
Changes of the Freshwater Microbial Community Structure and Assembly Processes during Different Sample Storage Conditions



Abstract
:1. Introduction
2. Materials and Methods
2.1. Experimental Design and Sampling
2.2. DNA Extraction, PCR Amplification and Sequencing
2.3. Sequence Data Processing and Taxonomic Assignment
2.4. Network Analysis
2.5. Neutral Community Model
2.6. Community Co-Occurrence Pattern
2.7. Statistical Analyses
3. Results
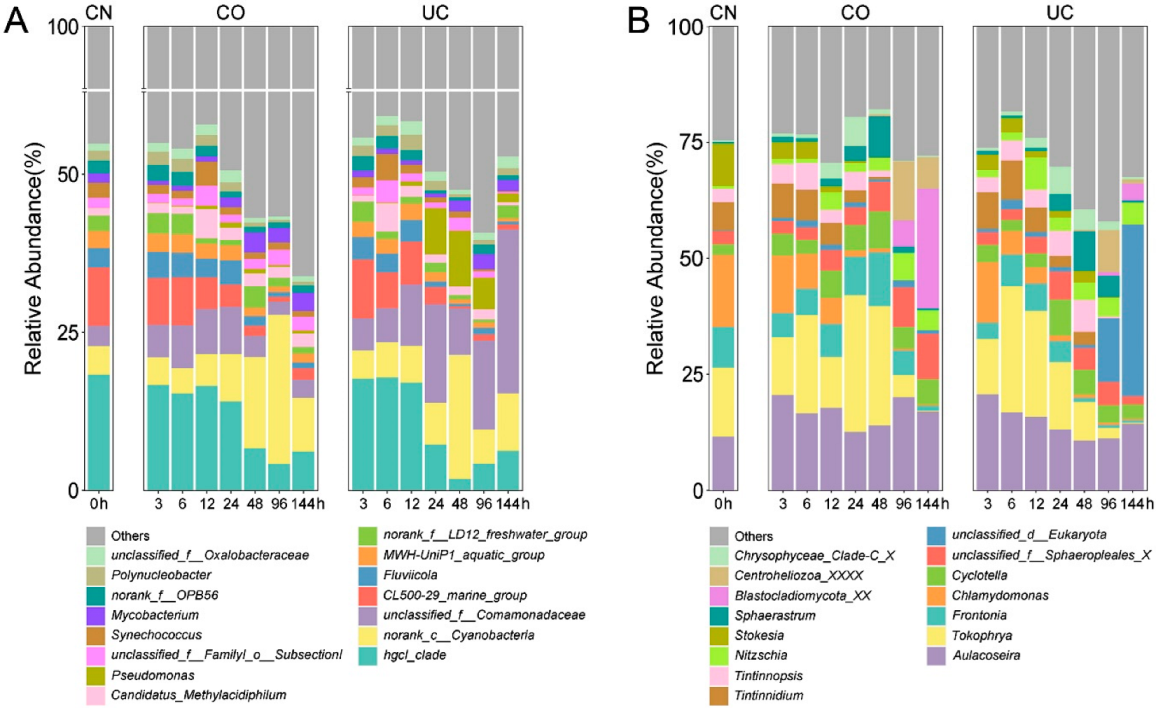
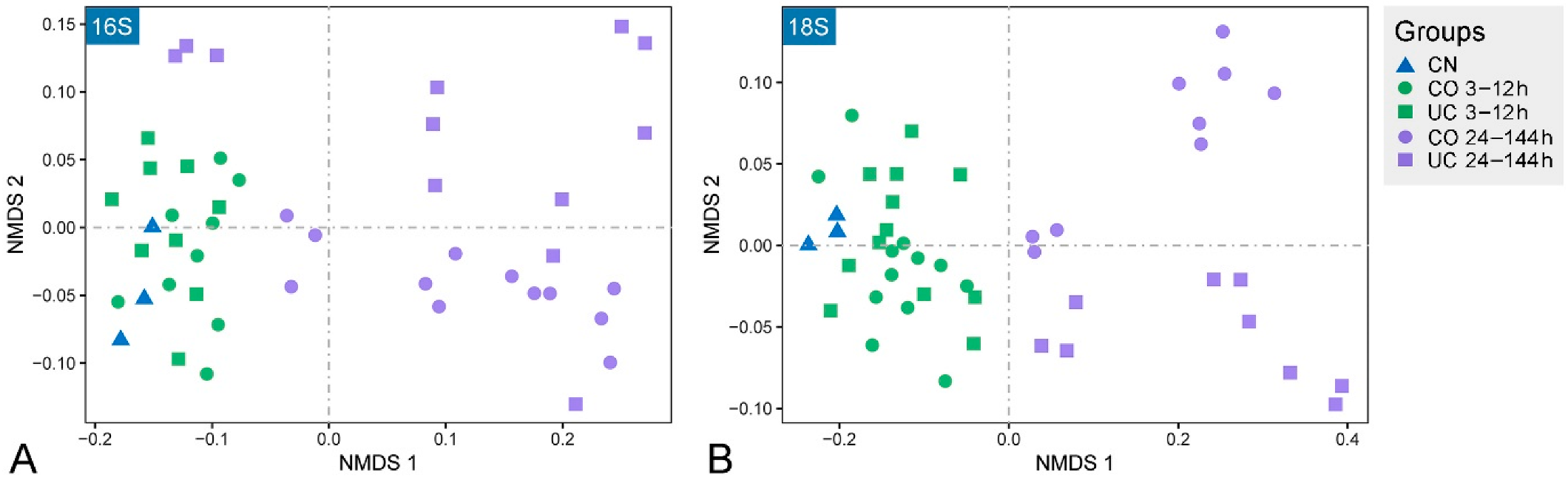
3.1. Effect of Sample Storage on Community Composition
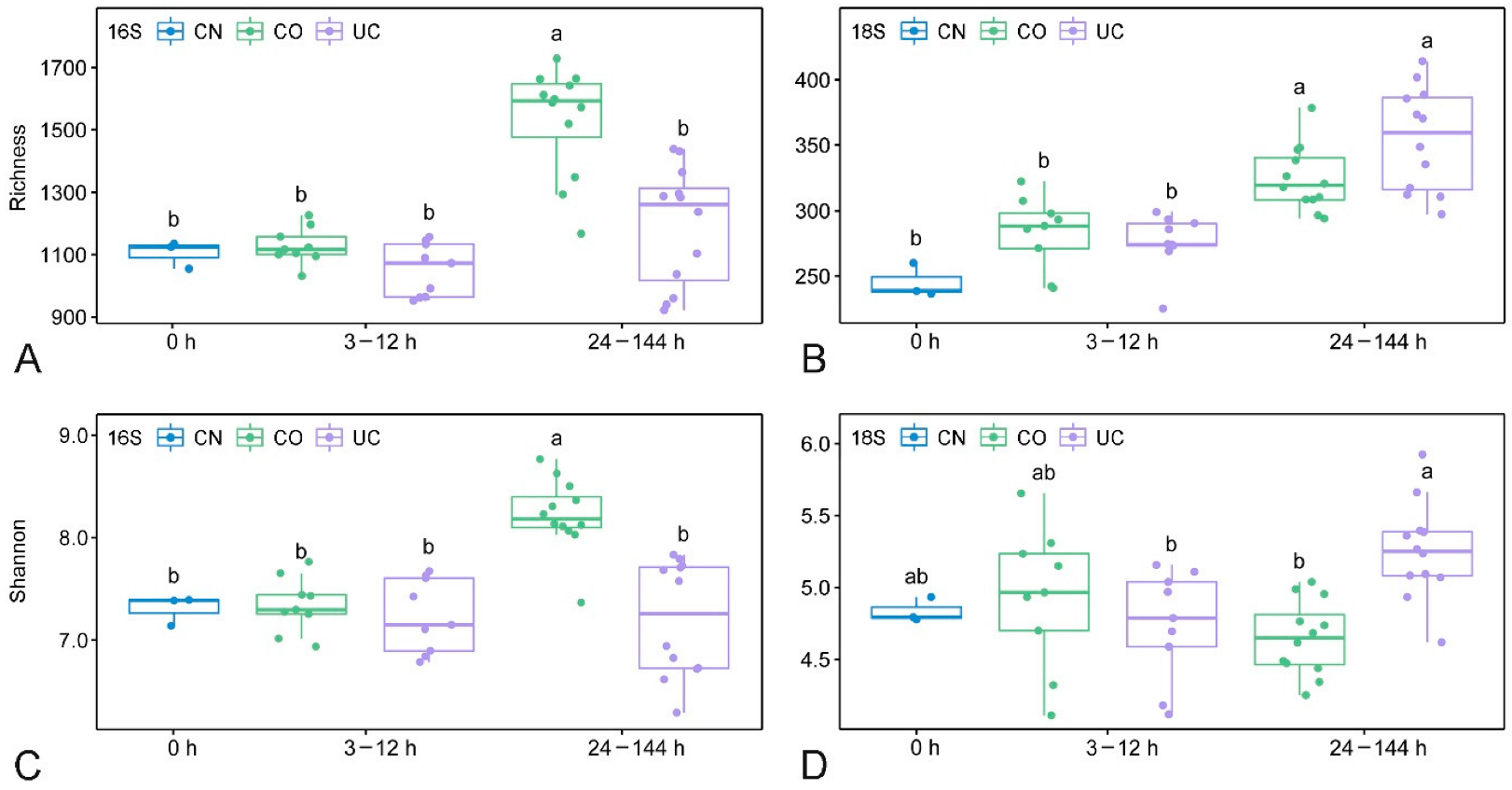
3.2. Effect of Sample Storage on Microbial Diversity
3.3. Effect of Sample Storage on Microbial Networks
3.4. Effect of Sample Storage on the Microbial Community Assembly
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Cavicchioli, R.; Ripple, W.J.; Timmis, K.N.; Azam, F.; Bakken, L.R.; Baylis, M.; Behrenfeld, M.J.; Boetius, A.; Boyd, P.W.; Classen, A.T.; et al. Scientists’ warning to humanity: Microorganisms and climate change. Nat. Rev. Microbiol. 2019, 17, 569–586. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Delgado-Baquerizo, M.; Oliverio, A.M.; Brewer, T.E.; Benavent-González, A.; Eldridge, D.J.; Bardgett, R.D.; Maestre, F.T.; Singh, B.K.; Fierer, N. A global atlas of the dominant bacteria found in soil. Science 2018, 359, 320–325. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yang, Z.F.; Xiao, X.; Zhang, Y. Microbial diversity of sediments from an inactive hydrothermal vent field, Southwest Indian Ridge. Mar. Life Sci. Technol. 2020, 2, 73–86. [Google Scholar] [CrossRef] [Green Version]
- Li, X.H.; Huang, J.; Filker, S.; Stoeck, T.; Bi, Y.H.; Yu, Y.H.; Song, W.B. Spatio-temporal patterns of zooplankton in a main-stem dam affected tributary: A case study in the Xiangxi River of the Three Gorges Reservoir, China. Sci. China Life Sci. 2019, 62, 1058–1069. [Google Scholar] [CrossRef]
- Liu, Q.; Zhao, Q.N.; McMinn, A.; Yang, E.J.; Jiang, Y. Planktonic microbial eukaryotes in polar surface waters: Recent advances in high-throughput sequencing. Mar. Life Sci. Technol. 2021, 3, 94–102. [Google Scholar] [CrossRef]
- Wang, B.H.; Zheng, X.F.; Zhang, H.J.; Yu, X.L.; Lian, Y.L.; Yang, X.Q.; Yu, H.; Hu, R.W.; He, Z.L.; Xiao, F.S.; et al. Metagenomic insights into the effects of submerged plants on functional potential of microbial communities in wetland sediments. Mar. Life Sci. Technol. 2021, 3, 405–415. [Google Scholar] [CrossRef]
- Eisenhofer, R.; Minich, J.J.; Marotz, C.; Cooper, A.; Knight, R.; Weyrich, L.S. Contamination in low microbial biomass microbiome studies: Issues and recommendations. Trends Microbiol. 2019, 27, 105–117. [Google Scholar] [CrossRef]
