
Metagenomic Analyses of the Soybean Root Mycobiome and Microbiome Reveal Signatures of the Healthy and Diseased Plants Affected by Taproot Decline

 , and
, and 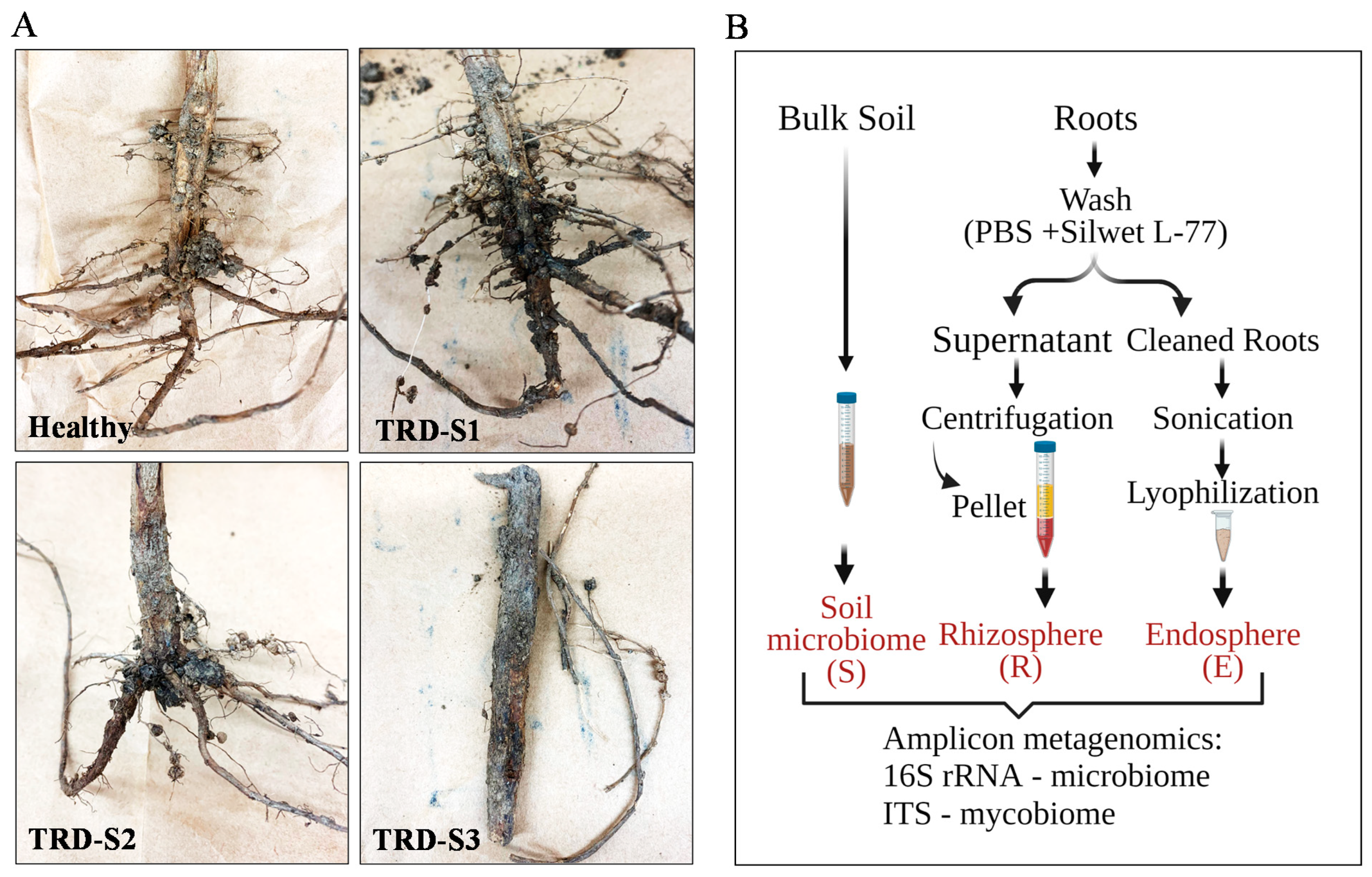{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Materials and Methods
3. Results
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Garcia-Aroca, T.; Price, P.P.; Tomaso-Peterson, M.; Allen, T.W.; Wilkerson, T.H.; Spurlock, T.N.; Faske, T.R.; Bluhm, B.; Conner, K.; Sikora, E. Xylaria necrophora, sp. nov., is an emerging root-associated pathogen responsible for taproot decline of soybean in the southern United States. Mycologia 2021, 113, 326–347. [Google Scholar] [CrossRef] [PubMed]
- Allen, T.; Bluhm, B.; Conner, K.; Doyle, V.; Price, T.; Sikora, E.; Singh, R.; Spurlock, T.; Tomaso-Peterson, M.; Wilkerson, T. First description of the causal agent of taproot decline of soybean, an emerging disease in the southern United States. Plant Health Prog. 2017, 18, 35–40. [Google Scholar] [CrossRef]
- Guyer, R.; Pate, S.; Garcia-Aroca, T.; Doyle, V.P.; Price, T.; Kelly, H. First Report of Taproot Decline Caused by Xylaria sp. on Soybean in Tennessee. Plant Dis. 2020, 104, 3267. [Google Scholar] [CrossRef]
- Aroca, T.G.; Doyle, V.; Price, P.P., III. Louisiana Plant Pathology Disease Identification and Management Series: Taproot Decline of Soybean (Xylaria necrophora). Available online: https://www.lsuagcenter.com/profiles/aiverson/articles/page1627567471819 (accessed on 17 April 2022).
