nutritional landscape and exclude competitors from the gut microbiota. Nat. Commun. 2021, 12, 462. [Google Scholar] [CrossRef]
- Best, E.L.; Freeman, J.; Wilcox, M.H. Models for the study of Clostridium difficile infection. Gut Microbes 2012, 3, 145–167. [Google Scholar] [CrossRef] [Green Version]
- Locher, H.H.; Seiler, P.; Chen, X.; Schroeder, S.; Pfaff, P.; Enderlin, M.; Klenk, A.; Fournier, E.; Hubschwerlen, C.; Ritz, D.; et al. In vitro and in vivo antibacterial evaluation of cadazolid, a new antibiotic for treatment of Clostridium difficile infections. Antimicrob. Agents Chemother. 2014, 58, 892–900. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, X.; Wang, H.; Zhang, Y.; Chen, K.; Davis, B.; Feng, H. Mouse relapse model of Clostridium difficile infection. Infect. Immun. 2011, 79, 2856–2864. [Google Scholar] [CrossRef] [PubMed]
- Theriot, C.M.; Koumpouras, C.C.; Carlson, P.E.; Bergin, I.I.; Aronoff, D.M.; Young, V.B. Cefoperazone-treated mice as an experimental platform to assess differential virulence of Clostridium difficile strains. Gut Microbes 2011, 2, 326–334. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Butler, M.M.; Shinabarger, D.L.; Citron, D.M.; Kelly, C.P.; Dvoskin, S.; Wright, G.E.; Feng, H.; Tzipori, S.; Bowlin, T.L. MBX-500, a hybrid antibiotic with in vitro and in vivo efficacy against toxigenic Clostridium difficile. Antimicrob. Agents Chemother. 2012, 56, 4786–4792. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- AbdelKhalek, A.; Seleem, M.N. Repurposing the Veterinary Antiprotozoal Drug Ronidazole for the Treatment of Clostridioides difficile Infection. Int. J. Antimicrob. Agents 2020, 56, 106188. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Katchar, K.; Goldsmith, J.D.; Nanthakumar, N.; Cheknis, A.; Gerding, D.N.; Kelly, C.P. A mouse model of Clostridium difficile-associated disease. Gastroenterology 2008, 135, 1984–1992. [Google Scholar] [CrossRef] [PubMed]
- Ames, S.K.; Hysom, D.A.; Gardner, S.N.; Lloyd, G.S.; Gokhale, M.B.; Allen, J.E. Scalable metagenomic taxonomy classification using a reference genome database. Bioinformatics 2013, 29, 2253–2260. [Google Scholar] [CrossRef] [Green Version]
- Wood, D.E.; Salzberg, S.L. Kraken: Ultrafast metagenomic sequence classification using exact alignments. Genome Biol. 2014, 15, R46. [Google Scholar] [CrossRef] [Green Version]
- Amrane, S.; Hocquart, M.; Afouda, P.; Kuete, E.; Pham, T.P.; Dione, N.; Ngom, I.I.; Valles, C.; Bachar, D.; Raoult, D.; et al. Metagenomic and culturomic analysis of gut microbiota dysbiosis during Clostridium difficile infection. Sci. Rep. 2019, 9, 12807. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lesniak, N.A.; Schubert, A.M.; Flynn, K.J.; Leslie, J.L.; Sinani, H.; Bergin, I.L.; Young, V.B.; Schloss, P.D. The gut bacterial community potentiates Clostridioides difficile infection severity. Mbio 2022, 13, e01183-22. [Google Scholar] [CrossRef]
- Yacyshyn, B. Pathophysiology of Clostridium difficile-Associated Diarrhea. Gastroenterol. Hepatol. 2016, 12, 558–560. [Google Scholar]
- Kim, J.; Cho, Y.; Seo, M.R.; Bae, M.H.; Kim, B.; Rho, M.; Pai, H. Quantitative characterization of Clostridioides difficile population in the gut microbiome of patients with C. difficile infection and their association with clinical factors. Sci. Rep. 2020, 10, 17608. [Google Scholar] [CrossRef]