- Knight, R.; Vrbanac, A.; Taylor, B.C.; Aksenov, A.; Callewaert, C.; Debelius, J.; Gonzalez, A.; Kosciolek, T.; McCall, L.I.; McDonald, D.; et al. Best practices for analysing microbiomes. Nat. Rev. Microbiol. 2018, 16, 410–422. [Google Scholar] [CrossRef] [Green Version]
- Małecka-Adamowicz, M.; Kubera, Ł. Patterns of structural and functional bacterioplankton metacommunity along a river under anthropogenic pressure. Sustainability 2021, 13, 11518. [Google Scholar] [CrossRef]
- Pollock, J.; Glendinning, L.; Wisedchanwet, T.; Watson, M. The madness of microbiome: Attempting to find consensus “best practice” for 16S microbiome studies. Appl. Environ. Microbiol. 2018, 84, e02627-17. [Google Scholar] [CrossRef] [Green Version]
- Calvo-Díaz, A.; Díaz-Pérez, L.; Suárez, L.Á.; Morán, X.A.G.; Teira, E.; Marañón, E. Decrease in the autotrophic-to-heterotrophic biomass ratio of picoplankton in oligotrophic marine waters due to bottle enclosure. Appl. Environ. Microbiol. 2011, 77, 5739–5746. [Google Scholar] [CrossRef] [Green Version]
- Countway, P.D.; Gast, R.J.; Savai, P.; Caron, D.A. Protistan diversity estimates based on 18S rDNA from seawater incubations in the Western North Atlantic. J. Eukaryot. Microbiol. 2005, 52, 95–106. [Google Scholar] [CrossRef]
- Kim, D.Y.; Countway, P.D.; Gast, R.J.; Caron, D.A. Rapid shifts in the structure and composition of a protistan assemblage during bottle incubations affect estimates of total protistan species richness. Aquat. Microb. Ecol. 2011, 62, 383–398. [Google Scholar] [CrossRef]
- Lauber, C.L.; Zhou, N.; Gordon, J.I.; Knight, R.; Fierer, N. Effect of storage conditions on the assessment of bacterial community structure in soil and human-associated samples. FEMS Microbiol. Lett. 2010, 307, 80–86. [Google Scholar] [CrossRef] [Green Version]
- Roesch, L.F.W.; Casella, G.; Simell, O.; Krischer, J.; Wasserfall, C.H.; Schatz, D.; Atkinson, M.A.; Neu, J.; Triplett, E.W. Influence of fecal sample storage on bacterial community diversity. Open Microbiol. J. 2009, 3, 40–46. [Google Scholar] [CrossRef] [Green Version]
- Amir, A.; McDonald, D.; Navas-Molina, J.A.; Debelius, J.; Morton, J.T.; Hyde, E.; Robbins-Pianka, A.; Knight, R. Correcting for microbial blooms in fecal samples during room-temperature shipping. mSystems 2017, 2, e00199-16. [Google Scholar] [CrossRef] [Green Version]
- Beardsley, C.; Moss, S.M.; Azam, F. Effect of storage temperature on prokaryotic cell counts and community composition analysis from fixed and filtered seawater samples. Helgol. Mar. Res. 2008, 62, 123–127. [Google Scholar] [CrossRef] [Green Version]