- Tolbert, A.C.; Spurlock, T.N.; Hoyle, R. Understanding taproot decline: A potentially yield limiting soybean disease in Arkansas. Res. Ser. Agric. Exp. Stn. 2019, 44–48. [Google Scholar]
- Husbands, D.R.; Urbina, H.; Lewis, S.M.; Aime, M.C. Xylaria karyophthora: A new seed-inhabiting fungus of Greenheart from Guyana. Mycologia 2018, 110, 434–447. [Google Scholar] [CrossRef]
- Smith, D.A. Fungicidal control and related studies on black root rot of apple (Malus pumila Mill.) caused by Xylaria mali Fromme. Master’s Thesis, Virginia Polytechnic Institute and State University, Blacksburg, VA, USA, 1973. [Google Scholar]
- Ab Rahman, S.F.S.; Singh, E.; Pieterse, C.M.J.; Schenk, P.M. Emerging microbial biocontrol strategies for plant pathogens. Plant Sci. 2018, 267, 102–111. [Google Scholar] [CrossRef] [Green Version]
- Stenberg, J.A.; Heil, M.; Åhman, I.; Björkman, C. Optimizing crops for biocontrol of pests and disease. Trends Plant Sci. 2015, 20, 698–712. [Google Scholar] [CrossRef] [Green Version]
- Badial, A.; Nejat, N.S.; Tomaso-Peterson, M.; Popescu, S. Advances in biological control of Xylaria sp., the causal agent of taproot decline of soybean. Plant Health 2020 Online 2020. Available online: https://apsnet.confex.com/apsnet/2020/meetingapp.cgi/Paper/16390 (accessed on 17 April 2022).
