
Gene Silencing through CRISPR Interference in Mycoplasmas

 ,
, 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Bacterial Strains and Growth Conditions
2.2. Plasmid Construction
2.3. Transformation
2.4. Western Blot Analysis
2.5. RNA Purification and cDNA Synthesis
2.6. Quantitative RT-PCR
2.7. Proteomic Analysis
2.8. In Silico Analysis
3. Results and Discussion
3.1. Construction of a CRISPRi System for Silencing Gene Expression in Mycoplasma
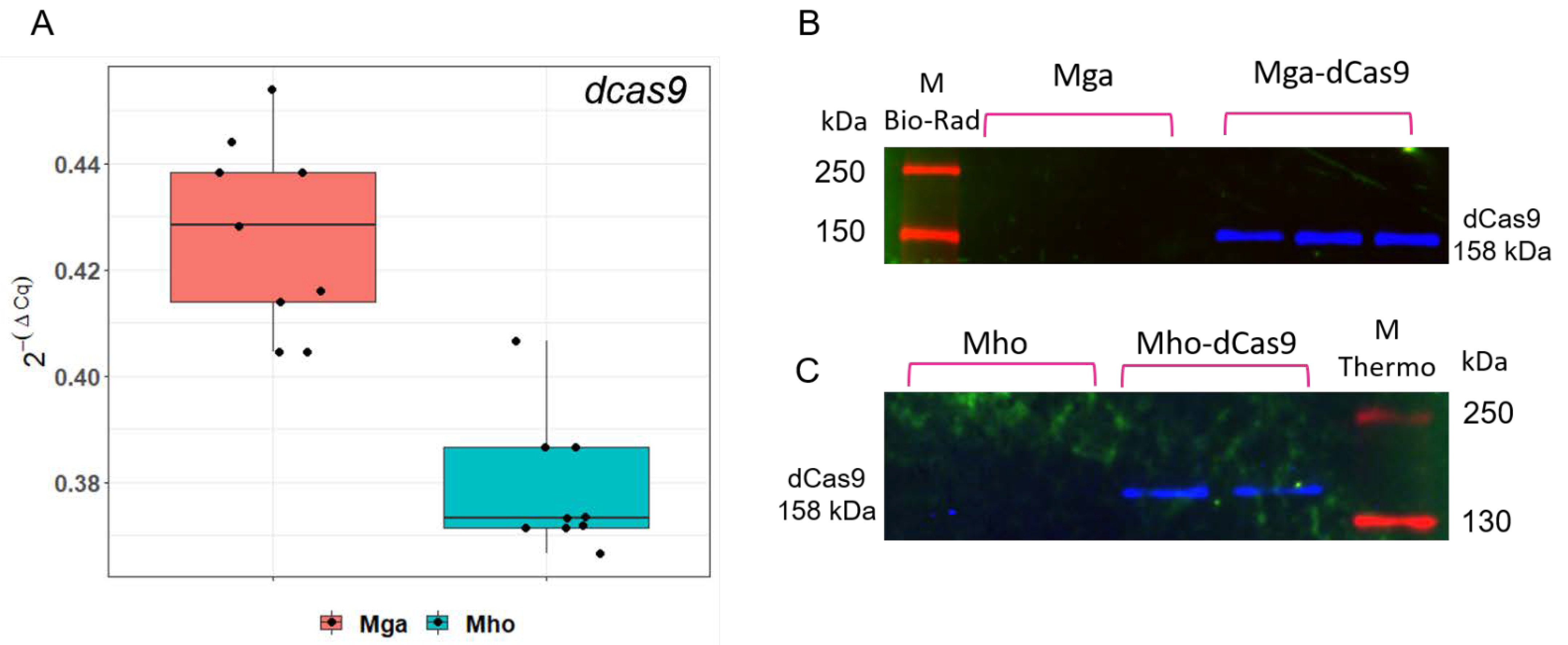
3.2. pRLM5L2-dcas9 Provides dcas9 Expression at Both mRNA and Protein Levels in Two Mycoplasma Species
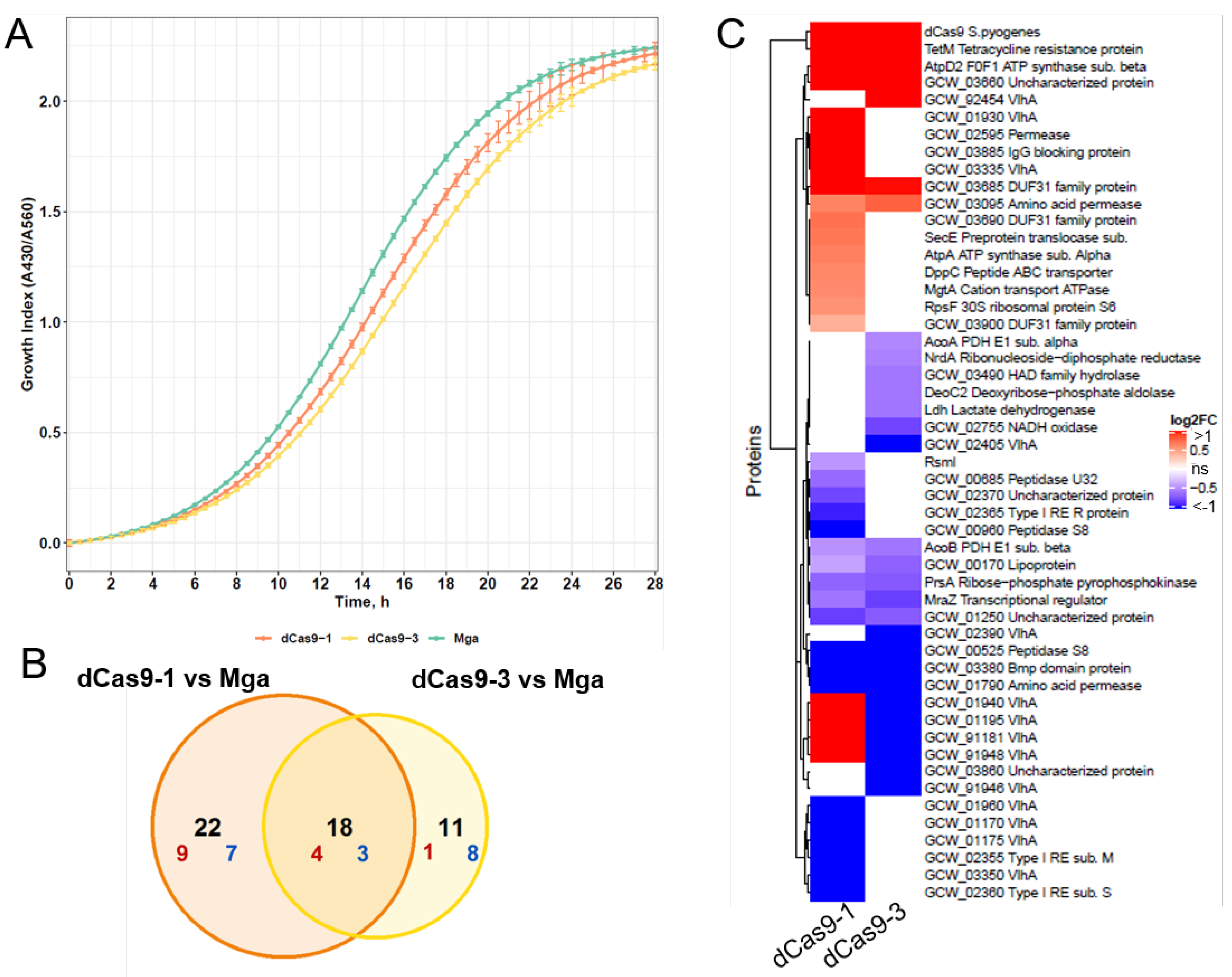
3.3. The Selected Expression Level of dCas9 Does Not Provide a Toxic Effect for M. gallisepticum
3.4. Design of sgRNA for Rapid Assembly and Cloning into pRLM5L2-dcas9
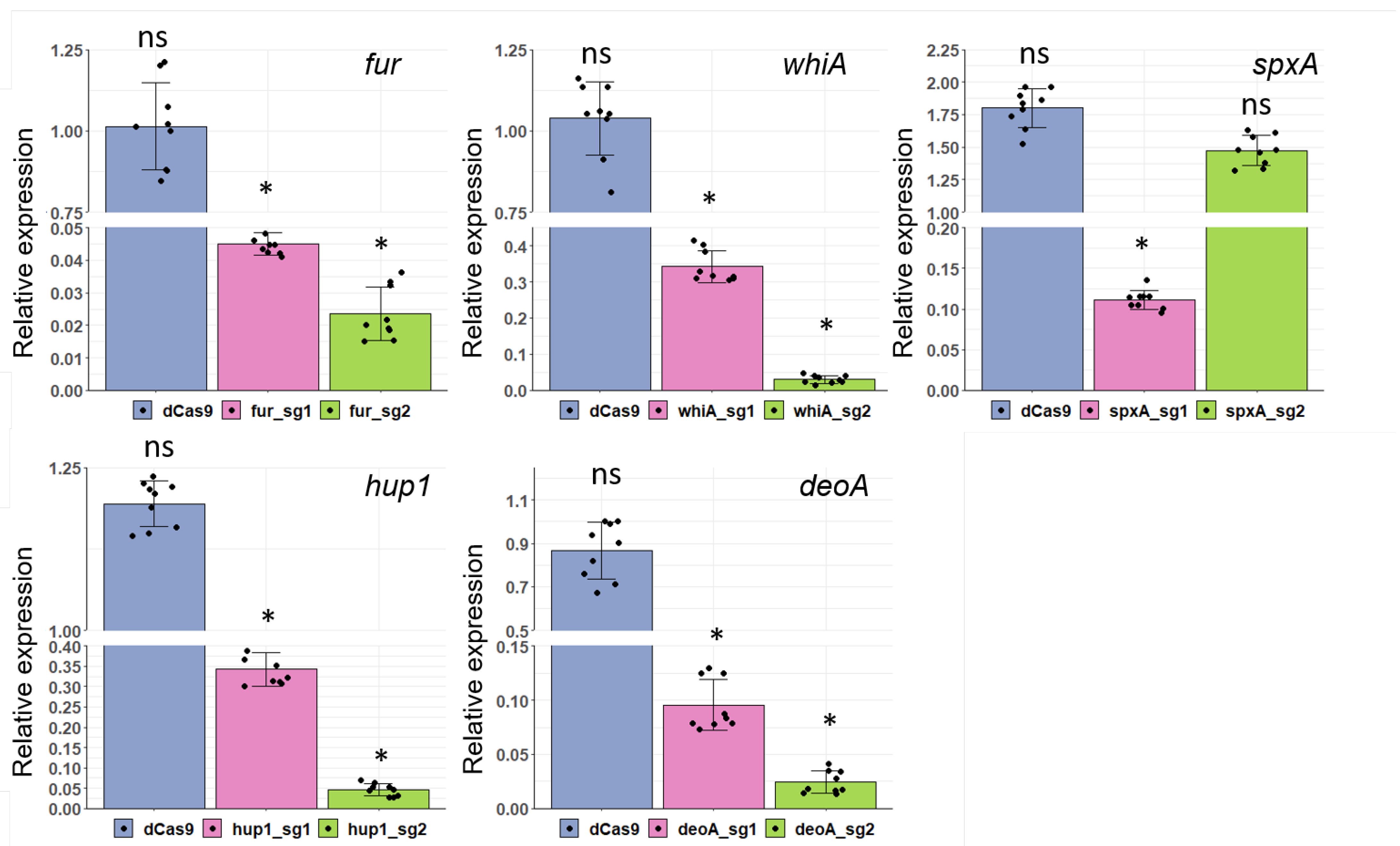
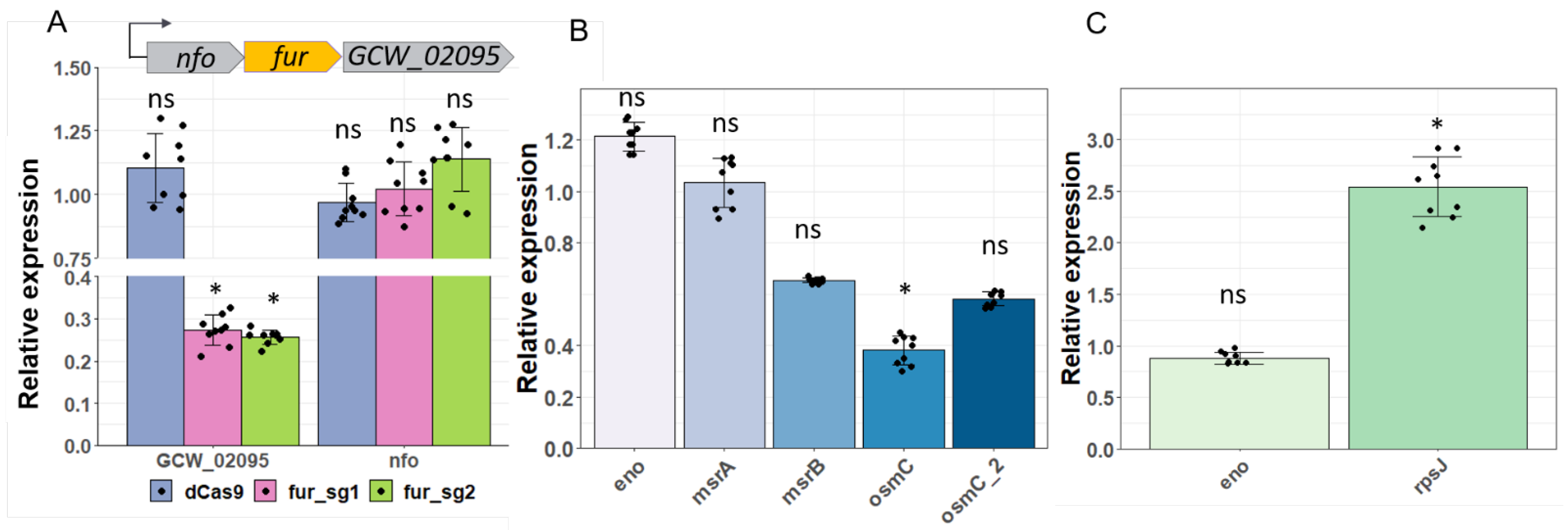
3.5. Application of CRISPRi for Gene Knockdown in Two Mycoplasma Species
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Razin, S. The mycoplasmas. Microbiol. Rev. 1978, 42, 414–470. [Google Scholar] [CrossRef] [PubMed]
- Pereyre, S.; Sirand-Pugnet, P.; Beven, L.; Charron, A.; Renaudin, H.; Barré, A.; Avenaud, P.; Jacob, D.; Couloux, A.; Barbe, V.; et al. Life on arginine for Mycoplasma hominis: Clues from its minimal genome and comparison with other human urogenital mycoplasmas. PLoS Genet 2009, 5, e1000677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Razin, S.; Yogev, D.; Naot, Y. Molecular Biology and Pathogenicity of Mycoplasmas. Microbiol. Mol. Biol. Rev. 1998, 62, 1094–1156. [Google Scholar] [CrossRef] [Green Version]