- Lee, Y.B.; Lorenz, N.; Dick, L.K.; Dick, R.P. Cold storage and pretreatment incubation effects on soil microbial properties. Soil Sci. Soc. Am. J. 2007, 71, 1299–1305. [Google Scholar] [CrossRef]
- Comte, J.; Monier, A.; Crevecoeur, S.; Lovejoy, C.; Vincent, W.F. Microbial biogeography of permafrost thaw ponds across the changing northern landscape. Ecography 2016, 39, 609–618. [Google Scholar] [CrossRef]
- Datry, T.; Malard, F.; Gibert, J. Dynamics of solutes and dissolved oxygen in shallow urban groundwater below a stormwater infiltration basin. Sci. Total Environ. 2004, 329, 215–229. [Google Scholar] [CrossRef]
- Gieskes, W.W.C.; Kraay, G.W.; Baars, M.A. Current 14C methods for measuring primary production: Gross underestimates in oceanic waters. Neth. J. Sea Res. 1979, 13, 58–78. [Google Scholar] [CrossRef]
- Massana, R.; Pedrόs-Aliό, C.; Casamayor, E.O.; Gasol, J.M. Changes in marine bacterioplankton phylogenetic composition during incubations designed to measure biogeochemically significant parameters. Limnol. Oceanogr. 2001, 46, 1181–1188. [Google Scholar] [CrossRef] [Green Version]
- Weber, F.; del Campo, J.; Wylezich, C.; Massana, R.; Jürgens, K. Unveiling trophic functions of uncultured protist taxa by incubation experiments in the brackish Baltic Sea. PLoS ONE 2012, 7, e41970. [Google Scholar]
- Bower, S.M.; Carnegie, R.B.; Goh, B.; Jones, S.R.M.; Lowe, G.J.; Mak, M.W.S. Preferential PCR amplification of parasitic protistan small subunit rDNA from metazoan tissues. J. Eukaryot. Microbiol. 2004, 51, 325–332. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Lozupone, C.A.; Turnbaugh, P.J.; Fierer, N.; Knight, R. Global patterns of 16S rRNA diversity at a depth of millions of sequences per sample. Proc. Natl. Acad. Sci. USA 2011, 108 (Suppl. 1), 4516–4522. [Google Scholar] [CrossRef] [Green Version]
- Simon, M.; Jardillier, L.; Deschamps, P.; Moreira, D.; Restoux, G.; Bertolino, P.; López-García, P. Complex communities of small protists and unexpected occurrence of typical marine lineages in shallow freshwater systems. Environ. Microbiol. 2015, 17, 3610–3627. [Google Scholar] [CrossRef] [Green Version]
- Magoč, T.; Salzberg, S.L. FLASH: Fast length adjustment of short reads to improve genome assemblies. Bioinformatics 2011, 27, 2957–2963. [Google Scholar] [CrossRef]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [Green Version]
- Caporaso, J.G.; Kuczynski, J.; Stombaugh, J.; Bittinger, K.; Bushman, F.D.; Costello, E.K.; Fierer, N.; Peña, A.G.; Goodrich, J.K.; Gordon, J. QIIME allows analysis of high-throughput community sequencing data. Nat. Methods 2010, 7, 335–336. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C.; Haas, B.J.; Clemente, J.C.; Quince, C.; Knight, R. UCHIME improves sensitivity and speed of chimera detection. Bioinformatics 2011, 27, 2194–2200. [Google Scholar] [CrossRef] [Green Version]