- de Almeida Lopes, K.B.; Carpentieri-Pipolo, V.; Fira, D.; Balatti, P.A.; López, S.M.Y.; Oro, T.H.; Stefani Pagliosa, E.; Degrassi, G. Screening of bacterial endophytes as potential biocontrol agents against soybean diseases. J. Appl. Microbiol. 2018, 125, 1466–1481. [Google Scholar] [CrossRef] [Green Version]
- Pollak, S.; Cordero, O.X. Rhizobiome shields plants from infection. Nat. Microbiol. 2020, 5, 978–979. [Google Scholar] [CrossRef]
- Compant, S.; Samad, A.; Faist, H.; Sessitsch, A. A review on the plant microbiome: Ecology, functions, and emerging trends in microbial application. J. Adv. Res. 2019, 19, 29–37. [Google Scholar] [CrossRef]
- Yeoh, Y.K.; Dennis, P.G.; Paungfoo-Lonhienne, C.; Weber, L.; Brackin, R.; Ragan, M.A.; Schmidt, S.; Hugenholtz, P. Evolutionary conservation of a core root microbiome across plant phyla along a tropical soil chronosequence. Nat. Commun. 2017, 8, 215. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; Sugiyama, S. Phylogenetic signal of host plants in the bacterial and fungal root microbiomes of cultivated angiosperms. Plant J. 2020, 104, 522–531. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, P.R.; Mauchline, T.H. Who’s who in the plant root microbiome? Nat. Biotechnol. 2012, 30, 961–962. [Google Scholar] [CrossRef] [PubMed]
- Berg, G.; Grube, M.; Schloter, M.; Smalla, K. The plant microbiome and its importance for plant and human health. Front. Microbiol. 2014, 5, 491. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottel, N.R.; Castro, H.F.; Kerley, M.; Yang, Z.; Pelletier, D.A.; Podar, M.; Karpinets, T.; Uberbacher, E.D.; Tuskan, G.A.; Vilgalys, R. Distinct microbial communities within the endosphere and rhizosphere of Populus deltoides roots across contrasting soil types. Appl. Environ. Microbiol. 2011, 77, 5934–5944. [Google Scholar] [CrossRef] [Green Version]
- Urbina, H.; Breed, M.F.; Zhao, W.; Gurrala, K.L.; Andersson, S.G.E.; Ågren, J.; Baldauf, S.; Rosling, A. Specificity in Arabidopsis thaliana recruitment of root fungal communities from soil and rhizosphere. Fungal Biol. 2018, 122, 231–240. [Google Scholar] [CrossRef]
- Lundberg, D.S.; Lebeis, S.L.; Paredes, S.H.; Yourstone, S.; Gehring, J.; Malfatti, S.; Tremblay, J.; Engelbrektson, A.; Kunin, V.; Del Rio, T.G. Defining the core Arabidopsis thaliana root microbiome. Nature 2012, 488, 86–90. [Google Scholar] [CrossRef] [Green Version]
- Trivedi, P.; Leach, J.E.; Tringe, S.G.; Sa, T.; Singh, B.K. Plant–microbiome interactions: From community assembly to plant health. Nat. Rev. Microbiol. 2020, 18, 607–621. [Google Scholar] [CrossRef]
- Berg, G.; Köberl, M.; Rybakova, D.; Müller, H.; Grosch, R.; Smalla, K. Plant microbial diversity is suggested as the key to future biocontrol and health trends. FEMS Microbiol. Ecol. 2017, 93. [Google Scholar] [CrossRef]
- Munoz-Ucros, J.; Zwetsloot, M.J.; Cuellar-Gempeler, C.; Bauerle, T.L. Spatiotemporal patterns of rhizosphere microbiome assembly: From ecological theory to agricultural application. J. Appl. Ecol. 2021, 58, 894–904. [Google Scholar] [CrossRef]
- Vetterlein, D.; Lippold, E.; Schreiter, S.; Phalempin, M.; Fahrenkampf, T.; Hochholdinger, F.; Marcon, C.; Tarkka, M.; Oburger, E.; Ahmed, M. Experimental platforms for the investigation of spatiotemporal patterns in the rhizosphere—laboratory and field scale. J. Plant Nutr. Soil Sci. 2021, 184, 35–50. [Google Scholar] [CrossRef]
- Moroenyane, I.; Tremblay, J.; Yergeau, É. Temporal and spatial interactions modulate the soybean microbiome. FEMS Microbiol. Ecol. 2021, 97, fiaa2062. [Google Scholar] [CrossRef] [PubMed]