- Hutchison, C.A.; Chuang, R.Y.; Noskov, V.N.; Assad-Garcia, N.; Deerinck, T.J.; Ellisman, M.H.; Gill, J.; Kannan, K.; Karas, B.J.; Ma, L.; et al. Design and synthesis of a minimal bacterial genome. Science 2016, 351, aad6253. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisunov, G.Y.; Zubov, A.I.; Pobeguts, O.V.; Varizhuk, A.M.; Galyamina, M.A.; Evsyutina, D.V.; Semashko, T.A.; Manuvera, V.A.; Kovalchuk, S.I.; Ziganshin, R.K.; et al. The Dynamics of Mycoplasma gallisepticum Nucleoid Structure at the Exponential and Stationary Growth Phases. Front. Microbiol. 2021, 12, 753760. [Google Scholar] [CrossRef]
- Yavlovich, A.; Rechnitzer, H.; Rottem, S. Enolase Resides on the Cell Surface of Mycoplasma fermentans and Binds Plasminogen. Infect. Immun. 2007, 75, 5716–5719. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Alvarez, R.A.; Blaylock, M.W.; Baseman, J.B. Surface localized glyceraldehyde-3-phosphate dehydrogenase of Mycoplasma genitalium binds mucin. Mol. Microbiol. 2003, 48, 1417–1425. [Google Scholar] [CrossRef] [PubMed]
- Balasubramanian, S.; Kannan, T.R.; Baseman, J.B. The Surface-Exposed Carboxyl Region of Mycoplasma pneumoniae Elongation Factor Tu Interacts with Fibronectin. Infect. Immun. 2008, 76, 3116–3123. [Google Scholar] [CrossRef] [Green Version]
- Glass, J.I.; Assad-garcia, N.; Alperovich, N.; Yooseph, S.; Lewis, M.R.; Maruf, M.; Iii, C.A.H.; Smith, H.O.; Venter, J.C. Essential genes of a minimal bacterium. Proc. Natl. Acad. Sci. USA 2006, 103, 425–430. [Google Scholar] [CrossRef] [Green Version]
- Lin, Y.; Zhang, R.R. Putative essential and core-essential genes in Mycoplasma genomes. Sci. Rep. 2001, 53, 1–7. [Google Scholar] [CrossRef] [Green Version]
- Dybvig, K.; French, C.T.; Voelker, L.R.L. Construction and use of derivatives of transposon Tn4001 that function in Mycoplasma pulmonis and Mycoplasma arthritidis. J. Bacteriol. 2000, 182, 4343–4347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rideau, F.; Le Roy, C.; Sagné, E.; Renaudin, H.; Pereyre, S.; Henrich, B.; Dordet-Frisoni, E.; Citti, C.; Lartigue, C.; Bébéar, C. Random transposon insertion in the Mycoplasma hominis minimal genome. Sci. Rep. 2019, 9, 1–11. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisunov, G.Y.; Evsyutina, D.V.; Semashko, T.A.; Arzamasov, A.A.; Manuvera, V.A.; Letarov, A.V.; Govorun, V.M. Binding site of MraZ transcription factor in Mollicutes. Biochimie 2016, 125, 59–65. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Matyushkina, D.; Pobeguts, O.; Butenko, I.; Vanyushkina, A.; Anikanov, N.; Bukato, O.; Evsyutina, D.; Bogomazova, A.; Lagarkova, M.; Semashko, T.; et al. Phase Transition of the Bacterium upon Invasion of a Host Cell as a Mechanism of Adaptation: A Mycoplasma gallisepticum Model. Sci. Rep. 2016, 6, 35959. [Google Scholar] [CrossRef] [Green Version]
- Maglennon, G.A.; Cook, B.S.; Matthews, D.; Deeney, A.S.; Bossé, J.T.; Langford, P.R.; Maskell, D.J.; Tucker, A.W.; Wren, B.W.; Rycroft, A.N. Development of a self-replicating plasmid system for Mycoplasma hyopneumoniae. Vet. Res. 2013, 44, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ishag, H.Z.A.; Xiong, Q.; Liu, M.; Feng, Z.; Shao, G. Development of oriC-plasmids for use in Mycoplasma hyorhinis. Sci. Rep. 2017, 7, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Qi, L.S.; Larson, M.H.; Gilbert, L.A.; Doudna, J.A.; Weissman, J.S.; Arkin, A.P.; Lim, W.A. Repurposing CRISPR as an RNA-γuided platform for sequence-specific control of gene expression. Cell 2013, 152, 1173–1183. [Google Scholar] [CrossRef] [Green Version]
- Mariscal, A.M.; Kakizawa, S.; Hsu, J.Y.; Tanaka, K.; González-González, L.; Broto, A.; Querol, E.; Lluch-Senar, M.; Piñero-Lambea, C.; Sun, L.; et al. Tuning Gene Activity by Inducible and Targeted Regulation of Gene Expression in Minimal Bacterial Cells. ACS Synth. Biol. 2018, 7, 1538–1552. [Google Scholar] [CrossRef]