- Cavagnaro, C.; Berezin, S.; Medow, M.S. Corticosteroid treatment of severe, non-responsive Clostridium difficile induced colitis. Arch. Dis. Child. 2003, 88, 342–344. [Google Scholar] [CrossRef]
- Dirweesh, A.; Alvarez, C.; Khan, M.; Ambreen, B.; Yelisetti, R.; Hamiz, S.F.; Zia, S.; Tahir, M.; DeBari, V.A.; Christmas, D.; et al. Lack of Association Between the Clinical Outcome of Clostridium difficile Infection and Current Steroids Use. Gastroenterol. Res. 2017, 10, 116–119. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cardoso, R.A.; Filho, A.A.; Melo, M.C.; Lyerly, D.M.; Wilkins, T.D.; Lima, A.A.; Ribeiro, R.A.; Souza, G.E. Effects of anti-inflammatory drugs on fever and neutrophilia induced by Clostridium difficile toxin B. Mediat. Inflamm. 1996, 5, 183–187. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Carneiro-Filho, B.A.; Souza, M.L.; Lima, A.A.; Ribeiro, R.A. The effect of tumour necrosis factor (TNF) inhibitors in Clostridium difficile toxin-induced paw oedema and neutrophil migration. Pharmacol. Toxicol. 2001, 88, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Sykes, E.; McDonald, P.; Flanagan, P.K. Corticosteroids in the Treatment of Pseudomembranous Colitis: A Report of 3 Cases. Gastroenterol. Res. 2012, 5, 211–214. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Maseda, D.; Zackular, J.P.; Trindade, B.; Kirk, L.; Roxas, J.L.; Rogers, L.M.; Washington, M.K.; Du, L.; Koyama, T.; Viswanathan, V.K.; et al. Nonsteroidal Anti-inflammatory Drugs Alter the Microbiota and Exacerbate Clostridium difficile Colitis while Dysregulating the Inflammatory Response. mBio 2019, 10, e02282-18. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- McDermott, A.J.; Higdon, K.E.; Muraglia, R.; Erb-Downward, J.R.; Falkowski, N.R.; McDonald, R.A.; Young, V.B.; Huffnagle, G.B. The role of Gr-1(+) cells and tumour necrosis factor-alpha signalling during Clostridium difficile colitis in mice. Immunology 2015, 144, 704–716. [Google Scholar] [CrossRef] [Green Version]
- Munoz-Miralles, J.; Trindade, B.C.; Castro-Cordova, P.; Bergin, I.L.; Kirk, L.A.; Gil, F.; Aronoff, D.M.; Paredes-Sabja, D. Indomethacin increases severity of Clostridium difficile infection in mouse model. Future Microbiol. 2018, 13, 1271–1281. [Google Scholar] [CrossRef]
- Henry, J.; Neal, J.; Swaminath, A.; Korelitz, B.; Alexandra, F. The Utility of Anti-TNF Therapy in Treating Clostridium difficile Infections in Inflammatory Bowel Disease: A Case Series. Am. J. Gastroenterol. 2015, 110, S297. [Google Scholar]
- Garcia, P.; Chebli, L.; Tarsila, R.; Castro, C.A.; Pedro, G.; Fábio, P.; Kátia, B.; Lívia, C.; Roberta, R.; Bernardo, M.; et al. Steroids Use Is the Major Factor Associated With Clostridium Difficile Infection During IBD Flares in the Outpatient Setting. Am. J. Gastroenterol. 2018, 113, S19. [Google Scholar]
- Novack, L.; Kogan, S.; Gimpelevich, L.; Howell, M.; Borer, A.; Kelly, C.P.; Leffler, D.A.; Novack, V. Acid suppression therapy does not predispose to Clostridium difficile infection: The case of the potential bias. PLoS ONE 2014, 9, e110790. [Google Scholar] [CrossRef] [Green Version]