- Bulgarelli, D.; Rott, M.; Schlaeppi, K.; van Themaat, E.V.L.; Ahmadinejad, N.; Assenza, F.; Rauf, P.; Huettel, B.; Reinhardt, R.; Schmelzer, E. Revealing structure and assembly cues for Arabidopsis root-inhabiting bacterial microbiota. Nature 2012, 488, 91–95. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Howell, K. Community succession of the grapevine fungal microbiome in the annual growth cycle. Environ. Microbiol. 2021, 23, 1842–1857. [Google Scholar] [CrossRef]
- Ma, Z.S.; Li, L.; Gotelli, N.J. Diversity-disease relationships and shared species analyses for human microbiome-associated diseases. ISME J. 2019, 13, 1911–1919. [Google Scholar] [CrossRef]
- Gilbert, J.A.; Blaser, M.J.; Caporaso, J.G.; Jansson, J.K.; Lynch, S.V.; Knight, R. Current understanding of the human microbiome. Nat. Med. 2018, 24, 392–400. [Google Scholar] [CrossRef]
- Anal, A.K.D.; Rai, S.; Singh, M.; Solanki, M.K. Plant Mycobiome: Current Research and Applications. In Phytobiomes: Current Insights and Future Vistas; Springer: Singapore, 2020; pp. 81–104. [Google Scholar]
- Cui, L.; Morris, A.; Ghedin, E. The human mycobiome in health and disease. Genome Med. 2013, 5, 63. [Google Scholar] [CrossRef] [Green Version]
- Allen, T.W.; Bradley, C.A.; Sisson, A.J.; Byamukama, E.; Chilvers, M.I.; Coker, C.M.; Collins, A.A.; Damicone, J.P.; Dorrance, A.E.; Dufault, N.S. Soybean yield loss estimates due to diseases in the United States and Ontario, Canada, from 2010 to 2014. Plant Health Prog. 2017, 18, 19–27. [Google Scholar] [CrossRef] [Green Version]
- Chong, J.; Liu, P.; Zhou, G.; Xia, J. Using MicrobiomeAnalyst for comprehensive statistical, functional, and meta-analysis of microbiome data. Nat. Protoc. 2020, 15, 799–821. [Google Scholar] [CrossRef]
- Kim, B.-R.; Shin, J.; Guevarra, R.B.; Lee, J.H.; Kim, D.W.; Seol, K.-H.; Lee, J.-H.; Kim, H.B.; Isaacson, R.E. Deciphering diversity indices for a better understanding of microbial communities. J. Microbiol. Biotechnol. 2017, 27, 2089–2093. [Google Scholar] [CrossRef] [Green Version]
- Peres-Neto, P.R.; Jackson, D.A.; Somers, K.M. How many principal components? Stopping rules for determining the number of non-trivial axes revisited. Comput. Stat. Data Anal. 2005, 49, 974–997. [Google Scholar] [CrossRef]
- Anderson, M.J. Permutational Multivariate Analysis of Variance (PERMANOVA); Balakrishnan, N., Colton, T., Everitt, B., Piegorsch, W., Ruggeri, F., Teugels, J.L., Eds.; John Wiley & Sons, Ltd.: Hoboken, NJ, USA, 2017; pp. 1–15. [Google Scholar]
- Friedman, J.; Alm, E.J. Inferring correlation networks from genomic survey data. PLoS Comput. Biol. 2012, 8, e1002687. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tu, Y.; Shen, H.-W. Visualizing changes of hierarchical data using treemaps. IEEE Trans. Vis. Comput. Graph. 2007, 13, 1286–1293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Little, A.E.F.; Robinson, C.J.; Peterson, S.B.; Raffa, K.F.; Handelsman, J. Rules of engagement: Interspecies interactions that regulate microbial communities. Annu. Rev. Microbiol. 2008, 62, 375–401. [Google Scholar] [CrossRef] [Green Version]
- Mannaa, M.; Seo, Y.-S. Plants under the attack of allies: Moving towards the plant pathobiome paradigm. Plants 2021, 10, 125. [Google Scholar] [CrossRef]
- Willis, J.R.; Gabaldón, T. The human oral microbiome in health and disease: From sequences to ecosystems. Microorganisms 2020, 8, 308. [Google Scholar] [CrossRef] [Green Version]