- Mahdizadeh, S.; Sansom, F.M.; Lee, S.; Browning, G.F. Targeted mutagenesis of Mycoplasma gallisepticum using its endogenous CRISPR/Cas system Targeted mutagenesis of Mycoplasma gallisepticum using its endogenous CRISPR/Cas system. Vet. Microbiol. 2020, 250, 108868. [Google Scholar] [CrossRef]
- Klose, S.M.; Wawegama, N.; Sansom, F.M.; Marenda, M.S.; Browning, G.F. Efficient disruption of the function of the mnuA nuclease gene using the endogenous CRISPR/Cas system in Mycoplasma gallisepticum. Vet. Microbiol. 2022, 269, 109436. [Google Scholar] [CrossRef]
- Ipoutcha, T.; Tsarmpopoulos, I.; Talenton, V.; Gaspin, C.; Moisan, A.; Walker, C.A.; Brownlie, J.; Blanchard, A.; Thebault, P.; Sirand-Pugnet, P. Multiple Origins and Specific Evolution of CRISPR/Cas9 Systems in Minimal Bacteria (Mollicutes). Front. Microbiol. 2019, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Levitskii, S.; Mazur, A.K.; Govorun, V. Mycoplasma gallisepticum Produces a Histone-like Protein That Recognizes Base Mismatches in DNA. Biochemistry 2011, 50, 8692–8702. [Google Scholar]
- Yus, E.; Maier, T.; Michalodimitrakis, K.; Van Noort, V.; Yamada, T.; Chen, W.-H.; Wodke, J.H.; Güell, M.; Martínez, S.; Bourgeois, R.; et al. Impact of genome reduction on bacterial metabolism and its regulation. Science 2009, 326, 1263–1268. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mazin, P.V.; Fisunov, G.Y.; Gorbachev, A.Y.; Kapitskaya, K.Y.; Altukhov, I.A.; Semashko, T.A.; Alexeev, D.G.; Govorun, V.M. Transcriptome analysis reveals novel regulatory mechanisms in a genome-reduced bacterium. Nucleic Acids Res. 2014, 42, 13254–13268. [Google Scholar] [CrossRef]
- Gorbachev, A.Y.; Fisunov, G.Y.; Izraelson, M.; Evsyutina, D.V.; Mazin, P.V.; Alexeev, D.G.; Pobeguts, O.V.; Gorshkova, T.N.; Kovalchuk, S.I.; Kamashev, D.E.; et al. DNA repair in Mycoplasma gallisepticum. BMC Genom. 2013, 14, 726. [Google Scholar] [CrossRef] [Green Version]