- Edgar, R.C. UPARSE: Highly accurate OTU sequences from microbial amplicon reads. Nat. Methods 2013, 10, 996–998. [Google Scholar] [CrossRef]
- Guillou, L.; Bachar, D.; Audic, S.; Bass, D.; Berney, C.; Bittner, L.; Boutte, C.; Burgaud, G.; De Vargas, C.; Decelle, J.; et al. The Protist Ribosomal Reference database (PR2): A catalog of unicellular eukaryote small sub-unit rRNA sequences with curated taxonomy. Nucleic Acids Res. 2012, 41, D597–D604. [Google Scholar] [CrossRef] [Green Version]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef] [Green Version]
- Deng, Y.; Jiang, Y.H.; Yang, Y.F.; He, Z.L.; Luo, F.; Zhou, J.Z. Molecular ecological network analyses. BMC Bioinform. 2012, 13, 113. [Google Scholar] [CrossRef] [Green Version]
- Lupatini, M.; Suleiman, A.K.A.; Jacques, R.J.S.; Antoniolli, Z.I.; de Siqueira Ferreira, A.; Kuramae, E.E.; Roesch, L.F.W. Network topology reveals high connectance levels and few key microbial genera within soils. Front. Environ. Sci. 2014, 2, 10. [Google Scholar] [CrossRef] [Green Version]
- Steele, J.A.; Countway, P.D.; Xia, L.; Vigil, P.D.; Beman, J.M.; Kim, D.Y.; Chow, C.E.T.; Sachdeva, R.; Jones, A.C.; Schwalbach, M.S.; et al. Marine bacterial, archaeal and protistan association networks reveal ecological linkages. ISME J. 2011, 5, 1414–1425. [Google Scholar] [CrossRef]
- Gephi: An Open Source Software for Exploring and Manipulating Networks. Available online: https://ojs.aaai.org/index.php/ICWSM/article/view/13937 (accessed on 15 January 2021).
- Roger, G.; Luís, A.N.A. Functional cartography of complex metabolic networks. Nature 2005, 433, 895–900. [Google Scholar]
- Olesen, J.M.; Bascompte, J.; Dupont, Y.L.; Jordano, P. The modularity of pollination networks. Proc. Natl. Acad. Sci. USA 2007, 104, 19891–19896. [Google Scholar] [CrossRef] [Green Version]
- Sloan, W.T.; Lunn, M.; Woodcock, S.; Head, I.M.; Nee, S.; Curtis, T.P. Quantifying the roles of immigration and chance in shaping prokaryote community structure. Environ. Microbiol. 2006, 8, 732–740. [Google Scholar] [CrossRef]
- Zhu, C.Y.; Liu, W.W.; Li, X.; Xu, Y.; El-Serehy, H.A.; Al-Farraj, S.A.; Ma, H.; Stoeck, T.; Yi, Z.Z. High salinity gradients and intermediate spatial scales shaped similar biogeographical and co-occurrence patterns of microeukaryotes in a tropical freshwater-saltwater ecosystem. Environ. Microbiol. 2021, 23, 4778–4796. [Google Scholar] [CrossRef]
- Nyirabuhoro, P.; Liu, M.; Xiao, P.; Liu, L.M.; Yu, Z.; Wang, L.; Yang, J. Seasonal variability of conditionally rare taxa in the water column bacterioplankton community of subtropical reservoirs in China. Microb. Ecol. 2020, 80, 14–26. [Google Scholar] [CrossRef]