- Armour, C.R.; Nayfach, S.; Pollard, K.S.; Sharpton, T.J. A metagenomic meta-analysis reveals functional signatures of health and disease in the human gut microbiome. mSystems 2019, 4, e00332-18. [Google Scholar] [CrossRef] [Green Version]
- Gao, M.; Xiong, C.; Gao, C.; Tsui, C.K.M.; Wang, M.-M.; Zhou, X.; Zhang, A.-M.; Cai, L. Disease-induced changes in plant microbiome assembly and functional adaptation. Microbiome 2021, 9, 187. [Google Scholar] [CrossRef]
- Cui, Z.; Huntley, R.B.; Zeng, Q.; Steven, B. Temporal and spatial dynamics in the apple flower microbiome in the presence of the phytopathogen Erwinia amylovora. ISME J. 2021, 15, 318–329. [Google Scholar] [CrossRef]
- Masenya, K.; Thompson, G.D.; Tekere, M.; Makhalanyane, T.P.; Pierneef, R.E.; Rees, D.J.G. Pathogen infection influences a distinct microbial community composition in sorghum RILs. Plant Soil 2021, 463, 555–572. [Google Scholar] [CrossRef]
- Noman, M.; Ahmed, T.; Ijaz, U.; Shahid, M.; Li, D.; Manzoor, I.; Song, F. Plant–Microbiome crosstalk: Dawning from composition and assembly of microbial community to improvement of disease resilience in plants. Int. J. Mol. Sci. 2021, 22, 6852. [Google Scholar] [CrossRef] [PubMed]
- Nuccio, E.E.; Starr, E.; Karaoz, U.; Brodie, E.L.; Zhou, J.; Tringe, S.G.; Malmstrom, R.R.; Woyke, T.; Banfield, J.F.; Firestone, M.K. Niche differentiation is spatially and temporally regulated in the rhizosphere. ISME J. 2020, 14, 999–1014. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Trivedi, C.; Reich, P.B.; Maestre, F.T.; Hu, H.-W.; Singh, B.K.; Delgado-Baquerizo, M. Plant-driven niche differentiation of ammonia-oxidizing bacteria and archaea in global drylands. ISME J. 2019, 13, 2727–2736. [Google Scholar] [CrossRef] [Green Version]
- Huo, C.; Luo, Y.; Cheng, W. Rhizosphere priming effect: A meta-analysis. Soil Biol. Biochem. 2017, 111, 78–84. [Google Scholar] [CrossRef] [Green Version]
- Xu, Y.; Ge, Y.; Song, J.; Rensing, C. Assembly of root-associated microbial community of typical rice cultivars in different soil types. Biol. Fertil. Soils 2020, 56, 249–260. [Google Scholar] [CrossRef]
- Asaff-Torres, A.; Armendáriz-Ruiz, M.; Kirchmayr, M.; Rodríguez-Heredia, R.; Orozco, M.; Mateos-Díaz, J.C.; Figueroa-Yáñez, L.; Baqueiro-Peña, I.; Verdín, J. Rhizospheric microbiome profiling of Capsicum annuum L. cultivated in amended soils by 16S and internal transcribed spacer 2 rRNA amplicon metagenome sequencing. Genome Announc. 2017, 5, e00626-17. [Google Scholar] [CrossRef] [Green Version]
- Cordero, J.; de Freitas, J.R.; Germida, J.J. Bacterial microbiome associated with the rhizosphere and root interior of crops in Saskatchewan, Canada. Can. J. Microbiol. 2020, 66, 71–85. [Google Scholar] [CrossRef]
- Qi, X.; Wang, E.; Xing, M.; Zhao, W.; Chen, X. Rhizosphere and non-rhizosphere bacterial community composition of the wild medicinal plant Rumex patientia. World J. Microbiol. Biotechnol. 2012, 28, 2257–2265. [Google Scholar] [CrossRef]
- Wen, X.; Dubinsky, E.; Yao, W.U.; Rong, Y.; Fu, C. Wheat, maize and sunflower cropping systems selectively influence bacteria community structure and diversity in their and succeeding crop’s rhizosphere. J. Integr. Agric. 2016, 15, 1892–1902. [Google Scholar] [CrossRef] [Green Version]
- Hodge, A.; Fitter, A.H. Microbial mediation of plant competition and community structure. Funct. Ecol. 2013, 27, 865–875. [Google Scholar] [CrossRef]