- Garanina, I.A.; Fisunov, G.Y.; Govorun, V.M. BAC-BROWSER: The tool for visualization and analysis of prokaryotic genomes. Front. Microbiol. 2018, 9, 1–6. [Google Scholar] [CrossRef]
- Semashko, T.A.; Evsyutina, D.V.; Ladygina, V.G.; Zubov, A.I.; Rakovskaya, I.V.; Kovalchuk, S.I.; Ziganshin, R.H.; Pobeguts, O.V. Data on proteome of Mycoplasma hominis cultivated with arginine or thymidine as a carbon source. Data Brief 2020, 31, 106034. [Google Scholar] [CrossRef]
- Kovalchuk, S.I.; Jensen, O.N.; Rogowska-Wrzesinska, A. FlashPack: Fast and simple preparation of ultrahigh-performance capillary columns for LC-MS. Mol. Cell Proteom. 2019, 18, 383–390. [Google Scholar] [CrossRef] [Green Version]
- Zhang, X.; Smits, A.H.; Van Tilburg, G.B.A.; Ovaa, H.; Huber, W.; Vermeulen, M. Proteome-wide identification of ubiquitin interactions using UbIA-MS. Nat. Protoc. 2018, 13, 530–550. [Google Scholar] [CrossRef]
- Ferretti, J.J.; McShan, W.M.; Ajdic, D.; Savic, D.J.; Savic, G.; Lyon, K.; Primeaux, C.; Sezate, S.; Suvorov, A.N.; Kenton, S.; et al. Complete genome sequence of an M1 strain of streptococcus pyogenes. Proc. Natl. Acad. Sci. USA 2001, 98, 4658–4663. [Google Scholar] [CrossRef] [Green Version]
- Cho, S.; Choe, D.; Lee, E.; Kim, S.C.; Palsson, B.; Cho, B.K. High-Level dCas9 Expression Induces Abnormal Cell Morphology in Escherichia coli. ACS Synth. Biol. 2018, 7, 1085–1094. [Google Scholar] [CrossRef] [PubMed]
- McArthur, J.V. Microbial Ecology: An Evolutionary Approach; Academic Press: Oxford, UK, 2006. [Google Scholar]
- Béven, L.; Charenton, C.; Dautant, A.; Bouyssou, G.; Labroussaa, F.; Sköllermo, A.; Persson, A.; Blanchard, A.; Sirand-Pugnet, P. Specific evolution of F 1-like ATPases in mycoplasmas. PLoS ONE 2012, 7, e38793. [Google Scholar] [CrossRef] [PubMed]