- Stone, L.; Roberts, A. The checkerboard score and species distributions. Oecologia 1990, 85, 74–79. [Google Scholar] [CrossRef]
- Connor, E.F.; Simberloff, D. The assembly of species communities: Chance or competition? Ecology 1979, 60, 1132–1140. [Google Scholar] [CrossRef]
- Gotelli, N.J. Null model analysis of species co-occurrence patterns. Ecology 2000, 81, 2606–2621. [Google Scholar] [CrossRef]
- Mo, Y.Y.; Peng, F.; Gao, X.F.; Xiao, P.; Logares, R.; Jeppesen, E.; Ren, K.; Xue, Y.Y.; Yang, J. Low shifts in salinity determined assembly processes and network stability of microeukaryotic plankton communities in a subtropical urban reservoir. Microbiome 2021, 9, 128. [Google Scholar] [CrossRef]
- Satdichanh, M.; Ma, H.X.; Yan, K.; Dossa, G.G.O.; Winowiecki, L.; Vågen, T.G.; Gassner, A.; Xu, J.C.; Harrison, R.D. Phylogenetic diversity correlated with above-ground biomass production during forest succession: Evidence from tropical forests in Southeast Asia. J. Ecol. 2019, 107, 1419–1432. [Google Scholar] [CrossRef]
- Kohli, B.A.; Terry, R.C.; Rowe, R.J. A trait-based framework for discerning drivers of species co-occurrence across heterogeneous landscapes. Ecography 2018, 41, 1921–1933. [Google Scholar] [CrossRef] [Green Version]
- Gotelli, N.J.; McCabe, D.J. Species co-occurrence: A meta-analysis of J. M. Diamond’s assembly rules model. Ecology 2002, 83, 2091–2096. [Google Scholar] [CrossRef]
- Villanueva, R.A.M.; Chen, Z.J. ggplot2: Elegant graphics for data analysis (2nd ed.). Meas. Interdiscip. Res. Perspect. 2019, 17, 160–167. [Google Scholar] [CrossRef]
- Breslow, N. A generalized Kruskal-Wallis test for comparing K samples subject to unequal patterns of censorship. Biometrika 1970, 57, 579–594. [Google Scholar] [CrossRef]
- Vegan: Community Ecology Package Version 2.0-10. Available online: https://CRAN.R-project.org/package=vegan (accessed on 28 December 2021).
- Parks, D.H.; Tyson, G.W.; Hugenholtz, P.; Beiko, R.G. STAMP: Statistical analysis of taxonomic and functional profiles. Bioinformatics 2014, 30, 3123–3124. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lennon, J.T.; Jones, S.E. Microbial seed banks: The ecological and evolutionary implications of dormancy. Nat. Rev. Microbiol. 2011, 9, 119–130. [Google Scholar] [CrossRef] [PubMed]
- Rubin, B.E.R.; Gibbons, S.M.; Kennedy, S.; Hampton-Marcell, J.; Owens, S.; Gilbert, J.A. Investigating the impact of storage conditions on microbial community composition in soil samples. PLoS ONE 2013, 8, e70460. [Google Scholar] [CrossRef] [PubMed]