- Vergine, M.; Meyer, J.B.; Cardinale, M.; Sabella, E.; Hartmann, M.; Cherubini, P.; De Bellis, L.; Luvisi, A. The Xylella fastidiosa-Resistant Olive Cultivar “Leccino” Has Stable Endophytic Microbiota during the Olive Quick Decline Syndrome (OQDS). Pathogens 2020, 9, 35. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Strom, N.; Hu, W.; Chen, S.; Bushley, K. Continuous Monoculture Shapes Root and Rhizosphere Fungal Communities of Corn and Soybean in Soybean Cyst Nematode-Infested Soil. Phytobiomes J. 2019, 3, 300–314. [Google Scholar] [CrossRef] [Green Version]
- Deyett, E.; Roper, M.C.; Ruegger, P.; Yang, J.-I.; Borneman, J.; Rolshausen, P.E. Microbial landscape of the grapevine endosphere in the context of Pierce’s disease. Phytobiomes 2017, 1, 138–149. [Google Scholar] [CrossRef]
- Sun, X.; Song, B.; Xu, R.; Zhang, M.; Gao, P.; Lin, H.; Sun, W. Root-associated (rhizosphere and endosphere) microbiomes of the Miscanthus sinensis and their response to the heavy metal contamination. J. Environ. Sci. 2021, 104, 387–398. [Google Scholar] [CrossRef]
- Harder, C.B.; Hesling, E.; Botnen, S.S.; Dima, B.; von Bonsdorff-Salminen, T.; Niskanen, T.; Jarvis, S.G.; Lorberau, K.E.; Ouimette, A.; Hester, A.; et al. Mycena species can be opportunist-generalist plant root invaders. bioRxiv 2021. [Google Scholar] [CrossRef]
- Niego, A.G.; Raspé, O.; Thongklang, N.; Charoensup, R.; Lumyong, S.; Stadler, M.; Hyde, K.D. Taxonomy, Diversity and Cultivation of the Oudemansielloid/Xeruloid Taxa Hymenopellis, Mucidula, Oudemansiella, and Xerula with Respect to Their Bioactivities: A Review. J. Fungi 2021, 7, 51. [Google Scholar] [CrossRef]
- Jen, W.-C.; Jones, G.A. Effects of chetomin on growth and acidic fermentation products of rumen bacteria. Can. J. Microbiol. 1983, 29, 1399–1404. [Google Scholar] [CrossRef]
- Mayhood, P.; Mirza, B.S. Soybean Root Nodule and Rhizosphere Microbiome: Distribution of Rhizobial and Nonrhizobial Endophytes. Appl. Environ. Microbiol. 2021, 87, e02884-20. [Google Scholar] [CrossRef]
- Gkarmiri, K.; Mahmood, S.; Ekblad, A.; Alström, S.; Högberg, N.; Finlay, R. Identifying the Active Microbiome Associated with Roots and Rhizosphere Soil of Oilseed Rape. Appl. Environ. Microbiol. 2022, 83, e01938-17. [Google Scholar] [CrossRef] [Green Version]
- Buckley, D.H.; Huangyutitham, V.; Hsu, S.-F.; Nelson, T.A. Stable isotope probing with 15N2 reveals novel noncultivated diazotrophs in soil. Appl. Environ. Microbiol. 2007, 73, 3196–3204. [Google Scholar] [CrossRef] [Green Version]
- Wang, P.-H.; Leu, Y.-L.; Ismail, W.; Tang, S.-L.; Tsai, C.-Y.; Chen, H.-J.; Kao, A.-T.; Chiang, Y.-R. Anaerobic and aerobic cleavage of the steroid core ring structure by Steroidobacter denitrificans [S]. J. Lipid Res. 2013, 54, 1493–1504. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sakai, M.; Hosoda, A.; Ogura, K.; Ikenaga, M. The growth of Steroidobacter agariperforans sp. nov., a novel agar-degrading bacterium isolated from soil, is enhanced by the diffusible metabolites produced by bacteria belonging to Rhizobiales. Microbes Environ. 2014, 29, ME13169. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alcaraz, L.D.; Peimbert, M.; Barajas, H.R.; Dorantes-Acosta, A.E.; Bowman, J.L.; Arteaga-Vázquez, M.A. Marchantia liverworts as a proxy to plants’ basal microbiomes. Sci. Rep. 2018, 8, 12712. [Google Scholar] [CrossRef] [PubMed]
- Rojas-Jimenez, K.; Hernandez, M.; Blanco, J.; Vargas, L.D.; Acosta-Vargas, L.G.; Tamayo, G. Richness of cultivable endophytic fungi along an altitudinal gradient in wet forests of Costa Rica. Fungal Ecol. 2016, 20, 124–131. [Google Scholar] [CrossRef]