- Arfi, Y.; Minder, L.; Di Primo, C.; Le Roy, A.; Ebel, C.; Coquet, L.; Claverol, S.; Vashee, S.; Jores, J.; Blanchard, A.; et al. MIB–MIP is a mycoplasma system that captures and cleaves immunoglobulin G. Proc. Natl. Acad. Sci. USA 2016, 113, 5406–5411. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hosie, A.H.F.; Poole, P.S. Bacterial ABC transporters of amino acids. Res. Microbiol. 2001, 152, 259–270. [Google Scholar] [CrossRef]
- Deka, R.K.; Brautigam, C.A.; Yang, X.F.; Blevins, J.S.; Machius, M.; Tomchick, D.R.; Norgard, M.V. The PnrA (Tp0319; TmpC) lipoprotein represents a new family of bacterial purine nucleoside receptor encoded within an ATP-binding cassette (ABC)-like operon in Treponema pallidum. J. Biol. Chem. 2006, 281, 8072–8081. [Google Scholar] [CrossRef] [Green Version]
- Howell, M.; Dumitrescu, D.G.; Blankenship, L.R.; Herkert, D.; Hatzios, S.K. Functional characterization of a subtilisin-like serine protease from Vibrio cholerae. J. Biol. Chem. 2019, 294, 9888–9900. [Google Scholar] [CrossRef]
- Pannkuk, E.L.; Risch, T.S.; Savary, B.J. Isolation and identification of an extracellular subtilisin-like serine protease secreted by the Bat Pathogen Pseudogymnoascus destructans. PLoS ONE 2015, 10, e0120508. [Google Scholar] [CrossRef] [Green Version]
- Pflaum, K.; Tulman, E.R.; Beaudet, J.; Liao, X.; Geary, S.J. Global Changes in Mycoplasma gallisepticum Phase-Variable Lipoprotein Gene vlhA Expression during In Vivo Infection of the Natural Chicken Host. Infect. Immun. 2016, 84, 351–355. [Google Scholar] [CrossRef] [Green Version]
- Pflaum, K.; Tulman, E.R.; Beaudet, J.; Canter, J.; Geary, S.J. Variable Lipoprotein Hemagglutinin A Gene (vlhA) Expression in Variant Mycoplasmagallisepticum Strains In Vivo. Infect Immun 2018, 86, 365–374. [Google Scholar] [CrossRef] [Green Version]