- Ressler, A.M.; Patel, A.; Rao, K. Evaluation of NSAID Exposure as a Risk Factor for Clostridium difficile infection: A Propensity-Score-Matched Case-Control Study. Open Forum Infect. Dis. 2020, 7, S439. [Google Scholar] [CrossRef]
- Bordon, Y. Cytokines: Oncostatin M-a new target in IBD? Nat. Rev. Immunol. 2017, 17, 280. [Google Scholar] [CrossRef] [PubMed]
- West, N.R.; Hegazy, A.N.; Owens, B.M.J.; Bullers, S.J.; Linggi, B.; Buonocore, S.; Coccia, M.; Gortz, D.; This, S.; Stockenhuber, K.; et al. Oncostatin M drives intestinal inflammation and predicts response to tumor necrosis factor-neutralizing therapy in patients with inflammatory bowel disease. Nat. Med. 2017, 23, 579–589. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whittaker, M.; Floyd, C.D.; Brown, P.; Gearing, A.J. Design and therapeutic application of matrix metalloproteinase inhibitors. Chem. Rev. 1999, 99, 2735–2776. [Google Scholar] [CrossRef] [PubMed]
- Ford, J.W.; McVicar, D.W. TREM and TREM-like receptors in inflammation and disease. Curr. Opin. Immunol. 2009, 21, 38–46. [Google Scholar] [CrossRef] [Green Version]
- Kokten, T.; Gibot, S.; Lepage, P.; D’Alessio, S.; Hablot, J.; Ndiaye, N.C.; Busby-Venner, H.; Monot, C.; Garnier, B.; Moulin, D.; et al. TREM-1 Inhibition Restores Impaired Autophagy Activity and Reduces Colitis in Mice. J. Crohn’s Colitis 2018, 12, 230–244. [Google Scholar] [CrossRef] [Green Version]
- MacFie, T.S.; Poulsom, R.; Parker, A.; Warnes, G.; Boitsova, T.; Nijhuis, A.; Suraweera, N.; Poehlmann, A.; Szary, J.; Feakins, R.; et al. DUOX2 and DUOXA2 form the predominant enzyme system capable of producing the reactive oxygen species H2O2 in active ulcerative colitis and are modulated by 5-aminosalicylic acid. Inflamm. Bowel Dis. 2014, 20, 514–524. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene ID | OSM 1 | MMP8 2 | Trem-1 3 | Duox2 4 | TNF-α 5 |
|---|---|---|---|---|---|
| Infected symptomatic versus uninfected | 11.0 | 15.3 | 11.9 | 9.8 | 7.3 |
| Infected symptomatic versus infected asymptomatic | 8.5 | 9.7 | 6.8 | 3.3 | 4.6 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
AbdelKhalek, A.; Narayanan, S.K. Comparison between Symptomatic and Asymptomatic Mice after Clostridioides difficile Infection Reveals Novel Inflammatory Pathways and Contributing Microbiota. Microorganisms 2022, 10, 2380. https://doi.org/10.3390/microorganisms10122380
AbdelKhalek A, Narayanan SK. Comparison between Symptomatic and Asymptomatic Mice after Clostridioides difficile Infection Reveals Novel Inflammatory Pathways and Contributing Microbiota. Microorganisms. 2022; 10(12):2380. https://doi.org/10.3390/microorganisms10122380
Chicago/Turabian StyleAbdelKhalek, Ahmed, and Sanjeev K. Narayanan. 2022. "Comparison between Symptomatic and Asymptomatic Mice after Clostridioides difficile Infection Reveals Novel Inflammatory Pathways and Contributing Microbiota" Microorganisms 10, no. 12: 2380. https://doi.org/10.3390/microorganisms10122380
APA StyleAbdelKhalek, A., & Narayanan, S. K. (2022). Comparison between Symptomatic and Asymptomatic Mice after Clostridioides difficile Infection Reveals Novel Inflammatory Pathways and Contributing Microbiota. Microorganisms, 10(12), 2380. https://doi.org/10.3390/microorganisms10122380