- Zhang, Y.; Crous, P.W.; Schoch, C.L.; Hyde, K.D. Pleosporales. Fungal Divers. 2012, 53, 1–221. [Google Scholar] [CrossRef] [Green Version]
- Ma, A.; Zhuang, X.; Wu, J.; Cui, M.; Lv, D.; Liu, C.; Zhuang, G. Ascomycota members dominate fungal communities during straw residue decomposition in arable soil. PLoS ONE 2013, 8, e66146. [Google Scholar] [CrossRef] [Green Version]
- Soman, A.G.; Gloer, J.B.; Wicklow, D.T. Antifungal and Antibacterial Metabolites from a Sclerotium-Colonizing Isolate of Mortierella vinacea. J. Nat. Prod. 1999, 62, 386–388. [Google Scholar] [CrossRef]
- Watson, T.T.; Strauss, S.L.; Desaeger, J.A. Identification and characterization of Javanese root-knot nematode (Meloidogyne javanica) suppressive soils in Florida. Appl. Soil Ecol. 2020, 154, 103597. [Google Scholar] [CrossRef]
- Palizi, P.; Goltapeh, E.M.; Pourjam, E.; Safaie, N. Potential of oyster mushrooms for the biocontrol of sugar beet nematode (Heterodera schachtii). J. Plant Prot. Res. 2009, 49, 27–33. [Google Scholar] [CrossRef]
- Wieczorek, A.S.; Schmidt, O.; Chatzinotas, A.; von Bergen, M.; Gorissen, A.; Kolb, S. Ecological Functions of Agricultural Soil Bacteria and Microeukaryotes in Chitin Degradation: A Case Study. Front. Microbiol. 2019, 10, 1293. [Google Scholar] [CrossRef] [PubMed]
- Carrión, V.J.; Perez-Jaramillo, J.; Cordovez, V.; Tracanna, V.; de Hollander, M.; Ruiz-Buck, D.; Mendes, L.W.; van Ijcken, W.F.J.; Gomez-Exposito, R.; Elsayed, S.S.; et al. Pathogen-induced activation of disease-suppressive functions in the endophytic root microbiome. Science 2019, 366, 606–612. [Google Scholar] [CrossRef] [PubMed]
- Manriquez, B.; Muller, D.; Prigent-Combaret, C. Experimental evolution in plant-microbe systems: A tool for deciphering the functioning and evolution of plant-associated microbial communities. Front. Microbiol. 2021, 12, 896. [Google Scholar] [CrossRef] [PubMed]
- Lyu, D.; Msimbira, L.A.; Nazari, M.; Antar, M.; Pagé, A.; Shah, A.; Monjezi, N.; Zajonc, J.; Tanney, C.A.S.; Backer, R. The coevolution of plants and microbes underpins sustainable agriculture. Microorganisms 2021, 9, 1036. [Google Scholar] [CrossRef]







Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Popescu, S.C.; Tomaso-Peterson, M.; Wilkerson, T.; Bronzato-Badial, A.; Wesser, U.; Popescu, G.V. Metagenomic Analyses of the Soybean Root Mycobiome and Microbiome Reveal Signatures of the Healthy and Diseased Plants Affected by Taproot Decline. Microorganisms 2022, 10, 856. https://doi.org/10.3390/microorganisms10050856
Popescu SC, Tomaso-Peterson M, Wilkerson T, Bronzato-Badial A, Wesser U, Popescu GV. Metagenomic Analyses of the Soybean Root Mycobiome and Microbiome Reveal Signatures of the Healthy and Diseased Plants Affected by Taproot Decline. Microorganisms. 2022; 10(5):856. https://doi.org/10.3390/microorganisms10050856
Chicago/Turabian StylePopescu, Sorina C., Maria Tomaso-Peterson, Teresa Wilkerson, Aline Bronzato-Badial, Uyen Wesser, and George V. Popescu. 2022. "Metagenomic Analyses of the Soybean Root Mycobiome and Microbiome Reveal Signatures of the Healthy and Diseased Plants Affected by Taproot Decline" Microorganisms 10, no. 5: 856. https://doi.org/10.3390/microorganisms10050856
APA StylePopescu, S. C., Tomaso-Peterson, M., Wilkerson, T., Bronzato-Badial, A., Wesser, U., & Popescu, G. V. (2022). Metagenomic Analyses of the Soybean Root Mycobiome and Microbiome Reveal Signatures of the Healthy and Diseased Plants Affected by Taproot Decline. Microorganisms, 10(5), 856. https://doi.org/10.3390/microorganisms10050856