- Lluch-Senar, M.; Delgado, J.; Chen, W.-H.; Llorens-Rico, V.; O’Reilly, F.J.; Wodke, J.A.; Unal, E.B.; Yus, E.; Martinez, S.; Nichols, R.J.; et al. Defining a minimal cell: Essentiality of small ORFs and ncRNAs in a genome-reduced bacterium. Mol. Syst. Biol. 2015, 11, 780. [Google Scholar] [CrossRef]
- Fisunov, G.Y.; Garanina, I.A.; Evsyutina, D.V.; Semashko, T.A.; Nikitina, A.S.; Govorun, V.M. Reconstruction of transcription control networks in mollicutes by high-throughput identification of promoters. Front. Microbiol. 2016, 7, 1–15. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yus, E.; Lloréns-Rico, V.; Martínez, S.; Gallo, C.; Eilers, H.; Blötz, C.; Stülke, J.; Lluch-Senar, M.; Serrano, L. Determination of the Gene Regulatory Network of a Genome-Reduced Bacterium Highlights Alternative Regulation Independent of Transcription Factors. Cell Syst. 2019, 9, 143–158.e13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fisunov, G.Y.; Pobeguts, O.V.; Ladygina, V.G.; Zubov, A.I.; Galyamina, M.A.; Kovalchuk, S.I.; Ziganshin, R.K.; Evsyutina, D.V.; Matyushkina, D.S.; Butenko, I.O.; et al. Thymidine utilisation pathway is a novel phenotypic switch of Mycoplasma hominis. J. Med. Microbiol. 2022, 71, 1468. [Google Scholar] [CrossRef] [PubMed]
- Peters, J.M.; Colavin, A.; Shi, H.; Czarny, T.L.; Larson, M.H.; Wong, S.; Hawkins, J.S.; Lu, C.H.S.; Koo, B.M.; Marta, E.; et al. A comprehensive, CRISPR-based functional analysis of essential genes in bacteria. Cell 2016, 165, 1493–1506. [Google Scholar] [CrossRef] [Green Version]
- Mateos, J.L.; Tilmes, V.; Madrigal, P.; Severing, E.; Richter, R.; Rijkenberg, C.W.M.; Krajewski, P.; Coupland, G. Divergence of regulatory networks governed by the orthologous transcription factors FLC and PEP1 in Brassicaceae species. Proc. Natl. Acad. Sci. USA 2017, 114, E11037–E11046. [Google Scholar] [CrossRef] [Green Version]