- Bartolomé, M.C.; D’Ors, A.; Sánchez-Fortún, S. Toxic effects induced by salt stress on selected freshwater prokaryotic and eukaryotic microalgal species. Ecotoxicology 2009, 18, 174–179. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.H.; Xie, Y.W.; Jeppe, K.; Long, S.; Pettigrove, V.; Zhang, X.W. Sensitive community responses of microbiota to copper in sediment toxicity test. Environ. Toxicol. Chem. 2018, 37, 599–608. [Google Scholar] [CrossRef] [PubMed]
- Fogel, G.B.; Collins, C.R.; Li, J.; Brunk, C.F. Prokaryotic genome size and SSU rDNA copy number: Estimation of microbial relative abundance from a mixed population. Microb. Ecol. 1999, 38, 93–113. [Google Scholar] [CrossRef]
- Polz, M.F.; Cavanaugh, C.M. Bias in template-to-product ratios in multitemplate PCR. Appl. Environ. Microbiol. 1998, 64, 3724–3730. [Google Scholar] [CrossRef] [Green Version]
- Schütte, U.M.E.; Abdo, Z.; Bent, S.J.; Shyu, C.; Williams, C.J.; Pierson, J.D.; Forney, L.J. Advances in the use of terminal restriction fragment length polymorphism (T-RFLP) analysis of 16S rRNA genes to characterize microbial communities. Appl. Microbiol. Biotechnol. 2008, 80, 365–380. [Google Scholar] [CrossRef]
- Yannarell, A.C.; Triplett, E.W. Geographic and environmental sources of variation in lake bacterial community composition. Appl. Environ. Microbiol. 2005, 71, 227–239. [Google Scholar] [CrossRef] [Green Version]
- Cui, H.L.; Duan, G.L.; Zhang, H.M.; Cheng, W.D.; Zhu, Y.G. Microbiota in non-flooded and flooded rice culms. FEMS Microbiol. Ecol. 2019, 95, fiz036. [Google Scholar] [CrossRef] [Green Version]
- Pomeroy, L.R.; Sheldon, J.E.; Sheldon, W.M. Changes in bacterial numbers and leucine assimilation during estimations of microbial respiratory rates in seawater by the precision winkler method. Appl. Environ. Microbiol. 1994, 60, 328–332. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ulseth, A.J.; Hall, R.O.; Canadell, M.B.; Madinger, H.L.; Niayifar, A.; Battin, T.J. Distinct air-water gas exchange regimes in low-and high-energy streams. Nat. Geosci. 2019, 12, 259–263. [Google Scholar] [CrossRef]
- Fan, L.M.; Hu, G.D.; Qiu, L.P.; Meng, S.L.; Wu, W.; Zheng, Y.; Song, C.; Li, D.D.; Chen, J.Z. Variations in bacterioplankton communities in aquaculture ponds and the influencing factors during the peak period of culture. Environ. Pollut. 2020, 258, 113656. [Google Scholar] [CrossRef] [PubMed]
- Luo, Z.X.; Li, S.J.; Hou, K.; Ji, G.D. Spatial and seasonal bacterioplankton community dynamics in the main channel of the Middle Route of South-to-North Water Diversion Project. Res. Microbiol. 2019, 170, 24–34. [Google Scholar] [CrossRef]