- Schäfer, H.; Turgay, K. Spx, a versatile regulator of the Bacillus subtilis stress response. Curr. Genet. 2019, 65, 871–876. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Name of sgRNA | 20-nt Base-Pairing Region | Tm, °C | GC, % | Average Repression Range, Fold ± SE |
|---|---|---|---|---|
| fur-sg1 | ACCTTCTAGGTTTGGTGATT | 58.5 | 40 | 23 ± 0.6 |
| fur-sg2 | ACGAATCAACAAGATTGAGT | 57.4 | 35 | 43 ± 8.8 |
| whiA-sg1 | CATTTTGCACCAACAAGTCG | 60.9 | 45 | 3 ± 0.2 |
| whiA-sg2 | AGATGATCGTTTTTGGGTGT | 59.9 | 40 | 33 ± 8.3 |
| spxA-sg1 | ATAATACTATATTGTTCGTC | 49.4 | 25 | 9 ± 0.2 |
| spxA-sg2 | GTTATTTTGATTATCTGTCA | 50.7 | 25 | 0.7 ± 0.02 |
| hup1-sg1 | AAACAAGCCTTAACAAGTTT | 56.2 | 30 | 3 ± 0.1 |
| hup1-sg2 | ACTCTAAAAGTACCAAGTTC | 54.5 | 35 | 22 ± 2.8 |
| deoA-sg1 | AAAGCGGCAGCTTGGTAGTC | 65.6 | 55 | 11 ± 1.2 |
| deoA-sg2 | GCCTGAATGCATCATAGCGT | 63.3 | 50 | 42 ± 8.5 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Evsyutina, D.V.; Fisunov, G.Y.; Pobeguts, O.V.; Kovalchuk, S.I.; Govorun, V.M. Gene Silencing through CRISPR Interference in Mycoplasmas. Microorganisms 2022, 10, 1159. https://doi.org/10.3390/microorganisms10061159
Evsyutina DV, Fisunov GY, Pobeguts OV, Kovalchuk SI, Govorun VM. Gene Silencing through CRISPR Interference in Mycoplasmas. Microorganisms. 2022; 10(6):1159. https://doi.org/10.3390/microorganisms10061159
Chicago/Turabian StyleEvsyutina, Daria V., Gleb Y. Fisunov, Olga V. Pobeguts, Sergey I. Kovalchuk, and Vadim M. Govorun. 2022. "Gene Silencing through CRISPR Interference in Mycoplasmas" Microorganisms 10, no. 6: 1159. https://doi.org/10.3390/microorganisms10061159
APA StyleEvsyutina, D. V., Fisunov, G. Y., Pobeguts, O. V., Kovalchuk, S. I., & Govorun, V. M. (2022). Gene Silencing through CRISPR Interference in Mycoplasmas. Microorganisms, 10(6), 1159. https://doi.org/10.3390/microorganisms10061159