- Fukunaga, Y.; Kurahashi, M.; Sakiyama, Y.; Ohuchi, M.; Yokota, A.; Harayama, S. Phycisphaera mikurensis gen. nov., sp. nov., isolated from a marine alga, and proposal of Phycisphaeraceae fam. nov., Phycisphaerales ord. nov. and Phycisphaerae classis nov. in the phylum Planctomycetes. J. Gen. Appl. Microbiol. 2009, 55, 267–275. [Google Scholar] [CrossRef] [Green Version]
- Crump, B.C.; Peranteau, C.; Beckingham, B.; Cornwell, J.C. Respiratory succession and community succession of bacterioplankton in seasonally anoxic estuarine waters. Appl. Environ. Microbiol. 2007, 73, 6802–6810. [Google Scholar] [CrossRef] [Green Version]
- Yang, L.; Chen, J.; Qin, S.; Zeng, M.; Jiang, Y.G.; Hu, L.; Xiao, P.; Hao, W.L.; Hu, Z.L.; Lei, A.P. Growth and lipid accumulation by different nutrients in the microalga Chlamydomonas reinhardtii. Biotechnol. Biofuels 2018, 11, 40. [Google Scholar] [CrossRef]
- Bojanić, N.; Vidjak, O.; Šolić, M.; Krstulović, N.; Brautović, I.; Matijević, S.; Kušpilić, G.; Šestanović, S.; Gladan, Ž.N.; Marasović, I. Community structure and seasonal dynamics of tintinnid ciliates in Kaštela Bay (middle Adriatic Sea). J. Plankton Res. 2012, 34, 510–530. [Google Scholar] [CrossRef]
- Almanza, V.; Pedreros, P.; Laughinghouse, H.D., IV; Félez, J.; Parra, O.; Azócar, M.; Urrutia, R. Association between trophic state, watershed use, and blooms of cyanobacteria in south-central Chile. Limnologica 2019, 75, 30–41. [Google Scholar] [CrossRef]
- Saros, J.E.; Anderson, N.J. The ecology of the planktonic diatom Cyclotella and its implications for global environmental change studies. Biol. Rev. 2015, 90, 522–541. [Google Scholar] [CrossRef]
- van Hannen, E.J.; Mooij, W.; van Agterveld, M.P.; Gons, H.J.; Laanbroek, H.J. Detritus-dependent development of the microbial community in an experimental system: Qualitative analysis by denaturing gradient gel electrophoresis. Appl. Environ. Microbiol. 1999, 65, 2478–2484. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paerl, H.W.; Xu, H.; McCarthy, M.J.; Zhu, G.W.; Qin, B.Q.; Li, Y.P.; Gardner, W.S. Controlling harmful cyanobacterial blooms in a hyper-eutrophic lake (Lake Taihu, China): The need for a dual nutrient (N & P) management strategy. Water Res. 2011, 45, 1973–1983. [Google Scholar] [PubMed]
- Zhao, D.Y.; Shen, F.; Zeng, J.; Huang, R.; Yu, Z.B.; Wu, Q.L. Network analysis reveals seasonal variation of co-occurrence correlations between Cyanobacteria and other bacterioplankton. Sci. Total Environ. 2016, 573, 817–825. [Google Scholar] [CrossRef] [PubMed]
- He, Q.; Wang, S.; Hou, W.G.; Feng, K.; Li, F.R.; Hai, W.M.; Zhang, Y.D.; Sun, Y.X.; Deng, Y. Temperature and microbial interactions drive the deterministic assembly processes in sediments of hot springs. Sci. Total Environ. 2021, 772, 145465. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Groups | ANOSIM | PERMANOVA | MRPP | |||||
|---|---|---|---|---|---|---|---|---|
| R | p | R2 | p | Delta | p | |||
| Prokaryotes | 3–12 h | CO vs. UC | −0.085 | 0.900 | 0.024 | 0.861 | 0.316 | 0.924 |
| CO vs. CN | −0.066 | 0.578 | 0.118 | 0.216 | 0.302 | 0.097 | ||
| UC vs. CN | −0.126 | 0.746 | 0.103 | 0.324 | 0.315 | 0.155 | ||
| 24–144 h | CO vs. UC | 0.571 | 0.001 | 0.245 | 0.001 | 0.504 | 0.001 | |
| CO vs. CN | 0.670 | 0.004 | 0.320 | 0.004 | 0.447 | 0.005 | ||
| UC vs. CN | 0.883 | 0.001 | 0.347 | 0.002 | 0.535 | 0.002 | ||
| Eukaryotes | 3–12 h | CO vs. UC | 0.048 | 0.245 | 0.066 | 0.330 | 0.302 | 0.256 |
| CO vs. CN | 0.464 | 0.011 | 0.276 | 0.009 | 0.311 | 0.005 | ||
| UC vs. CN | 0.479 | 0.006 | 0.281 | 0.006 | 0.325 | 0.006 | ||
| 24–144 h | CO vs. UC | 0.175 | 0.030 | 0.123 | 0.024 | 0.540 | 0.014 | |
| CO vs. CN | 0.433 | 0.013 | 0.299 | 0.008 | 0.510 | 0.001 | ||
| UC vs. CN | 0.367 | 0.038 | 0.265 | 0.003 | 0.571 | 0.005 | ||
| Network Topological Properties | 3–12 h | 24–144 h | ||
|---|---|---|---|---|
| CO | UC | CO | UC | |
| Nodes | 667 | 639 | 596 | 494 |
| Edges | 1435 | 1120 | 2372 | 1883 |
| R2 of power law | 0.918 | 0.905 | 0.780 | 0.799 |
| Average clustering coefficient (avgCC) | 0.204 | 0.204 | 0.165 | 0.150 |
| Average connectivity (avgK) | 4.303 | 3.505 | 7.960 | 7.623 |
| Average geodesic distance (GD) | 7.616 | 7.100 | 5.625 | 4.960 |
| Geodesic efficiency I | 0.185 | 0.181 | 0.237 | 0.264 |
| Positive edges | 0.815 | 0.828 | 0.536 | 0.492 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wang, Y.; Li, X.; Chi, Y.; Song, W.; Yan, Q.; Huang, J. Changes of the Freshwater Microbial Community Structure and Assembly Processes during Different Sample Storage Conditions. Microorganisms 2022, 10, 1176. https://doi.org/10.3390/microorganisms10061176
Wang Y, Li X, Chi Y, Song W, Yan Q, Huang J. Changes of the Freshwater Microbial Community Structure and Assembly Processes during Different Sample Storage Conditions. Microorganisms. 2022; 10(6):1176. https://doi.org/10.3390/microorganisms10061176
Chicago/Turabian StyleWang, Yunfeng, Xinghao Li, Yong Chi, Weibo Song, Qingyun Yan, and Jie Huang. 2022. "Changes of the Freshwater Microbial Community Structure and Assembly Processes during Different Sample Storage Conditions" Microorganisms 10, no. 6: 1176. https://doi.org/10.3390/microorganisms10061176
APA StyleWang, Y., Li, X., Chi, Y., Song, W., Yan, Q., & Huang, J. (2022). Changes of the Freshwater Microbial Community Structure and Assembly Processes during Different Sample Storage Conditions. Microorganisms, 10(6), 1176. https://doi.org/10.3390/microorganisms10061176